
Abstract
Pancreatic cancer exhibits the worst prognostic outcome among human cancers. Recently, we have described that depletion of RUNX2 enhances gemcitabine (GEM) sensitivity of p53-deficient pancreatic cancer AsPC-1 cells through the activation of TAp63-mediated cell death pathway. These findings raised a question whether RUNX2 silencing could also improve GEM efficacy on pancreatic cancer cells bearing p53 mutation. In the present study, we have extended our study to p53-mutated pancreatic cancer MiaPaCa-2 cells. Based on our current results, MiaPaCa-2 cells were much more resistant to GEM as compared with p53-proficient pancreatic cancer SW1990 cells, and there existed a clear inverse relationship between the expression levels of TAp73 and RUNX2 in response to GEM. Forced expression of TAp73α in MiaPaCa-2 cells significantly promoted cell cycle arrest and/or cell death, indicating that a large amount of TAp73 might induce cell death even in the presence of mutant p53. Consistent with this notion, overexpression of TAp73α stimulated luciferase activity driven by p53/TAp73-target gene promoters in MiaPaCa-2 cells. Similar to AsPC-1 cells, small interfering RNA-mediated knockdown of RUNX2 remarkably enhanced GEM sensitivity of MiPaCa-2 cells. Under our experimental conditions, TAp73 further accumulated in RUNX2-depleted MiaPaCa-2 cells exposed to GEM relative to GEM-treated non-silencing control cells. As expected, silencing of p73 reduced GEM sensitivity of MiPaCa-2 cells. Moreover, GEM-mediated Tyr phosphorylation level of TAp73 was much more elevated in RUNX2-depleted MiaPaCa-2 cells. Collectively, our present findings strongly suggest that knockdown of RUNX2 contributes to a prominent enhancement of GEM sensitivity of p53-mutated pancreatic cancer cells through the activation of TAp73-mediated cell death pathway, and also provides a promising strategy for the treatment of patients with pancreatic cancer bearing p53 mutation.
Similar content being viewed by others


Introduction
Human pancreatic cancer is a serious disease with 5-year survival rate of <5% and its incidence is increasing annually.1, 2 In this connection, pancreatic cancer is expected to be the second leading cause of cancer-related death by 2030.3 Although surgical resection is the preferred treatment for pancreatic cancer patients and it has been significantly improved, most cases are found at a late advanced unresectable stage. Nucleoside analog termed gemcitabine (GEM) has been used as a first-line standard chemotherapy for pancreatic cancer patients, however its efficacy is extremely limited.4, 5 To date, no validated biomarker is available that can allow the prediction of the prognostic outcome of the patients and also the treatment efficacy in pancreatic cancer. Therefore, a new attractive molecular target(s) for the early detection and the treatment of pancreatic cancer patients should be urgently required.
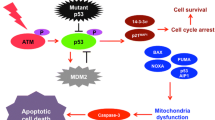
It has been well-established that tumor suppresser p53 has a critical role in tumor prevention.6, 7 Accumulating evidence strongly indicates that p53 is a nuclear transcription factor and transactivates numerous its target genes implicated in the induction of cell cycle arrest, cellular senescence and/or cell death in response to the exogenous as well as the endogenous stresses such as DNA damage.8, 9 Upon DNA damage, p53 is induced to accumulate in cell nucleus through the sequential post-translational modifications such as phosphorylation as well as acetylation and exerts its pro-apoptotic function.10 The amount of p53 is largely regulated at protein level. Under the physiological condition, p53 is kept at extremely low level through the interaction with a p53-specific E3 protein ubiquitin ligase MDM2, which subsequently targets p53 for ubiquitin-dependent degradation via the proteasome.11 When p53/MDM2 interaction is disrupted, p53 is rapidly stabilized in response to DNA damage.9 Recently, the additional E3 ubiquitin protein ligases including Pirh2, Trim24, COP1 and CHIP, which participate in the degradation of p53, have been identified.12, 13
Meanwhile, the extensive mutation search demonstrated that p53 is frequently mutated in a variety of human cancer tissues.14 Over 90% of mutations are localized within the genomic region encoding its core sequence-specific DNA-binding domain, suggesting that the majority of p53 mutants lack the sequence-specific transactivation ability and pro-apoptotic function.15 Of note, p53 is found to be mutated or lost in ~75% of pancreatic cancer.16 In contrast to the short-lived wild-type p53, mutant p53 has a longer half-life.17, 18 An increased stability of mutant p53 might be due to the interaction of mutant p53 with molecular chaperone HSP90, which has been shown to prevent mutant p53 degradation and thereby promoting its accumulation.19 In addition, Zheng et al.20 found that MDM2 isoforms prohibit MDM2-mediated degradation of mutant p53 and prolong its half-life. As described, 21 mutant p53 acts as a dominant-negative inhibitor against wild-type p53 and acquires pro-oncogenic potential. Indeed, mutant p53 is implicated in metastasis, resistance to anti-cancer drug and genomic instability.22
A small p53 family is composed of p53, p73 and p63. In a sharp contrast to p53, p73 and p63 are rarely mutated in human cancers.23 p73 and p63 encode two major isoforms such as transcriptionally active TA isoforms (TAp73 and TAp63) and N-terminally truncated ΔN ones (ΔNp73 and ΔNp63).24, 25 TA and ΔN isoforms are produced by alternative splicing and alternative promoter usage, respectively. As expected from their structural similarity, TA isoforms have an ability to transactivate overlapping set of p53-target genes and a pro-apoptotic function. Like p53, TAp73 and TAp63 are induced in response to a certain DNA damage.26, 27 By contrast, ΔN isoforms lose N-terminal transactivation domain and thereby lacking sequence-specific transactivation ability as well as pro-apoptotic function. Intriguingly, ΔN isoforms and mutant p53 display a dominant-negative behavior toward TA isoforms, and then acquire pro-oncogenic potential.25 Previously, it has been shown that deregulated expression of ΔNp63 stimulates pro-oncogenic β-catenin signaling pathway.28 Recently, Dolloo et al.29 demonstrated that ΔNp73 is able to induce the expression of angiogenic VEGF-A under tumor-relevant hypoxic condition. These observations indicate that ΔN isoforms might have their own target genes involved in carcinogenesis.
RUNX family, which is composed of RUNX1, RUNX2 and RUNX3, is a sequence-specific transcription factor and each of these family members has a distinct biological function. For example, RUNX1 has been originally identified as a part of the chromosome translocation in acute myeloid leukemia and is involved in the establishment of the hematopoietic stem cells.30, 31, 32 In a sharp contrast to RUNX1, RUNX2 is absolutely required for the osteoblast differentiation and bone formation. As described,33, 34 RUNX2-deficient mice failed to form mineralized bone. For RUNX3, a growing body of evidence implies that RUNX3 is tightly linked to gastoric development and also acts as a tumor suppressor toward gastoric cancer.35, 36
Recently, we have found that depletion of RUNX2 in p53-proficient human osteosarcoma-derived U2OS cells enhances adriamycin sensitivity in a p53/TAp73-dependent manner.37, 38 In addition, we have also described that GEM sensitivity is significantly improved in RUNX2-depleted p53-deficient human pancreatic cancer AsPC-1 cells in a TAp63-dependent fashion.39 These findings are consistent with the previous and recent reports showing that the expression level of RUNX2 in a variety of human cancer tissues including pancreatic cancer is higher than that of their corresponding normal ones, and RUNX2 transactivates various target genes implicated in carcinogenesis, indicating that, in addition to osteogenesis, RUNX2 has an pro-oncogenic potential.40
In the present study, we have examined whether silencing of RUNX2 in p53-mutated pancreatic cancer MiaPaCa-2 cells could enhance their GEM sensitivity. As p53 mutant acts as a strong dominant-negative inhibitor against wild-type p53, TAp73 and TAp63, it is important to adequately address this issue for the improvement of GEM efficacy in the presence of mutant p53.
Results
p53-mutated human pancreatic cancer MiaPaCa-2 cells exhibit GEM-resistant phenotype as compared with p53-proficient human pancreatic cancer SW1990 cells
As mutant p53 acquires pro-oncogenic activity and also contributes at least in part to drug resistance of aggressive cancers, we have sought to examine GEM sensitivity of p53-mutated human pancreatic cancer MiaPaCa-2 cells and p53-proficient human pancreatic cancer SW1990 cells. For this purpose, both cells were treated with the indicated concentrations of GEM. Forty-eight hours after treatment, cells were observed under phase-contrast microscope, analyzed by flow cytometry and trypan blue exclusion assay. For SW1990 cells, number of the attached cells was remarkably decreased and cells clearly underwent cell death in response to GEM (Supplementary Figure S1). For MiaPaCa-2 cells, number of the adherent cells remained almost unchanged regardless of GEM exposure and GEM-mediated cell death took place but to a lesser degree (Figure 1). Therefore, these observations indicate that, like AsPC-1 cells,39 MiaPaCa-2 cells are much more resistant to GEM relative to SW1990 cells.

p53-mutated human pancreatic cancer MiaPaCa-2 cells are resistant to GEM. (a) Phase-contrast micrographs. MiaPaCa-2 cells were treated with the indicated concentrations of GEM. Forty-eight hours after treatment, representative pictures were taken. (b, c) MiaPaCa-2 cells undergo cell death following GEM exposure but to a lesser degree. MiaPaCa-2 cells were treated as in (a). Forty-eight hours after treatment, floating and attached cells were harvested and processed for FACS analysis (b) and trypan blue exclusion assay (c), respectively.
Inverse relationship between the expression levels of TAp73 and RUNX2 in response to GEM
To understand the mechanistic basis of GEM-resistant phenotype of MiaPaCa-2 cells, we have checked the expression patterns of pro-apoptotic p53 family members and their target gene products in response to GEM. In these experiments, the accumulation of γH2AX and the proteolytic cleavage of PARP following GEM exposure were examined by immunoblotting as a molecular marker for DNA damage and a mitochondrial dysfunction-mediated cell death, respectively.
As shown in Figure 2, GEM-mediated accumulation of γH2AX was clearly observed in MiaPaCa-2 cells, indicating that MiaPaCa-2 cells receive GEM-mediated DNA damage. However, GEM-induced decrease in the amount of the native PARP was barely detectable in MiaPaCa-2 cells. The expression level of transcriptionally active form of p73 termed TAp73 was increased in response to GEM (Supplementary Figure S2), which might be due to GEM-mediated up- and down-regulation of E2F-1 and RUNX2, respectively. According to the previous and recent observations,38, 41, 42, 43 E2F-1 and RUNX2 act as a transcriptional activator and a repressor of TAp73, respectively. Thus, there exists an inverse relationship between pro-apoptotic TAp73 and pro-oncogenic RUNX2 in MiaPaCa-2 cells following GEM exposure.

Inverse relationship between the expression levels of TAp73 and RUNX2 in response to GEM. MiaPaCa-2 cells were treated as in Figure 1a. Forty-eight hours after treatment, total RNA and cell lysates were prepared and analyzed by RT–PCR (a) and immunoblotting (b), respectively. GAPDH and actin were used as an internal control and a loading control, respectively.
On the other hand, GEM treatment resulted in a massive reduction in another transcriptionally active p53 family member TAp63. At present, its functional significance is unclear. Consistent with the above-mentioned findings showing that TAp73 is induced to accumulate after GEM exposure, p53 family-target gene transcription such as p21WAF1, 14-3-3σ, BAX, NOXA and PUMA, was promoted in the presence of GEM. Considering that GEM displays a lower cytotoxic effect on MiaPaCa-2 cells, it is highly likely that mutant p53, which acts as a dominant-negative inhibitors against TAp73,25 weakens its pro-apoptotic ability. Intriguingly, forced expression of TAp73α in MiaPaCa-2 cells markedly promoted cell cycle arrest and/or cell death as examined by colony-formation assay (Supplementary Figure S3) and enhanced the luciferase activities driven by human p21WAF1 as well as NOXA promoter in a dose-dependent manner as examined by luciferase reporter analysis (Supplementary Figure S4). Thus, our observations strongly indicate that the intracellular balance between the amounts of mutant p53 and TAp73 has a vital role in the regulation of cell fate determination following GEM exposure.
For SW1990 cells, wild-type p53 was induced to accumulate and its phosphorylation level at Ser-15 was elevated after GEM exposure in association with the accumulation of γH2AX (Supplementary Figure S5). As expected, the amount of native PARP was significantly reduced following GEM treatment, implying that GEM-mediated induction of DNA damage triggers p53-dependent cell death in SW1990 cells. Under our experimental conditions, GEM treatment caused an increase in the transcription levels of TAp73, TAp63, E2F-1 and RUNX2, whereas the amounts of their gene products markedly reduced following GEM exposure. At present, we do not know the molecular mechanisms behind this discrepancy.
Knockdown of RUNX2 enhances GEM sensitivity of MiaPaCa-2 cells
Recently, we have found that depletion of RUNX2 remarkably enhances the sensitivity to GEM of p53-deficient pancreatic cancer AsPC-1 cells.39 As shown above (Figure 2), the expression level of RUNX2 reduced in MiaPaCa-2 cells exposed to GEM. These findings prompted us to ask whether further down-regulation of RUNX2 could improve the cytotoxic effect of GEM on MiaPaCa-2 cells. To this end, MiaPaCa-2 cells were transfected with control small interfering RNA (siRNA) or with siRNA against RUNX2 (Smart pool siRNA mixture) followed by the incubation with or without GEM. Under our experimental conditions, siRNA-mediated knockdown of RUNX2 was successful as examined by RT–PCR and immunoblotting (Figures 3a and b). Phase-contrast micrographs clearly showed that depletion of RUNX2 results in a massive reduction in number of attached cells following GEM exposure relative to non-silencing control cells exposed to GEM (Figure 3c).

Depletion of RUNX2 stimulates GEM-mediated reduction in number of viable cells. (a, b) siRNA-mediated silencing of RUNX2. MiaPaCa-2 cells were transfected with control siRNA or with siRNA targeting RUNX2. Twenty-four hours after transfection, cells were exposed to GEM (at a final concentration of 10 μm) or left untreated. Forty-eight hours after treatment, total RNA and cell lysates were extracted and subjected to RT–PCR (a) and immunoblotting (b), respectively. (c) Representative pictures.
To further confirm the above-mentioned observations, non-depleted and RUNX2-depleted cells were treated with GEM or left untreated. Forty-eight hours after treatment, the attached and floating cells were collected and analyzed by flow cytometry. As shown in Figure 4a, around two-fold increase in number of cells with sub-G1 DNA content was detectable in RUNX2-depleted cells relative to non-depleted cells in the presence of GEM. Consistent with these observations, GEM-mediated DNA fragmentation was further stimulated in RUNX2-silencing cells as compared with that in non-silencing cells (Figure 4b). These results were also supported by the findings obtained from trypan blue exclusion and WST cell survival assays (Figures 5a and b). Similar results were also obtained in RUNX2-depleted MiaPaCa-2 cells mediated by two independent RUNX2 siRNAs (Supplementary Figure S6). Together, our present results imply that silencing of RUNX2 enhances GEM sensitivity of p53-mutated pancreatic cancer cells.

Knockdown of RUNX2 enhances GEM sensitivity of MiaPaCa-2 cells. (a) FACS analysis. MiaPaCa-2 cells were treated as in Figure 3a. Forty-eight hours after treatment, floating and adherent cells were collected and analyzed by flow cytometry. (b) DNA fragmentation. MiaPaCa-2 cells were treated as in Figure 3a. Forty-eight hours after treatment, floating and adherent cells were collected and their genomic DNA was prepared according to the standard procedure. Genomic DNA was analyzed by 0.7% agarose gel electrophoresis and stained with ethidium bromide.

Depletion of RUNX2 enhances the sensitivity to GEM of MiaPaCa-2 cells. (a, b) Effects of RUNX2 knockdown on MiaPaCa-2 cells in the presence or absence of GEM. For trypan blue exclusion assay, MiaPaCa-2 cells were treated as in Figure 3a. Forty-eight hours after treatment, floating and attached cells were harvested and processed for trypan blue exclusion assay (a). For WST cell survival assay, MiaPaCa-2 cells were treated as in Figure 3a. Forty-eight hours after treatment, cells were subjected to WST cell survival assay (b). Results are presented as mean±s.d.
Silencing of RUNX2 augments TAp73-mediated cell death pathway
To gain an insight into understanding the precise molecular mechanisms how knockdown of RUNX2 could improve the efficacy of GEM on MiaPaCa-2 cells, non-silencing control cells and RUNX2-depleted cells were cultured in the presence or absence of GEM. Forty-eight hours after treatment, total RNA and cell lysates were prepared and analyzed by RT–PCR and immunoblotting, respectively. As expected, further stimulation of GEM-mediated TAp73 transcriptional induction in RUNX2-depleted cells was observed as compared with non-depleted cells exposed to GEM (Figure 6a). Consistent with these observations, a massive induction of p53/TAp73-target genes such as p21WAF1, 14-3-3σ and NOXA was detected in RUNX2-silencing cells in response to GEM. Similarly, immunoblotting experiments demonstrated that GEM-induced accumulation of TAp73 and cleaved PARP is obviously augmented by RUNX2 knockdown (Figure 6b). As the further stimulation of GEM-mediated induction of E2F-1 was also detectable in RUNX2-silencing cells, it is likely that E2F-1 is tightly linked to the upregulation of TAp73 under our experimental conditions.

Silencing of RUNX2 further stimulates GEM-mediated induction of TAp73 and cleavage of PARP. (a, b) Expression of TAp73 and its target genes in RUNX2-depleted MiaPaCa-2 cells in response to GEM. MiaPaCa-2 cells were treated as in Figure 3a. Forty-eight hours after treatment, total RNA and cell lysates were prepared and analyzed by RT–PCR (a) and immunoblotting (b), respectively.
Knockdown of p73 reduces GEM sensitivity of MiaPaCa-2 cells
To confirm our hypothesis that RUNX2/TAp73 regulatory axis could have a pivotal role in the modulation of GEM sensitivity of MiaPaCa-2 cells, MiaPaCa-2 cells were transfected with control siRNA or with siRNA targeting p73. As shown in Figure 7a, the endogenous TAp73 was successfully knocked down. Twenty-four hours after transfection, cells were treated with GEM or left untreated. Forty-eight hours after treatment, the attached and floating cells were collected and then subjected to the flow cytometric analysis. As seen in Figure 7b, GEM-mediated increase in number of cells with sub-G1 DNA content was attenuated by depletion of p73. In addition, trypan blue exclusion assay revealed that silencing of p73 results in a significant decrease and an increase in number of dead and viable cells in response to GEM, respectively (Figures 7c and d). Thus, our observations indicate that TAp73 has a vital role in the regulation of GEM sensitivity of MiaPaCa-2 cells.

Depletion of p73 reduces GEM sensitivity of MiaPaCa-2 cells. (a) siRNA-mediated knockdown of p73. MiaPaCa-2 cells were transfected with control siRNA or with siRNA against p73. Forty-eight hours after transfection, total RNA and cell lysates were prepared and analyzed by RT–PCR and immunoblotting, respectively. (b) FACS analysis. p73-depleted and non-depleted MiaPaCa-2 cells were exposed to GEM or left untreated. Forty-eight hours after treatment, floating and attached cells were harvested and subjected to flow cytometric analysis. (c, d) Trypan blue exclusion assay. MiaPaCa-2 cells were treated as in (b). Forty-eight hours after GEM exposure, floating and attached cells were collected and subjected to trypan blue exclusion assay.
GEM-mediated Tyr phosphorylation of TAp73 is augmented by RUNX2 knockdown
The next question to be solved is how RUNX2 depletion could enhance the transcriptional and pro-apoptotic activities of TAp73 following GEM exposure. As described previously,44, 45, 46 TAp73 is stabilized and activated through c-Abl-mediated phosphorylation at Tyr-99 in response to DNA damage. These observations prompted us to examine whether GEM treatment could promote Tyr phosphorylation of TAp73 in MiaPaCa-2 cells. For this purpose, MiaPaCa-2 cells were treated with or without GEM for 48 h and then cell lysates were prepared. Cell lysates were then subjected to immunoprecipitation with anti-p73 antibody or with control IgG. The immunoprecipitates were finally analyzed by immunoblotting with PY20. As clearly shown in Figure 8a, TAp73 was induced to be phosphorylated at a certain Tyr residue(s) in response to GEM.

GEM-mediated Tyr phosphorylation of TAp73 is further stimulated in RUNX2-depleted MiaPaCa-2 cells. (a) GEM-mediated Tyr phosphorylation of TAp73. MiaPaCa-2 cells were treated with or without GEM. Forty-eight hours after treatment, cell lysates were prepared and immunoprecipitated with anti-p73 antibody or with normal rabbit serum (NRS). The immunoprecipitates were analyzed by immunoblotting with PY20. (b) GEM-mediated Tyr phosphorylation of TAp73 is augmented in RUNX2-depleted cells. MiaPaCa-2 cells were transfected with control siRNA or with siRNA against RUNX2. Twenty-four hours after transfection, cells were exposed to GEM for 48 h. After treatment, cell lysates were analyzed by co-immunoprecipitation experiments as in (a).
Subsequently, we sought to assess whether depletion of RUNX2 could affect GEM-mediated Tyr phosphorylation of TAp73. Immunoprecipitation/immunoblotting experiments demonstrated that GEM-mediated Tyr phosphorylation level of TAp73 remarkably elevates in RUNX2-depleted MiaPaCa-2 cells relative to non-depleted MiaPaCa-2 cells (Figure 8b). Thus, these results suggest that RUNX2 attenuates GEM-dependent phosphorylation of TAp73 at Tyr residues, and thereby suppressing its pro-apoptotic activity.
Discussion
Our recent findings strongly indicate that depletion of RUNX2 improves the anti-cancer drug sensitivity of p53-proficient osteosarcoma U2OS cells as well as p53-deficient pancreatic cancer AsPC-1 cells in a p53 family-dependent manner.37, 38, 39 As described,25 mutant p53 exhibits a strong dominant-negative behavior against wild-type p53, TAp73 and TAp63, raising a question whether knockdown of RUNX2 could also enhance the anti-cancer drug sensitivity of cancerous cells bearing p53 mutation.
In the present study, we have found that silencing of RUNX2 contributes to a significant enhancement of GEM sensitivity of p53-mutated pancreatic cancer MiaPaCa-2 cells through the augmentation of TAp73-mediated cell death pathway. As depletion of RUNX2 improved adriamycin- and GEM sensitivity of U2OS cells and AsPC-1 cells, respectively,37, 38, 39 it is likely that knockdown of RUNX2 has a vital role in the improvement of chemo-sensitivity of cancer cells regardless of their p53 status.
Based on our current observations, MiaPaCa-2 cells were much more resistant to GEM as compared with p53-proficient SW1990 cells, which might be due to the presence of mutant p53. As described,21, 24, 25 mutant p53 displays a dominant-negative behavior against pro-apoptotic wild-type p53, TAp73 and TAp63. According to our results, the expression level of TAp73 was induced in MiaPaCa-2 cells exposed to GEM, however MiaPaCa-2 cells displayed a GEM-resistant phenotype. Therefore, it is possible that pro-apoptotic activity of TAp73 is weakened in the presence of a large amount of mutant p53 stably expressed in MiaPaCa-2 cells. For TAp63, its amount was markedly reduced at mRNA and protein level in GEM-exposed MiaPaCa-2 cells. At present, the precise molecular mechanisms behind GEM-mediated downregulation of TAp63 remain unknown. As MiaPaCa-2 cells underwent cell death in response to GEM but to a lesser degree, TAp63 might not contribute to GEM-mediated cell death observed in MiaPaCa-2 cells. Thus, it is suggestive that GEM sensitivity of MiaPaCa-2 cells might be determined at least in part by the intracellular balance between mutant p53 and TAp73 following GEM exposure.
As clearly shown in our colony-formation assay, forced expression of TAp73α significantly stimulated cell cycle arrest and/or cell death in MiaPaCa-2 cells. Consistent with these observations, overexpression of TAp73α in MiaPaCa-2 cells markedly enhanced the luciferase activities driven by human p21WAF1 and NOXA promoters in a dose-dependent manner. In support of our observations, it has been described that forced expression of TAp73 in p53-mutated cancerous cells results in a massive suppression of their proliferation rate.47 In addition, Muller et al.48 revealed that the intracellular balance between pro-apoptotic TAp73 and pro-oncogenic ΔNp73 is a critical determinant of chemo-sensitivity. Considering that mutant p53 acts as a dominant-negative inhibitor against TAp73,25 it is likely that, as mentioned above, a large amount of TAp73 might overcome the negative effect caused by mutant p53.
As shown in Figure 2, TAp73 was induced at mRNA and protein level following GEM exposure. Previously, it has been demonstrated that E2F-1 has as a transcriptional activator for TAp73, and E2F-1-mediated cell death is regulated at least in part in a TAp73-dependent manner.41, 42, 43 According to our present results (see Figure 6), knockdown of RUNX2 in MiaPaCa-2 cells stimulated GEM-mediated increase in TAp73 in association with the further upregulation of E2F-1. As RUNX2 has an ability to suppress the expression of TAp73,38 RUNX2 might be also involved in the negative regulation of TAp73 as well as E2F-1 in response to GEM. Notably, Berman et al. found that loss of pRB, a strong inhibitor of E2F-1, promotes RUNX2 expression.49 Collectively, it is conceivable that there could exist a negative-feedback loop regulatory system where E2F-1 up-regulates its negative regulator RUNX2.
Unexpectedly, GEM treatment significantly reduced the expression level of RUNX2 protein, whereas its mRNA level was markedly increased in MiaPaCa-2 cells after GEM exposure. It has been shown that RUNX2 undergoes ubiquitin-mediated proteasomal degradation.50 Shen et al.51 revealed that E3 ubiquitin ligase Smurf1 (Smad ubiquitin regulatory factor 1) stimulates proteasome-mediated degradation of RUNX2, which is enhanced by Smad6. Distinct from Smurf1-mediated proteolytic degradation of RUNX2, it has been demonstrated that cyclin D1/Cdk4 complex phosphorylates RUNX2 and promotes its degradation in an ubiquitin/proteasome-dependent manner.52 In addition, several lines of evidence suggest that Smurf1 has a pro-oncogenic potential.53, 54 In a good agreement with this notion, Shain et al.55 described that Smurf1 gene is amplified in certain subset of human pancreatic cancers and might contribute to their invasiveness. At present, it remains elusive whether Smurf1 could account for GEM-mediated degradation of RUNX2 in MiaPaCa-2 cells and contribute to the enhancement of their GEM sensitivity. Further studies should be required to adequately address this issue.
Another new finding of this study is that depletion of RUNX2 in MiaPaCa-2 cells elevates GEM-mediated Tyr phosphorylation level of TAp73 and the accumulation of γH2AX (Supplementary Figure S7). As Tyr phosphorylation of TAp73 was detectable by immunoblotting with PY20 antibody, we did not know which Tyr residue(s) of TAp73 could be phosphorylated in response to GEM. According to the previous observations,44, 45, 46 TAp73 was phosphorylated at Tyr-99 by non-receptor type tyrosine kinase c-Abl following DNA damage, and the phosphorylated TAp73 became stabilized and activated. It has been well known that c-Abl is activated through ataxia telangiectasia mutated (ATM)-mediated phosphorylation after DNA damage.56, 57 Given that silencing of RUNX2 further stimulates GEM-mediated accumulation of TAp73 as well as γH2AX, it is likely that TAp73 and/or RUNX2 participates in the regulation of DNA damage-dependent ATM phosphorylation. In accordance with this notion, we have recently found that TAp63 is tightly linked to GEM-mediated phosphorylation of ATM.39 Of note, Wang et al.58 indicating that c-Abl might act as an upstream regulator of ATM in response to DNA damage. Further studies should be required to clarify the functional interplay among p53 family, ATM/ATM Rad3-related protein, c-Abl and RUNX2 during DNA damage response. In addition, we have found putative Tyr phosphorylation sites other than Tyr-99 catalyzed by various protein tyrosine kinases including c-Abl and JAK within TAp73 amino-acid sequence (data not shown).
Taken together, our present results strongly suggest that depletion of RUNX2 enhances GEM sensitivity of p53-mutated pancreatic cancer cells through the stimulation of TAp73-dependent cell death pathway, and thus RUNX2 might be an attractive molecular target for the treatment of the patients bearing pancreatic cancer regardless of p53 status.
Materials and methods
Cell culture
The human pancreatic cancer SW1990 and MiaPaCa-2 cells were originated from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in Dulbecco’s Modified Eagle’s medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Life Technologies, Carlsbad, CA, USA) and penicillin–treptomycin in a 5% CO2 atmosphere at 37 °C.
FACS analysis
For sub-G1 analysis, cells were exposed to GEM (Sigma–Aldrich, St Louis, MO, USA) at the indicated concentrations. Forty-eight hours after treatment, floating and adherent cells were collected, resuspended in phosphate-buffered saline (PBS) and fixed with ice-cold 70% ethanol for 15 min. After centrifugation, ethanol was removed and cells were rehydrated in PBS for 10 min. Cells were treated with RNAase A at a final concentration of 1 μg/ml and incubated for 30 min at 37 °C. Propidium iodide was subsequently added at a final concentration of 1 μg/ml and then analyzed by flow cytometry (FACS Calibur, BD Biosciences, Franklin Lakes, NJ, USA).
WST cell survival assay
To examine cell viability, 1 × 103 cells/well were seeded in a 96-well plate in 100 μl of culture medium. Twelve-hours after seeding, cells were treated with the indicated concentrations of GEM. Forty-eight hours after treatment, viable cells were counted using the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) following the manufacturer’s instructions. Each experiment was carried out at least three times.
Trypan blue exclusion assay
Cells were seeded at 2 × 105 cells/six-well plate and allowed to attach overnight. Cells were then exposed to the indicated concentrations of GEM. Forty-eight hours after treatment, floating and adherent cells were collected and washed in PBS. After centrifugation, cells were resuspended in fresh medium, mixed with equal volume of 0.4% trypan blue solution and then analyzed by automatical cell counter (TC20, Bio-Rad, Hercules, CA, USA).
Reverse transcription
Total RNA was isolated from the indicated cells using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. Reverse transcription was performed with 1 μg of total RNA using SuperScript II reverse transcriptase (Life Technologies) according to the manufacturer’s instructions. The synthesized complementary DNA was amplified by PCR using specific primer sets. The PCR products were visualized by electrophoresis on agarose gels with ethidium bromide staining. GAPDH served as an internal control.
Immunoblotting
Cells were washed twice in ice-cold PBS and lysed in lysis buffer containing 25 mM Tris-HCl, pH 7.5, 137 mM NaCl, 2.7 mM KCl, and 1% Triton X-100 and protease inhibitor cocktail (Roche Applied Sciences, Indianapolis, IN, USA). Protein concentration of cell lysates was determined by the Bradford assay (Bio-Rad). Equal amounts of cell lysates (50 μg of protein) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membrane (Merck Millipore, Amsterdam, the Netherlands). The membrane was blocked with Tris-buffered saline containing 5% non-fat dry milk (Tris-buffered saline) and then incubated with anti-p53 (DO-1, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-TAp73 (GeneTex, Irvine, CA, USA), anti-TAp63 (Cell Signaling Technology, Beverley, CA, USA), anti-p21WAF1 (H164, Santa Cruz Biotechnology), anti-BAX (Cell Signaling Technology), anti-E2F-1 (Cell Signaling Technology), anti-RUNX2 (Cell Signaling Technology), anti-PARP (Cell Signaling Technology), anti-γH2AX (2F3, BioLegend, San Diego, CA, USA) anti-ATM (5C2, Santa Cruz Biotechnology) or with anti-actin (20-33, Sigma–Aldrich) antibody. The membrane was washed in Tris-buffered saline-T and incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies. After washing in Tris-buffered saline-T, immune-reactive signals were visualized with the enhanced chemiluminescence system (GE Healthcare Life Sciences, Piscataway, NJ, USA) according to the manufacturer’s instructions.
siRNA-mediated knockdown
MiaPaCa-2 cells were transfected with control siRNA or with a SMARTpool/ON-TARGETplus siRNA against RUNX2 (Dharmacon, Lafayette, CO, USA) using Lipofectamine 2000 reagent (Life Technologies) following the manufacturer’s protocols. Silencing of RUNX2 was evaluated by RT–PCR and immunoblotting.
Immunoprecipitation assay
Cell lysates were prepared and incubated with mouse control IgG (Cell Signaling Technology) or with monoclonal anti-phospho-Tyr antibody (PY20, Abcam, Cambridge, MA, USA) overnight at 4 °C. Antibody-bound proteins were precipitated with protein G-Sepharose beads (GE Healthcare Life Sciences). The beads were washed three times in lysis buffer and then eluted in 2 × sodium dodecyl sulfate sample buffer. The eluted proteins were subjected to immunoblotting with anti-TAp73 antibody.
Colony-formation assay
Cells were seeded at 2 × 105 cells/six-well plate and allowed to attach overnight. Cells were transfected with the empty plasmid or with the expression plasmid for TAp73α. Forty-eight hours after transfection, cells were transferred to fresh medium containing 400 μg/ml of G418 (Sigma–Aldrich). Two weeks after the selection, G418-resistant colonies were fixed, stained with Giemsa’s solution (Merck Millipore), air-dried and photographed.
Luciferase reporter assay
Cells were seeded at 5 × 104 cells/12-well plate and allowed to attach overnight. Cells were transfected with the constant amount of luciferase reporter plasmid bearing p21WAF1 or NOXA promoter and Renilla luciferase plasmid in the presence or absence of the increasing amounts of the expression plasmid for TAp73α. Total amount of plasmid DNA was kept constant by pcDNA3. Forty-eight hours after transfection, cell lysates were prepared and their luciferase activities were measured using Dual Luciferase Reporter System according to the manufacturer’s instructions (Promega, Madison, WI, USA).
Immunostaining
Cells were exposed to the indicated concentrations of GEM. Forty-eight hours after treatment, cells were fixed in 3.7% formaldehyde at room temperature for 30 min, permeabilized with 0.1% Triton X-100 at room temperature for 5 min and then blocked with 3% bovine serum albumin in PBS at room temperature for 1 h. After blocking, cells were washed in ice-cold PBS and incubated with mouse monoclonal anti-γH2AX antibody at room temperature for 1 h followed by the incubation with FITC-conjugated goat anti-mouse IgG (Life Technologies) at room temperature for 1 h. After the extensive washing, cell nuclei were stained with DAPI (Vector Laboratories, Peterborough, UK). Fluorescent images were captured using a confocal microscope.
DNA fragmentation assay
RUNX2-depleted and non-depleted cells were exposed to GEM or left untreated. Forty-eight hours after treatment, floating and attached cells were harvested and their genomic DNA was extracted according to the standard procedure. One microgram of genomic DNA was analyzed by 0.7% agarose gel electrophoresis and visualized with ethidium bromide.
Statistical analysis
All experiments were performed in triplicate. Data are presented as mean±s.d. Analysis utilized Student’s t-tests and analysis of variance. Values of P<0.05 were considered significant.
Abbreviations
- ADR:
-
adriamycin
- ATM:
-
ataxia telangiectasia mutated
- ATR:
-
ATM Rad3-related protein
- DAPI:
-
4′,6-diamidino-2-phenylindole
- DDR:
-
DNA damage response
- FACS:
-
fluorescence-activated cell sorting
- FITC:
-
fluorescein isothiocyanate
- GAPDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- GEM:
-
gemcitabine
- PARP:
-
poly ADP ribose polymerase
- PI:
-
propidium iodide
- RUNX2:
-
runt-related transcription factor 2
- siRNA:
-
small interfering RNA.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D . Global cancer statistics. CA Cancer J Clin 2011; 61: 69–90.
Xie D, Xie K . Pancreatic cancer stromal biology and therapy. Genes Dis 2015; 2: 133–143.
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM . Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2011; 74: 2913–2921.
Herreros-Villanueva M, Hijona E, Cosme A, Bujanda L . Adjuvant and neoadjuvant treatment in PDAC. World J Gastroenterol 2012; 18: 1565–1572.
Oettle H, Neuhaus P, Hochhaus A, Hartmann JT, Gellert K, Ridwelski K et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected PDAC: The conko-001 randomized trial. JAMA 2013; 310: 1473–1481.
Vousden KH, Lu X . Live or let die: the cell's response to p53. Nat Rev Cancer 2002; 2: 594–604.
Levine AJ, Oren M . The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009; 9: 749–758.
Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B . A model for p53-induced apoptosis. Nature 1997; 389: 300–305.
Horn HF, Vousden KH . Coping with stress: multiple ways to activate p53. Oncogene 2007; 26: 1306–1316.
Toledo F, Wahl GM . Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer 2006; 6: 909–923.
Kubbutat MH, Jones SN, Vousden KH . Regulation of p53 stability by Mdm2. Nature 1997; 387: 299–303.
Hock AK, Vousden KH . The role of ubiquitin modification in the regulation of p53. Biochim Biophys Acta 2014; 1843: 137–149.
Pant V, Lozano G . Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev 2014; 28: 1739–1751.
Hollstein M, Sidransky D, Vogelstein B, Harris CC . p53 mutations in human cancers. Science 1991; 253: 49–53.
Muller PA, Vousden KH . p53 mutations in cancer. Nat Cell Biol 2013; 15: 2–8.
Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R, Cleary K et al. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989; 342: 705–708.
Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004; 119: 861–872.
Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004; 119: 847–860.
Muller P, Hrstka R, Coomber D, Lane DP, Vojtesek B . Chaperone-dependent stabilization and degradation of p53 mutants. Oncogene 2008; 27: 3371–3383.
Zheng T, Wang J, Zhao Y, Zhang C, Lin M, Wang X et al. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat Commun 2013; 4: 2996.
Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, M Moore et al. Gain of function mutations in p53. Nat Genet 1993; 4: 42–46.
Soussi T, Wiman KG . TP53: an oncogene in disguise. Cell Death Differ 2015; 22: 1239–1249.
Ikawa S, Nakagawara A, Ikawa Y . p53 family genes: structural comparison, expression and mutation. Cell Death Differ 1999; 6: 1154–1161.
Levrero M, De Laurenzi V, Costanzo A, Gong J, Melino G, Wang JY . Structure, function and regulation of p63 and p73. Cell Death Differ 1999; 6: 1146–1153.
McKeon F, Melino G . Fog of war: the emerging p53 family. Cell Cycle 2007; 6: 229–232.
Katoh I, Aisaki KI, Kurata SI, Ikawa S, Ikawa Y . p51A (TAp63gamma), a p53 homolog, accumulates in response to DNA damage for cell regulation. Oncogene 2000; 19: 3126–3130.
Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG Jr . Chemosensitivity linked to p73 function. Cancer Cell 2003; 3: 403–140.
Patturajan M, Nomoto S, Sommer M, Fomenkov A, Hibi K, Zangen R et al. DeltaNp63 induces beta-catenin nuclear accumulation and signaling. Cancer Cell 2002; 1: 369–379.
Dulloo I, Hooi PB, Sabapathy K . Hypoxia-induced DNp73 stabilization regulates Vegf-A expression and tumor angiogenesis similar to TAp73. Cell Cycle 2015; 14: 3533–3539 in press.
Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M . t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci USA 1991; 88: 10431–10434.
Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR . AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996; 84: 321–330.
Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA . Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci USA 1996; 93: 3444–3449.
Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997; 89: 755–764.
Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997; 89: 765–771.
Taniuchi I, Osato M, Egawa T, Sunshine MJ, Bae SC, Komori T et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell 2002; 111: 621–633.
Li QL, Ito K, Sakakura C, Fukamachi H, Ki Inoue, Chi XZ et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 2002; 109: 113–124.
Ozaki T, Wu D, Sugimoto H, Nagase H, Nakagawara A . Runt-related transcription factor 2 (RUNX2) inhibits p53-dependent apoptosis through the collaboration with HDAC6 in response to DNA damage. Cell Death Dis 2013; 4: e610.
Ozaki T, Sugimoto H, Nakamura M, Hiraoka K, Yoda H, Sang M et al. Runt-related transcription factor 2 attenuates the transcriptional activity as well as DNA damage-mediated induction of pro-apoptotic TAp73 to regulate chemosensitivity. FEBS J 2015; 282: 114–128.
Sugimoto H, Nakamura M, Yoda H, Hiraoka H, Shinohara K, Sang M et al. Silencing of RUNX2 enhances gemcitabine sensitivity of p53-deficient human pancreatic cancer AsPC-1 cells through the stimulation of TAp63-mediated cell death. Cell Death Discov 2015; 1: 15010.
Ozaki T, Nakamura M, Shimozato O . Novel implications of DNA damage response in drug resistance of malignant cancers obtained from the functional interaction between p53 family and RUNX2. Biomolecules 2015; 5: 2854–2876.
Lissy NA, Davis PK, Irwin M, Kaelin WG, Dowdy SF . A common E2F-1 and p73 pathway mediates cell death induced by TCR activation. Nature 2000; 407: 642–645.
Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, Liu W et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature 2000; 407: 645–648.
Stiewe T, Pützer BM . Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat Genet 2000; 26: 464–469.
Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG Jr, Levrero M et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 1999; 399: 806–809.
Agami R, Blandino G, Oren M, Shaul Y . Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature 1999; 399: 809–813.
Yuan ZM, Shioya H, Ishiko T, Sun X, Gu J, Huang YY et al. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature 1999; 399: 814–817.
Willis AC, Pipes T, Zhu J, Chen X . p73 can suppress the proliferation of cells that express mutant p53. Oncogene 2003; 22: 5481–5495.
Muller M, Schilling T, Sayan AE, Kairat A, Lorenz K, Schulze-Bergkamen H et al. TAp73/ΔNp73 influences apoptotic response, chemosensitivity and prognosis in hepatocellular carcinoma. Cell Death Differ 2005; 12: 1564–1577.
Berman SD, Yuan TL, Miller ES, Lee EY, Caron A, Lees JA . The retinoblastoma protein tumor suppressor is important for appropriate osteoblast differentiation and bone development. Mol Cancer Res 2008; 6: 1440–1451.
Tintut Y, Parhami F, Le V, Karsenty G, Demer LL . Inhibition of osteoblast-specific transcription factor Cbfa1 by the cAMP pathway in osteoblastic cells. Ubiquitin/proteasome-dependent regulation. J Biol Chem 1999; 274: 28875–2889.
Shen R, Chen M, Wang YJ, Kaneki H, Xing L, O'keefe RJ et al. Smad6 interacts with Runx2 and mediates Smad ubiquitin regulatory factor 1-induced Runx2 degradation. J Biol Chem 2006; 281: 3569–3576.
Shen R, Wang X, Drissi H, Liu F, O'Keefe RJ, Chen D . Cyclin D1-cdk4 induce runx2 ubiquitination and degradation. J Biol Chem 2006; 281: 16347–16353.
Nie J, Liu L, Xing G, Zhang M, Wei R, Guo M et al. CKIP-1 acts as a colonic tumor suppressor by repressing oncogenic Smurf1 synthesis and promoting Smurf1 autodegradation. Oncogene 2014; 33: 3677–3687.
Xie P, Zhang M, He S, Lu K, Chen Y, Xing G et al. The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis. Nat Commun 2014; 5: 3733.
Shain AH, Salari K, Giacomini CP, Pollack JR . Integrative genomic and functional profiling of the pancreatic cancer genome. BMC Genomics 2013; 14: 624.
Baskaran R, Wood LD, Whitaker LL, Canman CE, Morgan SE, Xu Y et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature 1997; 387: 516–519.
Shafman T, Khanna KK, Kedar P, Spring K, Kozlov S, Yen T et al. Interaction between ATM protein and c-Abl in response to DNA damage. Nature 1997; 387: 520–523.
Wang X, Zeng L, Wang J, Chau JF, Lai KP, Jia D et al. A positive role for c-Abl in Atm and Atr activation in DNA damage response. Cell Death Differ 2011; 18: 5–15.
Acknowledgements
We thank Dr Hiroki Nagase (Laboratory of Cancer Genetics, Chiba Cancer Center Research Institute) for his valuable discussion. This work was supported in part by JSPS (MEXT) KAKENHI Grant Number 23501278.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogenesis website .
Supplementary information
Rights and permissions
Oncogenesis is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Nakamura, M., Sugimoto, H., Ogata, T. et al. Improvement of gemcitabine sensitivity of p53-mutated pancreatic cancer MiaPaCa-2 cells by RUNX2 depletion-mediated augmentation of TAp73-dependent cell death. Oncogenesis 5, e233 (2016). https://doi.org/10.1038/oncsis.2016.40
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/oncsis.2016.40
- Springer Nature Limited
This article is cited by
-
The role of diagnostic, prognostic, and predictive biomarkers in the management of early pancreatic cancer
Journal of Cancer Research and Clinical Oncology (2023)
-
Impact of RUNX2 on drug-resistant human pancreatic cancer cells with p53 mutations
BMC Cancer (2018)