
Abstract
The function of miR16 in multiforme glioblastoma multiforme (GBM) and its stem cells (GSCs) remains elusive. To this end, we investigated the patterns of miR16 expression in these cells and their correlation with malignant behaviors and clinical outcomes. The levels of miR16 and its targeted genes in tumor tissue of GBM and GBM SGH44, U87, U251 cells as well as their stem cell counterparts were measured by qRT–PCR or western blot or immunohistochemistry. Luciferase reporter assay was used to confirm the binding of miR16 to 3′-UTR of its target genes. The effects of miR16 on malignant behaviors were investigated, including tumor cell viability, soft-agar colony formation, GSCs Matrigel colony forming and migration and invasion as well as nude mice xenograft model. Differentially expression patterns of miR16 in glioblastoma cells and GSCs cells were found in this study. Changes of miR16 targeted genes, Bcl2 (B cell lymphoma 2), CDK6 (Cyclin-dependent kinase 6), CCND1 (cyclin D1), CCNE1 (cyclin E1) and SOX5 were confirmed in glioblastoma cell lines and tissue specimens. In vitro and in vivo studies showed that tumor cell proliferation was inhibited by miR16 mimic, but enhanced by miR16 inhibitor. The expression level of miR16 positively correlates with GSCs differentiation, but negatively with the abilities of migration, motility, invasion and colony formation in glioblastoma cells. The inhibitory effects of miR16 on its target genes were also found in nude mice xenograft model. Our findings revealed that the miR16 functions as a tumor suppressor in GSCs and its association with prognosis in GBM.
Similar content being viewed by others

Introduction
Glioblastoma multiforme (GBM) is a common aggressive brain malignancies and it has a very poor prognosis.1 Glial progenitor cells or astrocytes are considered as an origin of glioma, but pathogenesis of this disease remains unclear. Several studies revealed that glioblastoma stem cells (GSCs) are the driver of malignancy of glial cells and correlated with resistance to treatment.2, 3
MiRNAs belongs to non-coding small RNAs family that can silence gene expression at the post-transcriptional level, in a way to bind its complementary sequences in 3′-UTR regions of its target genes.4, 5 In recent years, emerging evidences indicate important roles of miRNAs in the regulation of a wide range of fundamental biological processes, including brain development and neuronal differentiation.6, 7 Dysfunction of miRNAs is correlated with human malignancies, including glioma,8, 9 implicating the potent function of miRNAs in tumorigenesis and tumor development. The involvement of the differentially expressed miRNAs, such as miR21 and miR16 in the malignant progression of gliomas has been reported.10, 11 It has been reported that miR16 inhibits migration and invasion of glioma cells.12, 13, 14 Overexpression of miR16 in GBM U87 and U251 cells, can inhibit adhesion and invasion of tumor cells as well as downregulate gene expression, which is related to epithelial–mesenchymal transition (EMT).15 Those results suggest that miR16 is an anti-apoptotic factor in GBM, which may be a potential therapeutic target and a prognostic indicator for glioblastoma therapy.
Recent studies found that GSCs are a sub-population of GBM cells that are involved in both initiation and maintenance of glioma. GSCs can extensively self-renew and differentiate into a heterogeneous population of endothelial cells (EC-GSCs), which may directly participate in the vascularization of GBM. Several markers, including prominin-1 (CD133), CD15/SSEA1, A2B5, L1CAM and endoglin (CD105) have been identified on the cell surfaces of GSCs and EC-GSCs. Those cell surface molecules that can be detected by flow cytometry and bio-imaging technology may be ideal markers for isolation of targeted cells from heterogeneous tumor cell populations.
However, miRNA’s roles in the development of GSCs are currently not fully clarified. Abnormal expression of miR-125b in human glioma16 confers resistance of GSCs to temozolomide by a mechanism related to the mitochondrial pathway of apoptosis.17, 18 To better understand the functions of miRNAs in human malignant glioma, our study investigated the expression levels of miR16 and its target genes in three types of human glioblastoma cells, its GSCs and tissue of GBM. In addition, the effects of miR16 on tumor cell proliferation, migration and invasion were also evaluated.
Results
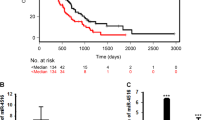
Expression levels of miR16 and its target genes correlate with overall survival of GMB patients
In a total of 132 GBM cases, 116 patients died after a 20 months-median follow-up (ranged from 5–50 months). Eleven months of the median overall survival (OS) was estimated. Overall, in 132 patients with GBM, miR16 expression level (1.81±0.72) in 77 cases (58.33%) was significantly lower than that (16.61±0.65) in other 55 cases (41.67%). However, the expression level of miR16 did not correlate with patient age, gender, tumor number, size and occurrence (P>0.05), but not with gender (Table 1). The expression of miR16 target genes, including Bcl2, CDK6, CCND1, CCNE1 and SOX5 were decreased in the cases with high miR16, but increased in those with low level of mRNA and protein of miR16 (Figures 1a and b). The difference of the expression of Bcl2, CDK6, CCND1, CCNE1 and SOX5 between low and high miR16 cases are statistically significant, indicating that miR16 expression negatively correlates with the expression level of its target genes. Moreover, GSCs marker (SOX2 and nestin) and proliferation marker Ki67 were significantly reduced in low-miR16 cases (Figure 1b). Finally, the correlation of GMB patients’ prognosis with expression levels of miR16 and its target genes was analyzed using Kaplan–Meier method. The cut-off value was best distinguished in the cases with high and low miR16. The median OS of the patients was 19±2.61 months in high miR16 cases, but 10±3.36 months in low-miR16 cases. The OS of the patients with high miR16 was longer than those with low miR16 (X2=5.33, P=0.02) (Figure 1c). In the patients with high or low expression of CCND1, the median OS were 11±3.05 and 18±1.33 months, respectively. The median OS of the cases with high and low SOX5 were 9±2.46 and 18±1.50 months, respectively. The OS of the cases with low CCND1 and SOX5 was also longer than those with either high CCND1 (X2=4.74, P=0.03) or SOX5 (X2=3.97, P<0.05) (Figure 1c). However, Bcl2, CDK6 and CCNE1 expression levels were not found to be related to the OS of patients with GMB.

The correlation of miR16 and its target genes expression with OS of GMB patients. (a) Comparison of difference of mRNA expression of miR16 target genes between miR16 low and high group by qRT–PCR. (b) Comparison of difference of miR16 target genes expression between GMB patients with low and high miR16 by immunohistochemistry (magnification, × 400). (c) The OS curves of the GMB patients with high miR16 or low miR16 (left). The OS curves of the GMB patients with high level of CCND1 or low level of CCND1 (middle). The OS curves of the GMB patients with high level of SOX5 or low level of SOX5 (right). The differences between the survival curves were tested by using Kaplan–Meier method.
Expression of miR16 and its target genes in glioblastoma and GSCs
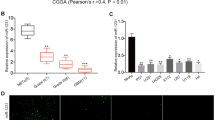
To investigate the expression patterns of miR16 in GSCs, microRNA microarray profiling of human glioblastoma SGH44, U87 and U251 cell lines as well as their stem cell counterparts (SGH44-GSC, U87-GSC and U251-GSC) was performed, using miRCURY LNA (locked nucleic acid) Array (Exiqon, Vedbæk, Denmark). As compared with normal neuroglial cell, seven downregulated miRNAs in glioblastoma cells and their GSCs were identified, including hsa-miR16, hsa-miR30b, hsa-miR374b, hsa-miR652, hsa-miR485-3p, hsa-miR30b and hsa-miR200b* (Figure 2a).


Expression of miR16 and its target genes in GBM cell lines SGH44, U87 and U251 and their GSCs. (a) MicroRNA microaary profiling using miRCURY LNA Array. Green block in heatmap were miRNA low expression areas, in which miR16 was found. (b) Identification of target genes of miR16 by luciferase reporter assay in U251 cells. Schematic diagram of Bcl2, CDK6, CCND1, CCNE1 and SOX5 3′-UTR pMIR-REPORT constructs (left). Comparison of sequences between mature miR16 and the wild-type (Wt) or mutant (Mut) putative target sites in the 3′-UTR of Bcl2, CDK6, CCND1, CCNE1 and SOX5. Cells were co-transfected with pMIR-REPORT containing the empty (Ctrl), WT or mutant target site of the Bcl2, CDK6, CCND1, CCNE1 and SOX5 3′-UTR plus miR16 or NC mimic for 48 h. The luciferase activity was normalized to the Renilla activity and presented as relative activity to the corresponding NC (assigned as value ‘1’). Values denote the mean±s.e.m. of three independent assays. *P<0.05.
Overall, 166 target genes of miR16 were predicated via miRBase and Target Scan 5.1 (http://www.targetscan.org/worm_12/), including the apoptosis repressor gene Bcl2, cell cycle-related genes (CCND1, CCNE1, CDK2 and CDK6), stem cell-related gene SOX5, neurofibromatosis type 1 (NF1) and genes involved in the Wnt signal pathway.19, 20, 21, 22, 23, 24 In the 3′-UTR region of the predicted genes, highly conserved binding sites of miR16 were found. To confirm Bcl2, CCND1, CCNE1, CDK6 and SOX5 as miR16 targeted genes in glioblastoma cells and GSCs, their mRNA expression in the cells transfected with miR16 mimic or miR16 inhibitor was examined by quantitative reverse transcription PCR (qRT–PCR). PCR data showed that the mRNA expression of Bcl2, CCND1, CCNE1, CDK6 and SOX5 were significantly downregulated by miR16 mimic, but upregulated by miR16 inhibitor (P<0.05; Figure 3a). Similar to PCR results, a decrease in protein level of Bcl2, CCND1, CCNE1, CDK6 and SOX5 in U251 cells transfected with miR16 mimic was also found by western-blotting analysis, (Figure 3b). Taken together, these data indicated that Bcl2, CCND1, CCNE1, CDK6 and SOX5 are target genes of miR16 in glioblastoma cells and their GSCs.

Expression of miR16 and its target genes in human GBM cell lines. (a) The levels of miR16 and mRNA of its target genes in glioblastoma cells SGH44, U87, U251 and their stem cell counterparts were evaluated by analysis of qRT–PCR. Comparison of the difference of miR16 expression between groups of miR16 inhibitor and negative control (NC) was performed. Comparison of the difference of mRNA expression of miR16 target genes, including Bcl2, CDK6, CCND1, CCNE1 and SOX5 between groups of miR16 mimic or inhibitor or NC was also performed. (b) Protein level of miR16 targeted genes: Bcl2, CDK6, CCND1, CCNE1 and SOX5 in groups of miR16 mimic or inhibitor or NC were determined by western blot analysis.
The luciferase reporters with Wt or Mut miR16 binding sites in 3′-UTR of those target genes were constructed and their activities in U251 cells were examined. Co-transfection of miR16 and Wt- Bcl2 reporter, rather than Mut- Bcl2 reporter in U251 cells resulted in a decreased reporter activity (Figure 2b). Similar results were observed after co-transfection of Wt-CCND1, CCNE1, CDK6, SOX5 reporters and miR16 (Figure 2b). Taken together, these data indicated that miR16 can directly regulate the expression of Bcl2, CCND1, CCNE1, CDK6 and SOX5 in glioblastoma cells.
Modulation of miR16 expression in glioblastoma and GSCs cells impacts on their proliferation, cell cycle and apoptosis
As shown in Figure 4, the growth of gliobalstoma cells and GSCs transfected with miR16 mimic was significantly inhibited at 72 h after transfection. Whereas, miR16 inhibitor significantly promoted cell proliferation in both glioblastoma cells and GSCs, relative to the control, suggesting the involvement of miR16 in the inhibition of glioblastoma cell proliferation. Flow cytometry results showed that overexpression of miR16 in glioblastoma cells can induce a G0/G1 arrest, leading to a decrease in S phase population (Figure 4b). Moreover, compare with the control group, the ratio of early apoptotic cells in glioblastoma cells and GSCs transfected with miR16 mimic was significantly higher (P<0.05), suggesting that the overexpression of miR16 can induce apoptosis in glioblastoma cells.

Effects of miR16 on GBM proliferation and apoptosis. (a) Changes of proliferation potential in GBM cell lines SGH44, U87, U251 and their stem cell counterparts after administration of miR16 mimic and inhibitor were evaluated by CCK8 assay. Comparison of the difference of proliferation potential at different time point between groups of miR16 mimic and miR16 inhibitor or NC was performed. (b) The cell cycle distribution in glioblastoma cell lines and their stem cell counterparts were examined using flow cytometry assay. (c) Apoptotic rate of glioblastoma cells and their stem cell counterparts was estimated by flow cytometry analysis. Comparison of the difference of apoptotic rate between groups of miR16 mimic and miR16 inhibitor or NC was also performed.
MiR16 inhibits the migration, motility and invasion of GBM cells
Wound scratch assay data revealed that miR16 mimic suppressed the migration ability of glioblastoma SGH44, U87 and U251 cells, as compared to that of the scramble control, whereas miR16 inhibitor promoted glioma cell migration (Figure 5a). The transwell experiment results also confirmed that the ability of motility and invasion were significantly lower in cells transfected with miR16 mimic than that of the control (P<0.01), but these characteristics were significantly higher in cells with miR16 inhibitor, as compared to those with miR16 mimic (P<0.01; Figure 5b).

Effects of miR16 abilities of GBM cell migration, motility and invasion. The abilities of migration and invasion of GBM cells were determined by wound scratch assay (a) and Transwell assay (b). Comparison of the difference of the abilities of migration and invasion between groups of miR16 mimic and miR16 inhibitor or NC was performed.
Inhibitory effects of miR16 on colony formation ability of glioblastoma cells and differentiation of GSCs
To illustrate the effect of miR16 on in vitro colony formation of glioblastoma cells, two assays, soft-agar colony formation and GSCs Matrigel colony formation were conducted in this study. Similar to miR16-mediated suppression of GBM cell proliferation, migration, motility and invasion, miR16 mimic significantly reduced colony formation in SGH44, U87 and U251 cells relative to the control (P<0.05; Figure 6a), whereas the number of colonies in miR16 inhibitor group was significantly increased, compared with control group (P<0.01). Similarly, in GSCs, the colony formation in miR16 mimic group was significantly less, compared with the control (P<0.05) or GSCs with miR16 inhibitor (P<0.001) (Figure 6b).

Effects of miR16 on colony formation ability of glioblastoma cells and differentiation of GSCs. (a) The ability of colony formation (number and size) in glioblastoma cells was evaluated by soft-agar assay (magnification, × 100). Comparison of the difference of this ability between groups of miR16 mimic and miR16 inhibitor or NC was performed. (b) The ability of sphere formation (number) in glioblastoma cells was examined by sphere-forming assay. The number of sphere between groups of miR16 mimic and miR16 inhibitor or NC was compared. The data are present as the mean±s.d. from three independent experiments with triplicate in each condition. (c) The differentiation of U87-GSCs was verified by the cell surface markers that can be identified by immunofluorescence (middle, next right, magnification, × 200) and flow cytometry (right). The suspension ball of U87-GSC (CD133+/GFAP−) (left) was gradually transformed into U87 cells (CD133−/GFAP+) (middle, next to left) in the medium with serum in 5 days.
Moreover, the effect of miR16 on the capability of in vitro-differentiation of GSCs was also investigated. Compared with miR16 mimic group, the differentiation time was significantly longer in miR16 inhibitor group (P<0.01; Figure 6c). CD133, a surface biomarker of GSCs was found in three stem-like cell lines, but not in differentiated cells (GFAP-positive) (Figure 6c). Therefore, the ratio of differentiated GSCs in miR16 mimic group was higher than that in miR16 inhibitor group.
Overexpression of miR16 inhibits xenograft tumor growth in vivo
Tumor xenograft were formed in nude mice by intracerebrally inoculating U251 cells or U251-GSCs, which were transfected with scrambled control, miR16 mimic and miR16 inhibitor, respectively (Figure 7). As compared to scrambled control, average mean of tumor size in miR16 inhibitor-treated mice was significantly larger, compared with the miR16 mimic group (P<0.05; Figure 7a), indicating that miR16 as a tumor suppressor might suppress glioma growth. However, average mean of tumor size in U251-GSCs inoculated mice was significantly larger, as compared with the U251 implanted mice (P<0.05; Figure 7a), suggesting oncogenic role of GSCs. The expression of targeted genes of miR16 and proliferation marker Ki67 in tumor xenograft tissues was also examined by immunohistochemistry (IHC). As shown in Figure 7b, the expression levels of Bcl2, CCND1, CCNE1, CDK6 and Ki67 were reduced in miR16 mimic mice, but SOX5 was overexpressed in miR16 inhibitor mice, implicating that miR16 can inhibit proliferation and induce apoptosis in both GBM cells and GSCs. Furthermore, miR16 may regulate differentiation of GBM cell.

miR16 inhibitor promoted the tumor growth in vivo. Nude mice received intracerebral transplantation of U251 or U251-GSCs transfected with miR16 mimic, inhibitor or scrambled control. (a) Comparison of tumor size between groups of miR16 inhibitor, miR16 mimic group and scrambled control; (b) Expression level of miR16 and its target genes, including Bcl2, CDK6, CCND1, CCNE1, SOX5 and Ki67 in tumor xenograft tissues were examined by qRT-PCR and immunohistochemistry. Green block were low expression areas.
Discussion
Recent studies found that there is a sub-population of glioblastoma cells, named GSCs, responsible for initiation and maintenance of glioma. GSCs can extensively self-renew and differentiate into a heterogeneous population of endothelial cells (EC-GSCs) that may directly participate in the vascularization of GBM. Several cell surface markers of GSCs and EC-GSCs, including prominin-1 (CD133), CD15/SSEA1, A2B5, L1CAM, and endoglin (CD105) have been identified.25 Those cell surface molecules that can be detected by flow cytometry and bio-imaging technology may be ideal markers for isolation of targeted cells from heterogeneous tumor cell populations.
As GSCs, a sub-population of glioma cells, might be the key reason for therapeutic resistance and recurrence of GBM, elimination of GSCs is widely accepted as important for achieving a better therapeutic outcome.26, 27 Previous studies have identified various differentially expressed miRNAs in glioma cells, including an increase in miR-10b, miR-130a, miR-221, miR-125b-1, miR-125b-2, miR-9-2, miR-21, miR-25 and miR-123 as well as a decrease in miR-128a, miR-181c, miR-181a and miR-181b.28, 29 In addition, it has been reported that miR-34a expression was decreased in GSCs.30, 31 MiR-125b expression was also greatly inhibited in CD133-positive GSCs, as compared with CD133-negative GSCs.32 However, the miRNAs-mediated regulation in the development of GSCs is still largely unknown. A recent report shows that miR-125b confers human glioblastoma stem cells resistance to temozolomide through the mitochondrial pathway of apoptosis,17 whereas miR-125b inhibitor can enhance the chemosensitivity of GSCs to temozolomide by targeting Bak1 and PIAS3.33, 34
Recently, miR16 was found to be a differentially expressed miRNA in glioma.10, 11 It has been reported that miR16 can inhibit migration and invasion of glioma cells.12, 13 Nevertheless, the expression of miR16 in GSCs remains unclear. To this end, we performed microRNA microarray profiling assay to examine the miR16 expression in human GBM cell lines SGH44, U87 and U251 as well as their GSCs. In the profiling data, several upregulated miRNAs, including hsa-miR16 were identified in GSCs, as compared to their counterpart glioblastoma cell lines, suggesting the involvement of miR16 in the regulation of GSCs function.
To address this question, in this study we examined the effects of the ectopic expressed miR16 mimic or miR16 on cell proliferation, migration and invasion in either glioblastoma cells or GSCs. Compared with the non-transfected controls, miR16 mimic inhibits cell proliferation, migration and invasion of GBM cells and its GSCs, but miR16 inhibitor exerted the reverse effects. In this study, CDK6, CCND1, CCNE1 and Bcl2 were identified as the target genes of miR16. Thus, we believe that miR16 may regulate cell proliferation of GBM cells by targeting cell cycle-related genes.
MiR15 and miR16 may suppress tumorigenesis by inhibiting the anti-apoptotic factor Bcl2, according to a miRNA profiling study.35 Our in vitro experiments found that miR16 inhibited migration, motility, invasion and colony formation of glioblastoma cells. Moreover, intracerebral inoculation of the miR16 inhibitor-transfected U251 cells in nude mice appeared to promote tumor growth. Our results are consistent with other reports of the inhibition of glioma cell migration and invasion by miR16.12, 13 Taken together, we consider miR16 may function as a tumor suppressor to regulate the development of glioblastoma.
Considering that miR16 can inhibit the abilities of proliferation, migration and invasion in both GBM cells and GSCs, we speculated the differentially expressed miR16 in GSCs may regulate GSCs differentiation. Therefore, we examined whether miR16 can regulate the capability of in vitro-differentiation of GSCs and found that the differentiation time in the cells treated with miR16 inhibitor was significantly longer, as compared to those with miR16 mimic, but the ratio of differentiated GSCs (CD133−, GFAP+) in miR16 mimic group was higher than that in miR16 inhibitor group (Figure 6c). These data support that overexpression of miR16 can promote GSCs differentiation, whereas inhibition of miR16 expression might suppress GSCs differentiation. Our findings revealed a novel biological function of miR16 related to its role in tumor suppression, especially for targeting GSCs. We are also interested to explore the mechanism underlying miR16-mediated GSCs differentiation and find novel targets of miR16 that contribute to the regulation of GSCs differentiation. Through bioinformatics analysis, we found that SOX5 is an interesting target of miR16, which is also associated with development of glioma.36, 37 In addition, SOX2 and SOX5 belong to SRY-related HMG-box (SOX) family. As SOX2 is a well-known gene associated with stem cells, it is interesting to explore the correlation of miR16 with SOX family members in GSCs. Previous study revealed that gliogenesis is dependent on SOXD including SOX5, SOX6 and SOX13 as well as the members of SOXE, such as SOX8, SOX9 and SOX10.30 SOX5 is one of the members of the SOX family, which is a transcription factor to participate in the gene regulation and maintenance of chromatin structure in a wide variety of developmental processes. Expression of ectopic SOX5 in human glioma cells inhibited their clone formation and proliferation.36, 37 Our results showed that the transfection of GSCs with the miR16 mimic appeared to promote the differentiation of GSCs. Furthermore, a poor prognosis of the patients of GMB correlates with the downregulated miR16 and high expression of SOX5, suggesting the possible involvement of miR16 in the differentiation of GSCs by targeting SOX5. Similarly, a previous report revealed that miR-124 and miR-137 can promote the differentiation of glioblastoma-derived GSCs, leading to cell-cycle arrest in glioblastoma.38
It has been reported that miR128-1, a brain-specific miRNA is downregulated in both GBM and GSCs due to a mechanism related to DNA methylation. The expression of miR128-1 is closely associated with the progression of GBM. Either forced expression of miR128-1 or treatment with the DNA methylation inhibitors Aza or PBA suppressed the potentials of proliferation, migration and invasion of cancer cells in vitro and transplant tumor growth in vivo, through direct targeting BMI1 and E2F3.39 However, the underlying molecular mechanism of the downregulation of miR16 in GBM and GSCs remains largely unknown. It is necessary to investigate whether it is related to gene methylation or other mechanism in our future study.
The correlation of miR16 expression and prognosis of patients with GMB was analyzed in our study, we found that the median OS of GMB patients in the low-miR16 group was significantly shorter (10±3.36 months), as compared to that (19±2.61 months) in the high-miR16 group. The median OS of the patients with high-expression levels of CCND1 or SOX5, both of which are target genes of miR16, was also significantly shorter, similar to the low-miR16 group. However, the expression levels of other miR16 target genes, such as Bcl2, CDK6 and CCNE1 were not found to be related to the OS of patients with GMB. The prognostic value of miR16 in GMB highlighted the complexity of miR16 functions.
In summary, we demonstrated the differentially expression of miR16 in glioblastoma cells and GSCs. Furthermore, the evidence indicates that miR16 may function as a tumor suppressor, possibly by inhibiting the genes expression of CDK6, CCND1, CCNE1 and Bcl2. Downregulation of miR16 expression might be associated with prognosis of patients with GMB.
Materials and methods
Patients and samples
Paraffin-embedded tumor specimens from 132 cases (male 84, female 48, median age 45 years, range: 12–78 yeas), were surgically collected during the period of September 2010–June 2014. Diagnosis of GBM in those cases was confirmed by pathological examination. All patients in this study did not receive any treatment of chemotherapy or radiotherapy, prior to the surgical excision. In addition, 11 cases of normal brain tissues (non-tumor tissues) were obtained as a control. All the patients provided signed written informed consent. A median of 20 months (range from 5–50 months, by July 2015) follow-up in all of 132 patients was included. Our study was approved by the ethics committee of Anhui provincial hospitals.
Cell culture
SGH44, U87 and U251 are human glioblastoma cell lines, which were obtained from Shanghai Life Sciences Research Institute Cell Resources Center, Chinese Academy of Science, Shanghai, China. Cells were cultured in DMEM (Gibco, Thermo Scientific, New York, NY, USA) plus 10% FBS (Fetal bovine serum, products of Gibco, Thermo Scientific). To induce the formation of stem cells, cells were maintained in serum-free DMEM/F12 (Gibco, Thermo Scientific) supplemented with 2% B27 purchased from Gibco, Thermo Scientific; recombinant leukemia inhibitory factor (10 ng/ml, from PeproTech, Rocky Hill, RI, USA); basic fibroblast growth factor (bFGF) (20 ng/ml, also from PeproTech); epidermal growth factor (EGF) (20 ng/ml, products of PeproTech); penicillin (100 U/ml, from Shijiazhuang, Hebei, China); and streptomycin (100 μg/ml, from Shijiazhuang). At 7–10th day after initial seeding, the number of gliospheres was measured by a bright field microscope. The gliospheres were fixed with the solution containing 2.5% glutaraldehyde and 1% osmic acid. After dehydration with an acetone gradient, samples were incubated with isoamyl acetate. Then, the spray-dried samples were used for the analysis using scanning electron microscopy (SEM) (FEI, USA).
MicroRNA microarray profiling
Trizol reagent (Invitrogen, Grand Island, New York, NY, USA) was used to isolate total RNA from glioblastoma cell lines and their GSCs. Normal glial cells were also used as a control. RNA samples were labeled and hybridized by using the miRCURY LNA (locked nucleic acid) Array v14.0 (Exiqon, Vedbaek, Denmark). The hybridization results were scanned by the Axon GenePix 4000B microarray scanner. The raw intensity of the image was reading GenePix pro V6.0 software (Axon Instruments; Foster City, CA, USA). The net intensity of green signals was obtained by subtracting background. Then, the median value of four replicates of each probe on the same slide was determined. Fold changes in expression of each type of miRNA were examined. In the microarray data analysis and cluster analysis, we defined the differentially expressed miRNAs, which was increased or decreased more than twofolds, as compared to normal control.
Ectopic expression of miR16 and inhibition of endogenous miR16
GBM cells and their GSCs were transfected with miR16 mimic or miR16 inhibitor or scrambled control transiently (200 nm each, form Shanghai GenePharma, Shanghai, China) (Supplementary Table S1) using Lipofectamine 3000 reagent, or infected with lentivirus particles of miR16 (Shanghai GenePharma) for stably expression. Stable expressed cells were selected by G418 treatment for 4 weeks.
Target-luciferase reporter assay
The constructs of target-luciferase reporters, including Bcl2, CCND1, CCNE1, CDK6 and SOX5 have either wt (wild-type) or mut (mutant) miR16 binding sites in 3′-UTR region of target genes were established. Co-transfection of miR16 and different target-corresponding luciferase reporters was performed in U251 cells. NC mimic was used as a negative control in the reporter assay. Forty-eight hours after transfection, the activity of luciferase reporter was measured by Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA), according to the manufacturer’s instructions. The luciferase activity was normalized to the Renilla activity. Its activity was presented as relative activity to the corresponding NC (assigned as value ‘1’). Values are the mean±SEM of three independent assays.
Quantitative reverse transcription PCR
RNA was isolated from paraffin-embedded tumor specimens using OMEGA FFPE Kit and examined using BioDrop μLite, Micro-Volume UV/Vis Spectrophotometer. Reaction of reverse transcription with High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, ABI Technologies, Foster City, CA, USA) was performed at 25 °C for 10 min, 37 °C for 120 min, 85 °C for 5 min, cooled at 4 °C, following the manufacturer’s instructions. MiRNA was reverse transcribed under the following condition: 16 °C (30 min), 42 °C (30 min), 85 °C (5 min) and cooled at 4 °C. Primers for qRT–PCR were synthesized by Applied Biosystems (Supplementary Table S2). Those primers were used to measure expression of miR16 and its target genes, using miR TaqMan real-time PCR detection kit (Applied Biosystems). U6 was served as internal control for PCR. The profile of qPCR was as follows: hot-start at 95 °C (10 min), then 40 cycles of denaturation at 95 °C (15 s), annealing and extension at 60 °C (1 min). β-actin was served as the internal control for PCR. The profile of qPCR for target genes was pre-denaturation at 50 °C for 2 min, 95 °C (10 min), followed by 50 cycles of denaturation at 95 °C (15 s), annealing and extension at 60 °C (1 min). The expression level of the specific gene between samples was expressed as relative expression by calculating 2−ΔΔCT of each sample.
Western blot analysis
Total protein of cell lysates was separated on a precast 4 to 15% sodium dodecyl sulfate polyacrylamide gel (Invitrogen) and transferred onto PVDF membrane, followed by blocking and probing with primary antibodies against Bcl2, CCND1, CCNE1, CDK2, CDK6 and SOX5 (GAPDH as an internal control). The bound antibodies were detected with HRP-conjugated secondary antibodies and visualized by an ECL plus chemiluminescence (Beyotime, Haimen, China). Densitometric analysis of protein bands was performed using Image J software (National Institutes of Health (NIH), Stapleton, NY, USA).
Anchorage-dependent and independent colony formation assays
Cells were seeded in a 6-well culture (200 cells per well) and cultured for about 10 or 12 days. Cells were fixed with 4% paraformaldehyde and stained with dye of 0.1% crystal violet. The number of anchorage-dependent colonies (defined as clusters of >50 cells) was measured by inverted microscope. Calculation of colony forming efficiency (%) was performed by a ratio of the number of colonies versus number of actual inoculated cells × 100%. Soft-agar assay for anchorage-independent growth was performed using 0.3% agarose with 1 × 103 suspended cells, which was plated over a layer of 0.7% agarose on a 60-mm plate. Two or 3 weeks after initial plating, colonies (clusters of >50 cells) were fixed, stained with Giemsa solution and counted.
GSCs assessment by Matrigel colony forming assay and differentiation assay
GSC-enriched cells were collected and then cultured in the serum-free DMEM/F12 medium with 0.2% Matrigel (BD Biosciences, New York, NY, USA) and overlaid onto 0.5 mm thick bottom Matrigel in a 6-well plate (30 cells per well). The GSC colonies (>10 cells) were counted under a light microscope after culture in Matrigel for 10-15 days. For differentiation assay, GCS-enriched cells were collected, suspended in DMEM medium and cultured till all cells grew as adherent cultures for 5 days. The differentiated cells that show positive expression of GFAP-PE, but negative expression of CD133/2-PE, were confirmed by immune fluorescence and flow cytometry.
Cell proliferation
Cell proliferative potential was determined by CCK8 assay (Dojindo Laboratories, Kumamoto, Japan) in 96-well plate, following the instruction provided by manufacturer. The potentials of cell proliferation was determined, based on absorbance at 450 nm (excitation) and 600 nm (emission), measured in a spectrophotometer (Spectra Max 190, from Molecular Devices, Sunnyvale, CA, USA). The data are presented as the mean±s.d. (three independent experiments with triplicate in each condition).
Assessment of cell motility and invasion or wound healing assays
The cell motility and invasion abilities was estimated by an assay, in which cells were cultured in 24-well transwell cell culture chambers supplied with polycarbonate membranes (6.5 mm diameter, 8 μm pore size, from Corning, New York, NY, USA). 24 or 48 h after incubation, fix the migrated cells in 4% paraformaldehyde. Then stain those cells with crystal violet. The number of stained cells was counted by a bright field microscope (five fields randomly selected). Wound healing assay was performed using 1 × 106 cells in each well in 6-well plates. Cell monolayer was gently scratched with a 100-μl pipette tip across the center of the well. Wound healing capacity of each sample was evaluated by cell movement at the scratched area (width of the wounded area) at time points of 0, 24, 48 and 72 h after the scratch. The rate of migration was calculated as a percentage of the control, that is, the proportion of the distance where is cell free between both borderlines caused by scratching. All assays were performed in triplicate.
Flow cytometric analysis, cellular immune fluorescence and IHC
Cells transfected with miR16 mimics /inhibitor or NC were harvested at 72 h after transfection. Suspended cells (1 × 106 per ml) were fixed with 70% ethanol and stained with propidium iodide (PI) for flow cytometric analysis (FCM) using flow cytometer (BD Biosciences) and FlowJo software. Apoptotic cells was determined by FCM and Cytosoft software (Guava Technologies, Chicago, IL, USA) using Alexa Fluor 488 annexin V/Dead Cell Apoptosis Kit (Thermo Fisher Scientific, Catalog # V13245), according to manufacturer’s instructions. DNA replication was analyzed using Click-iT Plus EdU Alexa Fluor 488 Flow Cytometry Assay Kit (Catalog # C10632, Thermo Fisher Scientific). Immune fluorescence was performed to examine cell surface markers using primary antibodies GFAP-PE (BD Biosciences) and CD133/2-PE (Miltenyi Biotec, Bergisch Gladbach, Cologne, Germany) and PE-conjugated secondary antibodies. Cells were also counterstained with 4′, 6-diamidino-2-phenylindole (DAPI) and examined under a fluorescent microscope. The protein expression of mi16 target genes in tumor tissues (paraffin-embedded tissue sections) was examined by IHC using BenchMark XT kit (Roche, Basle, Switzerland). Specific antibodies used in IHC were Bcl2 (ZM-0010), CCND1 (ZA-0101), nestin (ZM-0323), SOX2 (ZA-0571) and Ki67 (ZM-0167) from Zhongshan Golden Bridge Bio-technology, Beijing, China as well as CCNE1 (ab88259), CDK6 (ab54576) and SOX5 (ab26041) from Abcam, MA, USA), including secondary antibodies HRP-conjugated goat anti mouse or anti rabbit Ig G/Ig M (Roche). Positive cells were semi-quantitatively analyzed using score 0 (negative), 1 (pale yellow), 2 (yellow) and 3 (brown).
Orthotopic implantation of GBM cells or GSCs in nude mice
Approval of the animal treatment protocols by the Ethics Committee for the Care and Use of Laboratory Animals of Anhui Medical University was received before experiments in vivo. Forty female (6–8 weeks old) nude mice were randomly separated into 8 groups (5 mice per group). Two control groups were inoculated in bregma, where is 1 mm posterior and 3 mm ventral to the surface of the brain, with non-transfected U251 cells or U251-GSC, whereas other six groups were inoculated with 1 × 106 U251 cells or U251-GSC that had been transfected with either miR16 mimic, or miR16 inhibitor, or scrambled control. Nude mice body weight and their status were daily monitored after inoculation of tumor cells. When mice displayed obvious symptoms (weight loss >20% body mass, or limbs paralysis or movement disorder, or lethargy, and a hunched posture), they will be euthanized. The intact brains were removed to prepare paraffin-embedded tissue sections for H&E staining and IHC. The volume of tumor was calculated by the formula as flows, Tumor volume = (width) 2 × length × 0.5.
Statistical analysis
Analysis of statistical data was performed using SPSS 21.0 software (SPSS Inc., Chicago, IL, USA). The difference between individual samples was tested using t-test (two-tailed unpaired). One-way analysis of variance was used to test the difference among multiple groups. To test the difference between two groups, Bonferroni's multiple comparison test was performed. Results are shown as means±s.e.m. or ±s.d. P<0.05 means statistically significant. The differences between the survival curves were test by log-rank test, whereas survival was estimated by the Kaplan–Meier method.
References
Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro Oncol 2013; 15 (Suppl 2): : ii1–56.
Cuddapah VA, Robel S, Watkins S, Sontheimer H . A neurocentric perspective on glioma invasion. Nat Rev Neurosci 2014; 15: 455–465.
Suvà ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014; 157: 580–594.
Bartel DP . MicroRNAs: target recognition and regulatory functions. Cell 2009; 136: 215–233.
Bartel DP . MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–297.
Krichevsky AM, King KS, Donahue CP, Khrapko K, Kosik KS . A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 2003; 9: 1274–1281.
Sempere LF, Freemantle S, Pitharowe I, Moss E, Dmitrovsky E, Ambros V . Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol 2004; 5: R13.
Areeb Z, Stylli SS, Koldej R, Ritchie DS, Siegal T, Morokoff AP et al. MicroRNA as potential biomarkers in glioblastoma. J Neurooncol 2015; 125: 237–248.
Ouyang Q, Xu L, Cui H, Xu M, Liang Y . MicroRNAs and cell cycle of malignant glioma. Int J Neurosci 2016; 126: 1–9.
Li C, Sun J, Xiang Q, Liang Y, Zhao N, Zhang Z et al. Prognostic role of microRNA-21 expression in gliomas: a meta-analysis. J Neurooncol 2016; 130: 11–17.
Brower JV, Clark PA, Lyon W, Kuo JS . MicroRNAs in cancer: glioblastoma and glioblastoma cancer stem cells. Neurochem Int 2014; 77: 68–77.
Yang TQ, Lu XJ, Wu TF, Ding DD, Zhao ZH, Chen GL et al. MicroRNA-16 inhibits glioma cell growth and invasion through suppression of BCL2 and the nuclear factor-κB1/MMP9 signaling pathway. Cancer Sci 2014; 105: 265–271.
Li X, Ling N, Bai Y, Dong W, Hui GZ, Liu D et al. MiR-16-1 plays a role in reducing migration and invasion of glioma cells. Anat Rec (Hoboken) 2013; 296: 427–432.
He XY, Liao YD, Guo XQ, Wang R, Xiao ZY, Wang YG . Prognostic role of microRNA-21 expression in brain tumors: a meta-analysis. Mol Neurobiol 2016; 53: 1856–1861.
Wang Q, Li X, Zhu Y, Yang P . MicroRNA-16 suppresses epithelial–mesenchymal transition-related gene expression in human glioma. Mol Med Rep 2014; 10: 3310–3314.
Li X, Zheng J, Chen L, Diao H, Liu Y . Predictive and prognostic roles of abnormal expression of tissue miR-125b, miR-221, and miR-222 in glioma. Mol Neurobiol 2016; 53: 577–583.
Shi L, Zhang S, Feng K, Wu F, Wan Y, Wang Z et al. MicroRNA-125b-2 confers human glioblastoma stem cells resistance to temozolomide through the mitochondrial pathway of apoptosis. Int J Oncol 2012; 40: 119–129.
Wu N, Xiao L, Zhao X, Zhao J, Wang J, Wang F et al. miR-125b regulates the proliferation of glioblastoma stem cells by targeting E2F2. FEBS Lett 2012; 586: 3831–3839.
Yue J, Tigyi G . Conservation of miR-15a/16-1 and miR-15b/16-2 clusters. Mamm Genome 2010; 21: 88–94.
Blakaj A, Lin H . Piecing together the mosaic of early mammalian development through microRNAs. J Biol Chem 2008; 283: 9505–9508.
Aqeilan RI, Calin GA, Croce CM . miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ 2010; 17: 215–220.
Liu Q, Fu H, Sun F, Zhang H, Yi T, Zhu J et al. miR-16 family induces cell cycle arrest by regulating multiple cell cycle gene. Nucleic Acids Res 2008; 36: 5391–5404.
Santhi WS, Prathibha R, Charles S, Anurup KG, Reshmi G, Ramachandran S et al. Oncogenic microRNAs as biomarkers of oral tumorigenesis and minimal residual disease. Oral Oncol 2013; 49: 567–575.
Liu G, Keeler BE, Zhukareva V, Houlé JD . Cycling exercise affects the expression of apoptosis-associated microRNAs after spinal cord injury in rats. Exp Neurol 2010; 226: 200–206.
Pavon LF, Sibov TT, de Oliveira DM, Marti LC, Cabral FR, Malheiros SM et al. Mesenchymal stem cell-like properties of CD133+ glioblastomainitiating cells. Oncotarget 2016; 7: 40546–40557.
Najbauer J, Kraljik N, Németh P . Glioma stem cells: markers, hallmarks and therapeutic targeting by metformin. Pathol Oncol Res 2014; 20: 789–797.
Rinkenbaugh AL, Cogswell PC, Calamini B, Dunn DE, Persson AI, Weiss WA et al. IKK/NF-κB signaling contributes to glioblastoma stem cell maintenance. Oncotarget 2016; 7: 69173–69187.
Ciafrè SA, Galardi S, Mangiola A, Ferracin M, C-G L, Sabatino G et al. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun 2005; 334: 1351–1358.
Zhi F, Chen X, Wang S, Xia X, Shi Y, Guan W et al. The use of hsa-miR-21, hsa-miR-181b and hsa-miR-106a as prognostic indicators of astrocytoma. Eur J Cancer 2010; 46: 1640–1649.
Guessous F, Zhang Y, Kofman A, Catania A, Li Y, Schiff D et al. microRNA-34a is tumor suppressive in brain tumors and glioma stem cells. Cell Cycle 2010; 9: 1031–1036.
Li Y, Guessous F, Zhang Y, DiPierro C, Kefas B, Johnson E et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res 2009; 69: 7569–7576.
Shi L, Wan Y, Sun G, Gu X, Qian C, Yan W et al. Functional differences of miR-125b on the invasion of primary glioblastoma CD133-negative cells and CD133-positive cells. Neuromolecular Med 2012; 14: 303–316.
Chen J, Fu X, Wan Y, Wang Z, Jiang D, Shi L . miR-125b inhibitor enhance the chemosensitivity of glioblastoma stem cells to temozolomide by targeting Bak1. Tumor Biol 2014; 35: 6293–6302.
Shi L, Wan Y, Sun G, Zhang S, Wang Z, Zeng Y . miR-125b inhibitor may enhance the invasion-prevention activity of temozolomide in glioblastoma stem cells by targeting PIAS3. BioDrugs 2014; 28: 41–54.
Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, Dumitru CD et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci USA 2004; 101: 11755–11760.
Boije M, Krajisnik T, Jiang Y, Kastemar M, Uhrbom L . Upregulation of SOX5 perturbs human glioma cell proliferation and is associated with proneural glioblastoma. Cancer Res 2012; 72: 3343–3343.
Tchougounova E, Jiang Y, Bråsäter D, Lindberg N, Kastemar M, Asplund A et al. Sox5 can suppress platelet-derived growth factor B-induced glioma development in Ink4a-deficient mice through induction of acute cellular senescence. Oncogene 2009; 28: 1537–1548.
Silber J, Lim DA, Petritsch C, Persson AI, Maunakea AK, Yu M et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med 2008; 6: 14.
Shan ZN, Tian R, Zhang M, Gui ZH, Wu J, He J et al. miR128-1 inhibits the growth of glioblastoma multiforme and glioma stem-like cells via targeting BMI1 and E2F3. Oncotarget 2016; 7: 78813–78826.
Acknowledgements
This study was sponsored by National Natural Science of China (Grant Nos. 81272800, 81072057 and 8152323).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Tian, R., Wang, J., Yan, H. et al. Differential expression of miR16 in glioblastoma and glioblastoma stem cells: their correlation with proliferation, differentiation, metastasis and prognosis. Oncogene 36, 5861–5873 (2017). https://doi.org/10.1038/onc.2017.182
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2017.182
- Springer Nature Limited