
Abstract
In addition to alleviating depression, long-term adaptive changes induced by antidepressants may regulate neural plasticity in the diseased brain, providing symptomatic and disease-modifying effects in Parkinson’s disease. The present study investigated whether chronic treatment with a frequently prescribed tricyclic antidepressant was neuroprotective in a 6-hydroxydopamine (6-OHDA) rat model of parkinsonism. In lesioned animals, chronic amitriptyline (AMI; 5 mg/kg) treatment resulted in a significant sparing of tyrosine hydroxylase-immunoreactive (THir) neurons in the substantia nigra pars compacta (SNpc) compared with saline treatment. Additionally, striatal fibers were preserved and functional motor deficits were attenuated. Although 6-OHDA lesions did not induce anhedonia in our model, the dose of AMI utilized had antidepressant activity as demonstrated by reduced immobility. Recent in vitro and in vivo data provide evidence that trophic factors such as brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF) may be key mediators of the therapeutic response to antidepressants. Therefore, we investigated whether AMI mediates changes in these specific trophic factors in the intact and degenerating nigrostriatal system. Chronic AMI treatment mediates an increase in nigral BDNF both before and during ongoing degeneration, suggesting it may contribute to neuroprotection observed in vivo. However, over time, AMI reduced BDNF levels in the striatum, indicating tricyclic therapy differentially regulates trophic factors within the nigrostriatal system. Combined, these results suggest that AMI treatment attenuates dopamine neuron loss and elicits significant trophic changes relevant to dopamine neuron survival.
Similar content being viewed by others
INTRODUCTION
To date, there are no successful therapies to slow the progression of Parkinson’s disease (PD); however, accumulating evidence suggests that antidepressants may offer such a benefit. Support for this comes from a multitude of studies that demonstrate how antidepressants such as tricyclics (TCAs), serotonin and norepinephrine reuptake inhibitors, and selective serotonin reuptake inhibitors modulate the signaling pathways involved in cell survival and plasticity (Drzyzga et al, 2009). Antidepressants elicit their therapeutic effect, in part, by increasing trophic factors in the brain. This trophic response is not specific to a particular class of antidepressant, as a variety of antidepressant drugs elicit significant increases in brain-derived neurotrophic factor (BDNF) in the rodent hippocampus, prefrontal cortex, ventral tegmental area, and nucleus accumbens (Nibuya et al, 1995; De Foubert et al, 2004; Martinez-Turrillas et al, 2005; Rogoz and Legutko, 2005). Further, the TCA amitriptyline (AMI) has been shown to significantly increase glial cell line-derived neurotrophic factor (GDNF) mRNA expression and release in rat and human astrocytes and C6 glioblastoma cells (Hisaoka et al, 2007; Kajitani et al, 2012). A more recent study demonstrated that fluoxetine treatment attenuated behavioral and neuropathological deficits by reversing the deficiency of hippocampal BDNF and GDNF in a transgenic alpha-synucleinopathy model (Ubhi et al, 2012). There is also clinical evidence that antidepressants augment trophic factors in humans. For example, several studies report that depressed patients chronically treated with antidepressants have significantly higher plasma BDNF levels compared with non-treated subjects (Gonul et al, 2005; Aydemir et al, 2006). Furthermore, post-mortem studies confirm elevated BDNF expression in the dentate gyrus, hilus, and supragranular regions in patients treated with antidepressants compared with untreated patients (Chen et al, 2001). Collectively, these studies provide evidence that antidepressants augment levels of BDNF and GDNF, trophic factors known to be important to dopamine (DA) neuron survival and function (Sautter et al, 1998; Allen et al, 2013). However, it is not known whether trophic factor changes occur in brain regions relevant to PD, whether these changes are dependent on the type of antidepressant, or whether such changes might correlate with neuroprotection in the parkinsonian brain.
Although it is true that many classes of antidepressants may afford general neuroprotection, specificity of neuroprotection in the parkinsonian brain may be unique to TCAs. We recently reported that TCAs (specifically AMI), were associated with a delay in the need to start dopaminergic therapy in patients with PD, and this apparent slowing in progression was not attributable to symptomatic effects (Paumier et al, 2012). Others have also demonstrated the therapeutic potential of AMI as a neuroprotective agent (Xu et al, 2003; Han and Lee, 2009; Jang et al, 2009). Specifically, a repeated low dose of AMI has been shown to increase BDNF expression in rodent brain and inhibit apoptosis (Xu et al, 2003). AMI also elicits neuroprotection by binding directly to neurotrophic factor receptors TrkA and TrkB (Jang et al, 2009). More recently, AMI was shown to mediate changes in hippocampal BDNF and GDNF, resulting in reduced neuroinflammation and alpha-synuclein accumulation in a mouse model of multiple systems atrophy (Valera et al, 2014). Finally, nortriptyline (a metabolite of AMI), was shown to delay the onset of neurodegeneration in mouse models of ALS and Huntington's disease (Wang et al, 2007). Collectively, these studies provide evidence that antidepressants, and AMI specifically, afford neuroprotection in multiple animal models of neurodegeneration.
As trophic factors may be important in the cellular adaptations that occur with chronic antidepressant treatment, the present study was designed to examine whether AMI: (i) elicits DA neuroprotection in a rat model of PD and (ii) mediates changes in relevant trophic factor levels within the intact and degenerating nigrostriatal system. Findings from this study are the first to demonstrate that AMI significantly attenuates DA neuron loss, preserves striatal DA fibers, and prevents motor deficits in a rat model of parkinsonism. Furthermore, these studies show that chronic AMI treatment significantly alters trophic support within the nigrostriatal system.
MATERIALS AND METHODS
Animals
Adult male Wistar rats (200–225 g; Harlan Laboratory) were utilized in all experiments. Studies were conducted at both the University of Cincinnati (UC) and Michigan State University (MSU). Rats at MSU were housed in the Van Andel Research Institute vivarium. Both animal facilities are accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care and complied with all Federal animal care and use guidelines. The Institutional Animal Care and Use Committee at both institutions approved all protocols.
Experimental Design
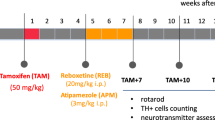
To assess whether AMI was neuroprotective, rats were randomized and divided into four groups (Saline-no lesion, AMI-no lesion, Saline+lesion, AMI+lesion (n=8–12 per group)). Owing to the delay in the therapeutic efficacy of antidepressants (ie, 2–3 weeks latency) (Malberg et al, 2000; Martinez-Turrillas et al, 2005), rats were pretreated 2 weeks prior to lesioning. Animals received daily i.p. injections of sterile saline (Henry Schein) or 5 mg/kg of AMI (Sigma) for 14 days. On day 15, rats did not receive saline or AMI injections; instead, rats in the appropriate groups (+lesion) received intrastriatal 6-hydroxydopamine (6-OHDA). The following day, all rats continued daily treatment over the course of 4 weeks. Behavioral testing was performed on day 14 to establish baseline levels and again 2 (day 28) and 4 weeks (day 42) later. Rats were killed on day 45 and brains were extracted and processed for immunohistochemical analyses.
An additional study was conducted using a separate cohort of animals (n=4–10 per group) to determine trophic changes within the nigrostriatal system. Rats received daily AMI (5 mg/kg) or saline injections over the same time course as the previous study, then select cohorts of rats from both groups were killed on days 14, 17, and 42 (time-points corresponding to the following days pre- or post-6-OHDA injection: day 0 (D0), day 3 (D3), and day 30 (D30)). Rats were killed at prescribed time-points, and then brains were extracted and processed for ELISA analysis.
6-OHDA Lesions
Anesthetized rats (0.3 mg/kg, equithesin, i.p.) were placed into a stereotaxic device and 1 μl of 6-OHDA (0.2% ascorbic acid-physiological saline; 5 μg/μl) was injected into two sites of the left striatum (AP +0.7, ML +3.0, DV −5.5; AP +0.7, ML +3.0, DV −5.0 from skull) at 0.5 μl per minute. The needle was left in place for 2 min and slowly withdrawn. Lesions were verified by needle tracks and morphological assessment of substantia nigra pars compacta (SNpc) neurons immunoreactive for tyrosine hydroxylase (THir).
Behavioral Evaluations
For all behavioral tasks, intact rats treated with saline (n=7) or AMI (n=9) served as controls and were compared with lesioned rats treated with saline (n=10) or AMI (n=11). Data presented are the averaged scores from two independent raters who were blind to treatment groups. All behavioral testing was repeated with two cohorts of animals on two separate occasions.
Cylinder task
Forelimb asymmetry during spontaneous exploration of the walls of a cylindrical enclosure is a common test of motor function after unilateral 6-OHDA lesions (Schallert, 2006). During the dark cycle, rats were placed in a clear, 16.5 cm tall plexiglass cylinder and videotaped for 5 min. The number of weight-bearing forepaw touches (left, right, or both paws) was recorded (up to 20) in a given trial. Data are reported as the percentage of contralateral forelimb use: ((contra+1/2 both) divided by (ipsi+contra+both)) × 100 as described previously (Schallert, 2006).
Tail suspension test
We utilized a modified version of the tail suspension test commonly used for mice (Chermat et al, 1986). Rats were suspended by their tails within a large plexiglass cylinder for a total of four minutes (240 s). Each rat was videotaped and the amount of time spent immobile, a correlate of depression-like behavior, was recorded. Additionally, the number of freezing bouts was recorded. Results are reported as mean duration of immobility±SEM.
Saccharin intake
Saccharin (SACC) preference is frequently used as measure of anhedonia in rodents (Pijlman et al, 2003; Gronli et al, 2005). Single-housed rats were trained to consume a palatable SACC solution (0.1%) 3 days prior to commencing antidepressant treatment. Each rat was exposed to two pre-weighed bottles for 30 min: one with water and one with a 0.1% SACC solution. The sum of water and SACC consumption was defined as the total intake. The percentage of SACC intake was calculated: % sucrose preference=sucrose intake × 100/total intake.
Immunohistochemistry
Animals were killed via pentobarbital overdose (60 mg/kg) and intracardially perfused with saline, then 4% paraformaldehyde. Brains were post-fixed in 4% paraformaldehyde for 24 h and sunk in 30% sucrose. A 1:6 series of free-floating coronal sections cut (40 μm) on a freezing microtome platform was double labeled for TH (DA) and NeuN (neurons). Tissue was incubated in 0.3% H202 for 45 min, rinsed and blocked in 10% goat serum (1 h) then incubated in primary mouse anti-NeuN (1:400; Chemicon) and rabbit-anti-TH (1:8000; Immunostar) antibodies overnight at 4 °C. Then, sections were incubated in biotinylated secondary antisera against mouse IgG (1:400, Chemicon) and rabbit (1:400, Chemicon) followed by Vector ABC detection kit (Vector Labs). Antibody labeling was visualized by exposure to 0.5 mg/ml 3,3' diaminobenzidine (DAB), 2.5 mg/ml nickel ammonium sulfate and 0.03% H2O2 in Tris buffer followed by incubation with the NovaRed kit (Vector Labs). Sections were mounted on subbed slides, dehydrated to xylene, and coverslipped with Cytoseal (Richard-Allan Scientific).
ELISA
Animals were anesthetized (60 mg/kg, pentobarbital, i.p.) and perfused with heparinized saline (0.9%). Brains were flash-frozen in 3-methyl butane (Fisher) then stored at −80 °C. Coronal slabs (1–2 mm) were blocked utilizing a brain blocker (Zivic) and both hemispheres of the SN, striatum, and hippocampus were microdissected at a constant −12 °C on a cold plate (Teca) and stored at—80 °C until analysis. Samples were homogenized on ice in 200 ul of lysis buffer (M-PER; Pierce) with protease inhibitors (Sigma-Aldrich) and protein concentration was determined (BCA Kit, Pierce). The homogenate was centrifuged at 4 °C for 20 min at 14 000 g and the supernatant was collected. ELISAs were run using the Immunolon 4 HBX 96-well plate according to manufacturer’s instructions (GDNF/BDNF Emax ImmunoAssay Kit, Promega). Samples were read (<30 min) on a spectrophotometer (Multiskan Spectrum, Thermolab Systems) with unknown values interpolated against standard curves. BDNF and GDNF pg/mg protein levels are expressed as a percentage of values obtained for the intact saline-treatment group at baseline.
Stereology
Stereology software (MBF Bioscience) was used for cell counts. Low magnification ( × 1.25) was used to outline the SNpc and 20% of the designated area was sampled via a random series of counting frames (50 μm × 50 μm) systematically distributed across a grid (183 μm × 112 μm) placed over the SNpc. An investigator blinded to experimental conditions counted neurons using the optical fractionator probe ( × 60 with oil). A marker was placed on each NeuNir/THir or NeuNir/non-THir neuron within the counting frame while focusing through a z-stack of images (1–2 μm). The total number of stained neurons (N) was calculated using the following formula: N=NV · VROI where NV is the numerical density and VROI is the volume of the region of interest. The volume was calculated according to the Cavalieri method and variability within animals was assessed via the Gundersen Coefficient of Error (<0.1) (Gundersen et al, 1999).
Volumetric Analysis of Striatal Lesions
Neurolucida software (MBF Bioscience) was used to estimate lesion volume within the striatum. Quantitative analysis on traced data was carried out using he Neuroexplorer software (MBF Bioscience). An investigator blinded to the experimental conditions traced two contours for each section under low magnification ( × 1.25); first a contour of the complete striatum, then a contour of the lesioned area. The lesioned area is delineated by lack of TH staining intensity. Contours were generated throughout the entire striatal series (1:6). Measurements included the first section caudal to the forceps minor of the corpus callosum (+2.7 mm anterior to Bregma) and seven additional caudal sections (40 μm apart). The total volume was calculated using Cavalieri's formula: V=A*F*T, where A is the sum of areas calculated across all measured sections; F is the sampling frequency (ie, 6 for a 1-in-6 series); and T is the section thickness. The total lesion volume was determined by manually calculating the percentage of total lesioned volume within each striatal volume for each animal.
Western Blotting
To determine whether AMI pre-treatment downregulated DA transporter levels, male rats were injected daily (i.p.) with either saline (n=6) or AMI (5 mg/kg; n=6) for 2 weeks and then killed and perfused with physiological saline. The striatum was dissected on ice and homogenized in RIPA (Sigma) containing protease inhibitors (Complete Mini tabs, Roche) and left on ice (30 min) prior to centrifugation (14 000 g/20 min) at 4 °C. Protein concentration was determined with a BCA protein assay kit (Pierce). Lysates containing 50 μg of proteins were separated on 4–12% Bis-Tris gels (Invitrogen), transferred onto nitrocellulose membranes (Invitrogen), and probed with the following antibodies: mouse anti-DA transporter (DAT) clone mAb16 (1:1000; Millipore), mouse anti-α-syn (1:1000, LB509; Covance), rabbit anti-beta tubulin (1:15 000; Cell Signaling) followed by goat anti-rabbit (1:10 000; Novus) and goat-anti-mouse (1:10 000; Novus). Protein bands were detected and quantified with the OdysseyClx infrared scanning system (Li Cor, Nebraska).
Statistical Analysis
Differences between two groups were analyzed by a two-tailed independent t-test. Behavioral measures were analyzed using a two-way ANOVA for repeated measures. AMI-mediated differences in trophic factor levels were analyzed by two-way ANOVA followed by Holm-Sidak multiple comparison post hoc analysis comparing AMI treatment to the saline treatment. When appropriate, post hoc comparisons were made between groups using the Student-Newman-Keuls or Bonferroni method.
RESULTS
AMI Hydrochloride Protects Nigral DA Neurons
Estimated total population counts revealed a significant sparing (F (3, 14)=7.729, p=0.0028) of THir neurons in the SNpc of rats that received chronic AMI treatment (intact: 15 948±1401 vs lesion: 12 693±1633) compared with saline control rats (intact: 16 341±2302 vs lesion: 5863±1296) measured at 4 weeks following 6-OHDA (Figure 1a–c).

Chronic amitriptyline treatment (AMI) prevents dopamine (DA) neuron loss. Rats treated chronically with saline had a substantial loss (−64%, a) of tyrosine hydroxylase immunoreactive (THir) neurons in the SN compared with rats treated with AMI (−25%, b). Stereological assessment revealed a significant sparing of THir neurons in the substantia nigra pars compacta (SNpc) of rats that received chronic AMI treatment compared with saline control animals (c). To ensure that AMI prevented THir neuron loss and not simply a downregulation of the TH phenotype, we quantified both NeuNir/nonTH (nondopaminergic; black arrows) and NeuNir/THir (dopamine: red arrows) neurons in the SNpc ipsilateral to 6-OHDA injection (d). Results indicate both populations of cells declined in comparable numbers, but overall there was less cell death in AMI treated animals (e). This is consistent with AMI promoting cell survival, not merely preservation of phenotype. Graph represents mean±SEM; *p<0.05, **p<0.01. Scale bar=250 μM.
Intrastriatal 6-OHDA Induces DA Neuron Death, Not Reversible Suppression of TH Phenotype
Results show an overall effect for treatment (F (1, 16)=6.94, p=0.018), indicating the total neuronal loss (NeuNir/nonTHir+NeuNir/THir) was significantly less in the AMI (−4612±13.7) compared with the saline group (−10 343±635.3). The average loss of total SNpc NeuNir neurons corresponds to loss of SNpc THir neurons in both AMI (−4613±1461 NeuN vs −4610±1092 TH) and saline (−9707.85± 4391 NeuN vs −10 978.40±1482 TH) treatment groups (Figure 1d). Post hoc analysis revealed that AMI rats possessed significantly more NeuNir/THir neurons compared with saline-treated rats (p<0.05). This pattern is consistent with our 6-OHDA parameters resulting in neuron death, not phenotype suppression, and demonstrates that AMI prevented the loss of SNpc DA neurons.
AMI Preserves Striatal THir Fibers and Prevents Forelimb Asymmetry
In saline-treated rats, intrastriatal 6-OHDA resulted in ≈50% loss in the volume of striatal THir innervation. Striatal fibers were partially spared in AMI-treated rats compared with saline controls (Figure 2a and b). Also, the volume of striatal denervation was significantly reduced in AMI-treated compared with saline-treated rats (t(11)=4.306, p=0.001) (Figure 2c). Cylinder task assessment of forelimb asymmetry indicated a significant overall effect of treatment demonstrating that AMI also provided functional benefit (F(3,33)=5.13; p=0.0051). Specifically, saline-treated lesioned rats exhibited a deficit in contralateral forelimb use at 2 (p<0.05) and 4 (p<0.01) weeks post lesion (Figure 2d).

Chronic amitriptyline partially preserves striatal THir fibers and prevents forelimb asymmetry. AMI-mediated nigrostriatal protection was verified with immunohistochemistry. THir staining in the striatum shows a substantial sparing of striatal fibers in AMI-treated rats compared with saline controls (a and b). Neurolucida volume estimates show that the volume of THir denervation was significantly reduced relative to the total striatal volume in AMI-treated rats (−32%) compared with controls (−50%; c). 6-OHDA lesions induced a significant deficit in contralateral forelimb use at 2 and 4 weeks post lesion compared with saline controls. There was no difference in forelimb use between lesioned AMI-treated animals, saline-treated control animals, or AMI-treated control animals, demonstrating that AMI prevents forelimb asymmetry in lesioned rats. Graphs represent mean±SEM; *p<0.05, **p<0.01, scale bar=250 μM. AMI, amitriptyline; L, lesion; SAL, saline; 6-OHDA, 6-hydroxydopamine.
Nigrostriatal Lesions Do Not Elicit Anhedonia, but a Low Dose of AMI Reduces Immobility
We examined two standard tests of depression-like behavior in rodents. No significant differences in SACC preference were observed between treatment groups at any time point (Figure 3a), thus demonstrating that the degree of lesion was not sufficient to induce anhedonia. However, in the tail suspension test, results from a two-way ANOVA show immobility was significantly decreased with time (F(2, 70)=25.02, p<0.0001) and treatment (F(3, 35)=5.54, p=0.019). Post hoc analyses indicated that animals treated with AMI spent significantly less time immobile at the 2- and 4-week time points compared with lesioned animals treated with saline (AMI compared with SAL+L, p<0.001; AMI+L compared with SAL+L, p<0.001; and AMI compared with SAL, p<0.05) (Figure 3b). Furthermore, the frequency of freezing bouts was significantly reduced in AMI-treated animals (t(10)=5.567, p=0.0002)(Figure 3c).

Partial dopamine depletion does not elicit a full depressive phenotype, but a low dose of amitriptyline reduces immobility. Rats were chronically treated with a low dose of AMI (5 mg/kg) or saline for a period of 6 weeks. Prior to beginning treatment, baseline scores were obtained, and animals were retested 2 and 4 weeks post lesion. Despite treatment group, all rats have equal preference for saccharin at all time points (p>0.05; a), suggesting that partial lesions do not produce this behavior consistent with anhedonia. However, results on a second measure of depression-like behavior indicate the duration of immobility was significantly reduced in AMI-treated intact and lesioned animals at the 2- and 4-week time point compared with saline-treated intact and lesioned animals (b). The number of freezing bouts was significantly reduced in AMI-treated animals compared with saline controls (c), suggesting that the low dose of AMI used in this study has antidepressant activity. Graphs represent mean±SEM; $p<0.05=AMI compared with SAL; **p<0.01, ***p<0.001=AMI compared with SAL +L; ##p<0.01, ###p<0.001=AMI +L compared with SAL+L. AMI, amitriptyline; H2O, water; kg, kilogram; L, lesion; mg, milligram; s, seconds.
Chronic AMI Does Not Downregulate DAT Levels
Owing to our pre-treatment design, it was important to assess whether chronic AMI treatment decreased DAT levels, which could impact the rate of 6-OHDA uptake. Western blot results show no difference in DAT levels after 2 weeks of treatment (t(10)=0.4299, p>0.05; Figure 4), suggesting that AMI does not downregulate DAT.

AMI does not alter dopamine transporter levels. To determine whether AMI-mediated neuroprotection was due to an effect on DAT levels, we treated rats for 2 weeks with either saline or AMI (5 mg/kg) and then examined DAT levels in the striatum. Western blot results indicate no significant differences between saline- and AMI-treated animals, suggesting that chronic AMI does not downregulate DAT. Graph represents mean±SEM. AMI, Amitriptyline; DAT, dopamine transporter.
Chronic AMI Significantly Increases SN BDNF Pre- and Post- 6-OHDA Insult
ELISAs were used to analyze BDNF/GDNF levels in the hippocampus, SN, and striatum of AMI- or saline-treated rats killed at specific time points relative to 6-OHDA lesions (Figure 5a). The hippocampus served as a positive control because antidepressant treatment is known to induce BDNF in this region over time (Nibuya et al, 1995; Nibuya et al, 1996). Results from a two-way ANOVA show a significant effect in the hippocampus in response to time (F(2, 47)=80.49, p<0.0001) and treatment (F(1, 47)=22.09, p<0.0001). Post hoc analyses indicate AMI treatment significantly increased BDNF (553%, p<0.0001) compared with saline controls in the hippocampus 30-days post lesion (Figure 5b). Results from a two-way ANOVA show a significant effect in the SN in response to treatment (F(1, 38)=5.707, p<0.05). Post hoc analyses show AMI treatment was associated with a significant increase in SN BDNF immediately post lesion (D3; 193%, p<0.05), which returned to baseline by day 30 (D30; Figure 5c). There was a substantial, but non-significant decrease (−56%, p=0.06) in BDNF in the striatum 30 days post lesion (Figure 5d). ANOVA results indicate a trend for an effect of treatment on GDNF in the hippocampus (F(1, 33)=3.733, p=0.06) and post hoc analysis shows GDNF levels are significantly elevated in response to AMI treatment pre-lesion (D0; 190%); however, levels were not significantly different between groups post lesion (Figure 5e). Nigrostriatal GDNF was not significantly altered (Figure 5f and g).

Chronic AMI differentially regulates nigrostriatal trophic support pre- and post 6-OHDA insult. Adult rats were treated daily with saline or AMI (5 mg/kg) for the duration of the study (6 weeks). Animals were killed at specific time points before (day 0; D0) and after (day 3; D3 and day 30; D30) DA depletion to assess the temporal course of AMI-mediated trophic changes in the nigrostriatal system (substantia nigra, SN and striatum, STR). The hippocampus was used as a positive control as antidepressant treatment induces BDNF in this region over time. Outlines of specific regions dissected for BDNF and GDNF ELISA analyses (a). AMI treatment significantly increased BDNF (553%) within the hippocampus (HIPP) 30 days post lesion (b). In the SN, AMI induced a non-significant increase in BDNF (148%) after 2 weeks of treatment in animals prior to the lesion (D0), and mediated a significant increase in BDNF (194%) immediately post lesion (D3), which returned to levels comparable with lesioned, saline-treated animals, by day 30 (D30). There was a substantial, but non-significant decrease in BDNF (−56%) in the striatum 30 days post lesion (d). All regions showed a slight increase in GDNF in response to AMI treatment (e–g); however, GDNF in the HIPP was significantly increased at D0 (187%; e). Values are expressed as a percentage of the baseline control (saline treated) group at day 0 (D0). Data represent mean±SEM and are combined from two separate experiments. Asterisk (*) indicates groups that differed significantly from saline-treated animals at each time point (*p⩽0.05; **p<0.01; ****p<0.0001). BDNF, brain derived neurotrophic factor; D0, day 0 (14 days of treatment, no lesion); D3, day 3 (17 days of treatment, 3 days post lesion); D30, day 30 (44 days of treatment, 30 days post lesion); GDNF, glial derived neurotrophic factor; HIPP, hippocampus; SN or Nigra, substantia nigra; STR, striatum.
DISCUSSION
The present study demonstrates that daily treatment with a low dose of AMI was sufficient to protect the vulnerable DA neurons in the SN and fibers in the striatum against a neurotoxic insult in a rat model of parkinsonism. Unbiased stereological assessment demonstrated a significant preservation of nigral DA neurons and striatal DA fibers after chronic AMI treatment. This AMI-mediated preservation of the nigrostriatal system translated into significant functional benefit as measured by forelimb use in the cylinder task. Furthermore, we provide the first evidence that chronic AMI treatment is associated with increased expression of BDNF within the nigrostriatal system. Taken together, these results suggest that AMI is neuroprotective in a toxin model of PD and increases trophic factors essential for DA neuron survival within the nigrostriatal system.
A variety of PD animal models (including unilateral toxin models) exhibit comorbid-depressive-like behaviors (Winter et al, 2007, Taylor et al, 2009; Santiago et al, 2010). However, the expression of these behaviors is dependent both upon the extent of DA depletion and the dose of antidepressant drug administered. Throughout the depression literature, antidepressants are administered to animals at doses ranging from 10 to 50 mg/kg (Nibuya et al, 1996; Malberg et al, 2000; Martinez-Turrillas et al, 2005); however, we opted to use a lower dose (5 mg/kg) that was more analogous to that commonly administered to a geriatric population (Antonini et al, 2006; Berger et al, 2007). The extent of striatal DA depletion induced in our model was not extensive enough to induce a full spectrum of depression-like behavior (ie, anhedonia); however, reduced immobility and freezing bouts in AMI-treated compared with saline-treated parkinsonian rats, suggests that the dose utilized in this study elicits antidepressant activity. This supports the possibility that in some biological systems, AMI can serve dual functions, ameliorating depression and neurodegeneration simultaneously. This idea is supported by our previous finding that TCAs delay the need for DA therapy in PD patients (Paumier et al, 2012) and the controlled clinical trial demonstrating that TCAs are more efficacious at treating depression within the PD population (Menza et al, 2009).
Although acute antidepressant treatment elicits immediate biochemical changes in the brain, adaptive processes such as gene regulation and increased neurotrophic factor levels occur after a latency of 2–3 weeks (Malberg et al, 2000; Martinez-Turrillas et al, 2005); therefore, we chose to use a pre-treatment experimental design to assess neuroprotection. However, this design raises the possible caveat of AMI treatment interfering with the uptake of 6-OHDA based upon the potential interaction of both substances with DAT. Although AMI has very low affinity for DAT (Tatsumi et al, 1997), even a slight affinity could prevent the toxin from entering the cell and ultimately prevent DA degeneration. Therefore, we assessed whether chronic AMI downregulates striatal DAT levels, potentially altering 6-OHDA uptake. We found no significant differences in DAT between saline- and AMI-treated animals after chronic treatment, suggesting DAT protein levels were equivalent when animals received 6-OHDA. Additionally, the plasma and brain elimination kinetics for AMI are rapid; in fact, brain levels for AMI and its metabolites (even after multiple i.p. injections) are undetectable 12 h after the last injection (Coudore et al, 1996). Therefore, animals were not injected with saline or AMI on the day of surgery to ensure that AMI was not present in the system to impede 6-OHDA uptake. Taken together, we are confident that the results presented represent true neuroprotection and not merely a prevention of toxin uptake.
In the intrastriatal 6-OHDA lesion model, the nigral DA neuron loss is delayed, beginning about 1 week after the initial insult and continues for several weeks afterwards (Sauer and Oertel, 1994). We report that AMI induced a robust increase in BDNF in the SN after 2 weeks of treatment, which was further enhanced 3 days post lesion. This increase was maintained in the AMI-treated rats despite lesioning, whereas BDNF levels fell by 35% in saline-treated lesioned rats, suggesting that the increased trophic support elicited by AMI may be a likely contributor to the nigral DA neuroprotection observed in this study. However, the pattern and magnitude of the effect suggests that other factors are likely involved. In contrast to the increase in nigral trophic support, we found chronic AMI mediated a substantial, albeit not statistically significant, decrease in striatal BDNF that occurred over time and independent of the lesion. Interestingly, the decrease in striatal BDNF at D30 post lesion occurs at a time at which SN BDNF levels are preserved in saline-treated animals. It may be that preservation of SN BDNF by AMI is at the expense of STR BDNF. Mechanisms by which this occurs are entirely unclear and beyond the scope of this report. However, it has been reported that AMI can inhibit axonal transport of BDNF (Edstrom et al, 1987; D'Sa and Duman, 2002), and can modulate a cholinergic tone that regulates BDNF and represents an interneuron population in STR (Cooper et al, 1996). Whatever the underlying cause for the AMI-mediated divergent regulation of BDNF, these results indicate that AMI treatment is associated with trophic changes within the nigrostriatal system and may have a significant impact on the survival and maintenance of neurons in patients who take antidepressants daily.
To our knowledge, this is the first study to show that chronic administration of a low-dose of AMI provides nigrostriatal neuroprotection in a toxin rat model of PD. Although we did not test other antidepressants, the combination of preclinical and clinical data suggest that TCAs may have dual therapeutic value by providing therapy that extends beyond alleviating depressive symptoms (Frisina et al, 2008; Menza et al, 2009) and that may include slowing ongoing neurodegeneration in PD patients. Although the etiology of depression in PD is unclear, there is evidence implicating a dysfunction of multiple transmitter systems (Remy et al, 2005), suggesting that drugs that simultaneously act on multiple targets (ie, ‘dirty drugs’) may prove superior to single target drugs for treating complex diseases like PD (Piau et al, 2011). In addition to interacting with multiple neurotransmitter systems, TCAs have also been shown to induce changes in trophic and inflammatory factors, which also underlie clinical depression and have been associated with PD. Thus, it is reasonable to suggest that chronic AMI treatment, which increases trophic support in the SN and hippocampus, may provide both relief from depression and neuroprotection for the DA system.
FUNDING AND DISCLOSURE
Dr Paumier was supported by funding from the Davis Phinney Foundation for Parkinson’s disease. Drs Paumier, Madhavan, and Collier were supported by the Morris K. Udall Center of Excellence for Parkinson’s Disease Research at Michigan State University and the University of Cincinnati (NS 058830). Dr Collier also receives support from the Michael J. Fox Foundation and is compensated as an associate editor for the European Journal of Neuroscience. Dr Terpstra, Dr Steece-Collier, Ms Celano, Mr Green, Ms Imus, Mr Marckini, and Mr Daley have nothing to disclose. All funding sources provided unrestricted support and had no role in the oversight or review of the research data or reporting.
References
Allen SJ, Watson JJ, Shoemark DK, Barua NU, Patel NK (2013). GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol Ther 138: 155–175.
Antonini A, Tesei S, Zecchinelli A, Barone P, De Gaspari D, Canesi M et al (2006). Randomized study of sertraline and low-dose amitriptyline in patients with Parkinson's disease and depression: effect on quality of life. Mov Disord 21: 1119–1122.
Aydemir C, Yalcin ES, Aksaray S, Kisa C, Yildirim SG, Uzbay T et al (2006). Brain-derived neurotrophic factor (BDNF) changes in the serum of depressed women. Prog Neuropsychopharmacol Biol Psychiatry 30: 1256–1260.
Berger A, Dukes E, Edelsberg J, Stacey B, Oster G (2007). Use of tricyclic antidepressants in older patients with diabetic peripheral neuropathy. Clin J Pain 23: 251–258.
Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT (2001). Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry 50: 260–265.
Chermat R, Thierry B, Mico JA, Steru L, Simon P (1986). Adaptation of the tail suspension test to the rat. J Pharmacol 17: 348–350.
Cooper JD, Skepper JN, Berzaghi MD, Lindholm D, Sofroniew MV (1996). Delayed death of septal cholinergic neurons after excitotoxic ablation of hippocampal neurons during early postnatal development in the rat. Exp Neurol 139: 143–155.
Coudore F, Besson A, Eschalier A, Lavarenne J, Fialip J (1996). Plasma and brain pharmacokinetics of amitriptyline and its demethylated and hydroxylated metabolites after one and six half-life repeated administrations to rats. Gen Pharmacol 27: 215–219.
D'Sa C, Duman RS (2002). Antidepressants and neuroplasticity. Bipolar Disord 4: 183–194.
De Foubert G, Carney SL, Robinson CS, Destexhe EJ, Tomlinson R, Hicks CA et al (2004). Fluoxetine-induced change in rat brain expression of brain-derived neurotrophic factor varies depending on length of treatment. Neuroscience 128: 597–604.
Drzyzga LR, Marcinowska A, Obuchowicz E (2009). Antiapoptotic and neurotrophic effects of antidepressants: a review of clinical and experimental studies. Brain Res Bull 79: 248–257.
Edstrom A, Ekstrom P, Kanje M, Sjoberg J (1987). The use of the regenerating frog sciatic nerve for pharmacological studies of orthograde and retrograde axonal transport. Brain Res 401: 34–42.
Frisina PG, Tenenbaum HR, Borod JC, Foldi NS (2008). The effects of antidepressants in Parkinson's disease: a meta-analysis. Int J Neurosci 118: 667–682.
Gonul AS, Akdeniz F, Taneli F, Donat O, Eker C, Vahip S (2005). Effect of treatment on serum brain-derived neurotrophic factor levels in depressed patients. Eur Arch Psychiatry Clin Neurosci 255: 381–386.
Gronli J, Murison R, Fiske E, Bjorvatn B, Sorensen E, Portas CM et al (2005). Effects of chronic mild stress on sexual behavior, locomotor activity and consumption of sucrose and saccharine solutions. Physiol Behav 84: 571–577.
Gundersen HJ, Jensen EB, Kieu K, Nielsen J (1999). The efficiency of systematic sampling in stereology—reconsidered. J Microsc 193: 199–211.
Han YS, Lee CS (2009). Antidepressants reveal differential effect against 1-methyl-4-phenylpyridinium toxicity in differentiated PC12 cells. Eur J Pharmacol 604: 36–44.
Hisaoka K, Takebayashi M, Tsuchioka M, Maeda N, Nakata Y, Yamawaki S (2007). Antidepressants increase glial cell line-derived neurotrophic factor production through monoamine independent activation of protein tyrosine kinase and extracellular signal-regulated kinase in glial cells. J Pharmacol Exp Ther 321: 148–157.
Jang SW, Liu X, Chan CB, Weinshenker D, Hall RA, Xiao G et al (2009). Amitriptyline is a TrkA and TrkB receptor agonist that promotes TrkA/TrkB heterodimerization and has potent neurotrophic activity. Chem Biol 16: 644–656.
Kajitani N, Hisaoka-Nakashima K, Morioka N, Okada-Tsuchioka M, Kaneko M, Kasai M et al (2012). Antidepressant acts on astrocytes leading to an increase in the expression of neurotrophic/growth factors: differential regulation of FGF-2 by noradrenaline. PLoS One 7: e51197.
Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20: 9104–9110.
Martinez-Turrillas R, Del Rio J, Frechilla D (2005). Sequential changes in BDNF mRNA expression and synaptic levels of AMPA receptor subunits in rat hippocampus after chronic antidepressant treatment. Neuropharmacology 49: 1178–1188.
Menza M, Dobkin RD, Marin H, Mark MH, Gara M, Buyske S et al (2009). A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 72: 886–892.
Nibuya M, Morinobu S, Duman RS (1995). Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci 15: 7539–7547.
Nibuya M, Nestler EJ, Duman RS (1996). Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci 16: 2365–2372.
Paumier KL, Siderowf AD, Auinger P, Oakes D, Madhavan L, Espay AJ et alParkinson Study Group Genetics Epidemiology Working G (2012). Tricyclic antidepressants delay the need for dopaminergic therapy in early Parkinson's disease. Mov Disord 27: 880–887.
Piau A, Nourhashemi F, Hein C, Caillaud C, Vellas B (2011). Progress in the development of new drugs in Alzheimer's disease. J Nutr Health Aging 15: 45–57.
Pijlman FT, Wolterink G, Van Ree JM (2003). Physical and emotional stress have differential effects on preference for saccharine and open field behaviour in rats. Behav Brain Res 139: 131–138.
Remy P, Doder M, Lees A, Turjanski N, Brooks D (2005). Depression in Parkinson's disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain 128: 1314–1322.
Rogoz Z, Legutko B (2005). Combined treatment with imipramine and metyrapone induces hippocampal and cortical brain-derived neurotrophic factor gene expression in rats. Pharmacol Rep 57: 840–844.
Santiago RM, Barbieiro J, Lima MM, Dombrowski PA, Andreatini R, Vital MA (2010). Depressive-like behaviors alterations induced by intranigral MPTP, 6-OHDA, LPS and rotenone models of Parkinson's disease are predominantly associated with serotonin and dopamine. Prog Neuropsychopharmacol Biol Psychiatry 34: 1104–1114.
Sauer H, Oertel WH (1994). Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6-hydroxydopamine: a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience 59: 401–415.
Sautter J, Meyer M, Spenger C, Seiler RW, Widmer HR (1998). Effects of combined BDNF and GDNF treatment on cultured dopaminergic midbrain neurons. Neuroreport 9: 1093–1096.
Schallert T (2006). Behavioral tests for preclinical intervention assessment. NeuroRx 3: 497–504.
Tatsumi M, Groshan K, Blakely RD, Richelson E (1997). Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol 340: 249–258.
Taylor TN, Caudle WM, Shepherd KR, Noorian A, Jackson CR, Iuvone PM et al (2009). Nonmotor symptoms of Parkinson's disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci 29: 8103–8113.
Ubhi K, Inglis C, Mante M, Patrick C, Adame A, Spencer B et al (2012). Fluoxetine ameliorates behavioral and neuropathological deficits in a transgenic model mouse of alpha-synucleinopathy. Exp Neurol 234: 405–416.
Valera E, Ubhi K, Mante M, Rockenstein E, Masliah E (2014). Antidepressants reduce neuroinflammatory responses and astroglial alpha-synuclein accumulation in a transgenic mouse model of multiple system atrophy. Glia 62: 317–337.
Wang H, Guan Y, Wang X, Smith K, Cormier K, Zhu S et al (2007). Nortriptyline delays disease onset in models of chronic neurodegeneration. Eur J Neurosci 26: 633–641.
Winter C, von Rumohr A, Mundt A, Petrus D, Klein J, Lee T et al (2007). Lesions of dopaminergic neurons in the substantia nigra pars compacta and in the ventral tegmental area enhance depressive-like behavior in rats. Behav Brain Res 184: 133–141.
Xu H, Steven Richardson J, Li XM (2003). Dose-related effects of chronic antidepressants on neuroprotective proteins BDNF, Bcl-2 and Cu/Zn-SOD in rat hippocampus. Neuropsychopharmacology 28: 53–62.
Acknowledgements
This work was supported by the Davis Phinney Foundation for Parkinson’s, the Morris K. Udall Center of Excellence for Parkinson’s disease Research at Michigan State University, and the University of Cincinnati (NS 058830). We wish to acknowledge the intellectual contributions of Nick Kanaan, Katherine Soderstrom, and technical expertise of Brian Daley, Allison Cole-Strauss, Emeline Tolod-Kemp, and Nate Levine. Special thanks to Sara Gombash and Valerie Thompson for their late night support in the lab.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Paumier, K., Sortwell, C., Madhavan, L. et al. Chronic Amitriptyline Treatment Attenuates Nigrostriatal Degeneration and Significantly Alters Trophic Support in a Rat Model of Parkinsonism. Neuropsychopharmacol 40, 874–883 (2015). https://doi.org/10.1038/npp.2014.262
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.262
- Springer Nature Switzerland AG
This article is cited by
-
The Brain-Derived Neurotrophic Factor: Missing Link Between Sleep Deprivation, Insomnia, and Depression
Neurochemical Research (2020)
-
Minocycline prevents the development of depression-like behavior and hippocampal inflammation in a rat model of Alzheimer’s disease
Psychopharmacology (2019)
-
Neuroprotective Effects of Antidepressants via Upregulation of Neurotrophic Factors in the MPTP Model of Parkinson’s Disease
Molecular Neurobiology (2018)