

Abstract
Random mutagenesis methods only partially cover the mutational space and are constrained by DNA synthesis length limitations. Here we demonstrate programmed allelic series (PALS), a single-volume, site-directed mutagenesis approach using microarray-programmed oligonucleotides. We created libraries including nearly every missense mutation as singleton events for the yeast transcription factor Gal4 (99.9% coverage) and human tumor suppressor p53 (93.5%). PALS-based comprehensive missense mutational scans may aid structure-function studies, protein engineering, and the interpretation of variants identified by clinical sequencing.


Similar content being viewed by others


Accession codes
Change history
10 April 2017
In the version of this article initially published, the unit (nM) for the dUTP concentration described in the "Wild-type template preparation" section of the Online Methods was incorrect. The correct unit should be μM. The error has been corrected in the HTML and PDF versions of the article as of 10 April 2017.
References
Botstein, D. & Shortle, D. Science 229, 1193–1201 (1985).
Kunkel, T.A. Proc. Natl. Acad. Sci. USA 82, 488–492 (1985).
Cunningham, B.C. & Wells, J.A. Science 244, 1081–1085 (1989).
Fowler, D.M. & Fields, S. Nat. Methods 11, 801–807 (2014).
Starita, L.M. et al. Proc. Natl. Acad. Sci. USA 110, E1263–E1272 (2013).
Wong, T.S., Roccatano, D., Zacharias, M. & Schwaneberg, U. J. Mol. Biol. 355, 858–871 (2006).
Roscoe, B.P., Thayer, K.M., Zeldovich, K.B., Fushman, D. & Bolon, D.N.A. J. Mol. Biol. 425, 1363–1377 (2013).
Qi, H. et al. PLoS Pathog. 10, e1004064 (2014).
Firnberg, E., Labonte, J.W., Gray, J.J. & Ostermeier, M. Mol. Biol. Evol. 31, 1581–1592 (2014).
Firnberg, E. & Ostermeier, M. PLoS ONE 7, e52031 (2012).
Hietpas, R.T., Jensen, J.D. & Bolon, D.N.A. Proc. Natl. Acad. Sci. USA 108, 7896–7901 (2011).
Jain, P.C. & Varadarajan, R. Anal. Biochem. 449, 90–98 (2014).
Hiatt, J.B., Patwardhan, R.P., Turner, E.H., Lee, C. & Shendure, J. Nat. Methods 7, 119–122 (2010).
Ma, J. & Ptashne, M. Cell 48, 847–853 (1987).
Johnston, M. & Dover, J. Genetics 120, 63–74 (1988).
Marmorstein, R., Carey, M., Ptashne, M. & Harrison, S.C. Nature 356, 408–414 (1992).
Hong, M. et al. Structure 16, 1019–1026 (2008).
Chou, P.Y. & Fasman, G.D. Annu. Rev. Biochem. 47, 251–276 (1978).
Ferdous, A. et al. Mol. Biosyst. 4, 1116–1125 (2008).
Patwardhan, R.P. et al. Nat. Biotechnol. 27, 1173–1175 (2009).
Melnikov, A., Rogov, P., Wang, L., Gnirke, A. & Mikkelsen, T.S. Nucleic Acids Res. 42, e112 (2014).
Wang, T., Wei, J.J., Sabatini, D.M. & Lander, E.S. Science 343, 80–84 (2014).
Shalem, O. et al. Science 343, 84–87 (2014).
Findlay, G.M., Boyle, E.A., Hause, R.J., Klein, J.C. & Shendure, J. Nature 513, 120–123 (2014).
Maurer, K. et al. PLoS ONE 1, e34 (2006).
Nakamura, Y., Gojobori, T. & Ikemura, T. Nucleic Acids Res. 28, 292 (2000).
Mumberg, D., Müller, R. & Funk, M. Gene 156, 119–122 (1995).
Gill, G. & Ptashne, M. Nature 334, 721–724 (1988).
Li, H. Preprint at http://arxiv.org/abs/1303.3997 (2013).
James, P., Halladay, J. & Craig, E.A. Genetics 144, 1425–1436 (1996).
Gietz, R.D. & Woods, R.A. Methods Enzymol. 350, 87–96 (2002).
Melamed, D., Young, D.L., Gamble, C.E., Miller, C.R. & Fields, S. RNA 19, 1537–1551 (2013).
Remmert, M., Biegert, A., Hauser, A. & Söding, J. Nat. Methods 9, 173–175 (2012).
Crooks, G.E., Hon, G., Chandonia, J.-M. & Brenner, S.E. Genome Res. 14, 1188–1190 (2004).
Sanner, M.F., Olson, A.J. & Spehner, J.C. Biopolymers 38, 305–320 (1996).
Acknowledgements
We thank P. Brzovic, R. Monnat and members of the Fields and Shendure Labs for helpful discussions. This work was supported by a graduate student research fellowship DGE-0718124 from the US National Science Foundation (to J.O.K.), a US National Institutes of Health (NIH) Pioneer Award #DP1HG007811 (to J.S.) and an NIH Biomedical Technology Research Resource project #P41GM103533 to S.F. S.F. is supported by the Howard Hughes Medical Institute as an investigator.
Author information
Authors and Affiliations
Contributions
J.O.K., L.M.S., S.F. and J.S. designed the study and wrote the manuscript. J.O.K., L.M.S. and R.S.L. performed experiments. All authors contributed to and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The University of Washington has filed a provisional patent application on this method, with J.O.K., L.M.S., S.F., and J.S. as inventors.
Integrated supplementary information
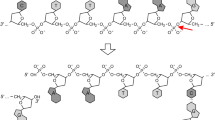
Supplementary Figure 1 Detailed schematic of PALS workflow.
Multiplex mutagenesis is carried out in a single volume that is subjected to eight steps, beginning with preparation of mutagenic primers from a DNA microarray. Next, strand extension, strand selection, and PCR are carried out twice to copy the wild-type sequence upstream and then downstream of each mutagenic primer.
Supplementary Figure 2 Coverage and uniformity of mutagenesis for PALS (targets: Gal4 DBD and p53), and randomized doped synthesis (Ube4b).
(a) The percentage of possible single-amino acid substitutions observed as singleton mutations at different thresholds of multiplicity (i.e., observed in one or more clones, two or more clones, etc.). Mutations are stratified and shaded by the minimum required number of base-pair edits to yield the corresponding amino acid substitution (1, 2, or 3). (b) Effects of clone library sequencing depth upon mutational coverage. Using a threshold of observation in at least three clones, the fraction of amino acid substitutions observed as singleton mutations in three or more clones is plotted against the depth of coverage (clones per residue), for random subsets of increasing size from each library. Protein length and final coverage by clones differ for each target, and dotted lines indicate equivalent clone coverage level across each, with the percent of all edit distance 1, 2 and 3 mutations covered at that shared level (1646 clones per residue).
Supplementary Figure 3 Regional coverage for PALS and doped oligonucleotide random mutagenesis.
Count of single coding mutant clones carrying each possible codon replacement is plotted against codon position. Each point represents a single codon replacement, shaded by number of base-pair differences, for mutant libraries for (a) Gal4 DBD, (b) Tp53 (both PALS mutagenesis), and (c) Ube4b (doped oligo synthesis).
Supplementary Figure 4 Purity and coverage for PALS and random mutagenesis.
For PALS and simulated random mutagenesis at various mutation rates, the percentage of possible amino acid substitutions carried on singleton clones (i.e., without any other missense mutations or frame-shift deletions) is plotted versus the percentage of possible substitutions carried on any clone. Simulated randomized mutagenesis was performed with different per-base substitution rates (point color indicates rate) and assuming various per-base deletion rates (rows; for Ube4b mutations introduced by doped oligonucleotide synthesis, a per-base deletion error rate of 8.9x10-4 was observed). Single-base substitution and deletion counts were sampled for each sequence from Poisson distributions with the indicated rates multiplied by sequence lengths, and missense and frame-shifting mutations were tallied. For substitutions, each of the three alternative bases was sampled with equal probability. Red points indicate the observed performance of PALS libraries in this study. Columns indicate the threshold minimum number of clones containing each mutation. Simulated clones were generated equal to the number of sequenced clones for (a) Gal4 DBD (n=704,973) and (b) p53 (n=646,939)
Supplementary Figure 6 Pairwise correlation scatter plots of effect size.
Per-mutation effect scores (log2-scaled) are plotted for each pair of selection stringencies and time points. Black line indicates y=x, and Spearman rank correlation measure is inset. Effect sizes under selective conditions (lower five rows) are strongly positively correlated. Without selection (top row), wild-type and functional alleles of Gal4 are mildly toxic28 compared with loss-of-function variants such as PTCs (premature termination codons, median log2E = +1.33 under non-selective conditions). As a result, effect sizes are negatively correlated between selective and non-selective conditions, although the magnitude of effect sizes in the former are much larger (90th percentile of absolute log2E values is 11.5 versus 1.66, a >900-fold difference in linear terms).
Supplementary Figure 7 Validation of effects for selected Gal4 mutant alleles.
Plates were spotted with 10-fold serial dilutions starting from approximately equal numbers of cells carrying Gal4 1-196, either wild-type or with one of eight specifically introduced missense alleles. Growth on non-selective media (left) was uniform, while specific growth effects on selective media (right) qualitatively agreed with effect sizes observed by large-scale selection (for each variant, top bar indicates effect size from non-selective culture and subsequent bars indicate effect size from selective outgrowth, Supplementary Table 2, shaded as in Supplementary Fig. 2). Qualitative activity as measured by Johnston and Dover15 is indicated alongside each allele (+++, wild-type activity; hypo, hypomorphic; N.D., not determined).
Supplementary Figure 8 Amino acid substitutions observed in orthologs are significantly less deleterious to Gal4 function than most mutations.
Mutation effect size distributions are shown in each of six selection timepoints (NONSEL, nonselective; others are selective). Premature truncations were excluded and remaining mutations are divided into three categories: (1, in blue) all substitutions observed in aligned Gal4 ortholog sequences, (2, in green) substitutions not observed in Gal4 orthologs, at sites that did vary within the alignment, and (3, in orange): substitutions at residues that were fixed among aligned Gal4 orthologs. Orthologs were identified by NCBI tblastx query of the wgs and genbank chromosomes databases at a cutoff of E<10-20, from genera Saccaromyces (n=11), Zygosaccharomyces (n=1), and Kluyveromyces (n=1). * denotes P<2.0x10-3, ** P<10-20 and *** P<10-50, Mann-Whitney U. Under every selective condition but not under non-selective outgrowth, mutations at fixed residues (group 3) were significantly more deleterious (more negative log2 effect size) than mutations in either other group, and at residues that did vary among Gal4 orthologs, mutations that were not observed in those orthologs (group 2) were significantly less deleterious than those that were (group 1).
Supplementary Figure 9 Functional scores mapped to the Gal4 DBD structure.
The crystal structure for Gal4 residues 8-100, PDB accession 3COQ (ref. 17) is shown, with each amino acid (through residue 65) shaded by the median effect size across variants, following selective growth for 64 h in SC–ura–his+1.5mM 3-AT (excluding mutations to proline and premature truncation). Several key residues including zinc-coordinating cysteines, are highlighted with median effect size indicated.
Supplementary Figure 10 Subassembly strategy.
Plasmid maps for PALS libraries constructed for (a) Gal4 DBD and (b) p53. The expected recircularization products are shown, with PCR primer names and amplicons inset. As implemented, subassembly extends ~800 bp inward from each end of a clone insert, but could be extended by subassembling from additional fixed points within the insert. Alternatively, with improved single-molecule long-read platforms (e.g., Pacific Biosciences), it may soon be possible to directly sequence clone libraries to associate each insert with its paired barcode.
Supplementary Figure 11 Examples of PALS amplification substrates and products.
Images of 6% TBE polyacrylamide gels stained by SYBR Gold (Invitrogen). Microarray-derived mutagenic primers are shown following (a) amplification (four different subsets of the library) and (b) adaptor clipping (* indicates desired, 84 bp product). Nested amplification products following the first round of mutagenic primer extension and adaptor-mediated PCR are shown (one replicate PCR product in each lane) in (c) for Gal4 and in (d) for p53. “25bpl” is 25 bp ladder (Invitrogen) and “100bpl” is 100bp ladder (NEB).
Supplementary Figure 12 Subassembly validation example.
Read pileup and resulting subassembly consensus for a representative p53 clone (tag ACCCTAAGAGAATACGAGCT, consensus haplotype K120L). Shown below are the capillary sequencing traces through the insert showing the K120L mutation (middle), and the clone-identifying tag sequence (bottom).
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–12, Supplementary Tables 1–8 and Supplementary Note (PDF 2290 kb)
Supplementary Data
Per-allele enrichment scores for Gal4 functional selection. Excelformatted spreadsheet containing enrichment log2-ratio values for each mutant allele and round of selection. (XLSX 127 kb)
Rights and permissions
About this article

Cite this article
Kitzman, J., Starita, L., Lo, R. et al. Massively parallel single-amino-acid mutagenesis. Nat Methods 12, 203–206 (2015). https://doi.org/10.1038/nmeth.3223
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmeth.3223
- Springer Nature America, Inc.
This article is cited by
-
DIMPLE: deep insertion, deletion, and missense mutation libraries for exploring protein variation in evolution, disease, and biology
Genome Biology (2023)
-
Discovering functionally important sites in proteins
Nature Communications (2023)
-
Purification of multiplex oligonucleotide libraries by synthesis and selection
Nature Biotechnology (2022)
-
Quantifying the Mutational Robustness of Protein-Coding Genes
Journal of Molecular Evolution (2021)
-
Bridging non-overlapping reads illuminates high-order epistasis between distal protein sites in a GPCR
Nature Communications (2020)