
Abstract
In chronic lymphocytic leukemia (CLL), the worst prognosis is associated with TP53 defects with the affected patients being potentially directed to alternative treatment. Therapy administration was shown to drive the selection of new TP53 mutations in CLL. Using ultra-deep next-generation sequencing (NGS), we performed a detailed analysis of TP53 mutations’ clonal evolution. We retrospectively analyzed samples that were assessed as TP53-wild-type (wt) by FASAY from 20 patients with a new TP53 mutation detected in relapse and 40 patients remaining TP53-wt in relapse. Minor TP53-mutated subclones were disclosed in 18/20 patients experiencing later mutation selection, while only one minor-clone mutation was observed in those patients remaining TP53-wt (n=40). We documented that (i) minor TP53 mutations may be present before therapy and may occur in any relapse; (ii) the majority of TP53-mutated minor clones expand to dominant clone under the selective pressure of chemotherapy, while persistence of minor-clone mutations is rare; (iii) multiple minor-clone TP53 mutations are common and may simultaneously expand. In conclusion, patients with minor-clone TP53 mutations carry a high risk of mutation selection by therapy. Deep sequencing can shift TP53 mutation identification to a period before therapy administration, which might be of particular importance for clinical trials.
Similar content being viewed by others

Introduction
In chronic lymphocytic leukemia (CLL), patients harboring TP53 defects represent a major challenge concerning the effective treatment.1 TP53 mutation and/or 17p deletion severely impede response to chemotherapy,2, 3 and affected patients also manifest short clinical responses to its combination with rituximab.4, 5 Although alemtuzumab is supposed to act independently on p53, the response rates in monotherapy are far from satisfactory in chemorefractory patients.6 The inability of mutated p53 protein to induce apoptosis properly seems to be a primary reason for the observed resistance to treatment.7 The p53 dysfunction is also the major cause of genomic instability in CLL cells,8 which leads to the acquisition of other genomic variants available for further selection.
TP53 gene defects have been observed as primarily subclonal events in CLL patients, often emerging at later disease stages.9 The frequency of TP53 defects at diagnosis or before first therapy is only between 5 and 15%,2, 3, 10, 11 but the proportion of affected patients is significantly higher after treatment and has been reported to be as high as 44% in a fludarabine-refractory cohort.12 Clonal evolution of genetic abnormalities including TP53 defects is well evidenced in CLL. Recent studies have illustrated the development of 17p and 11q deletions during the disease course, and associated clonal evolution of new 17p deletions with the presence of foregoing therapy.13, 14 Concerning TP53 mutations, well-documented cases of their acquirement under the pressure of chemotherapy have also been reported by us and independently by others.15, 16, 17, 18 This led to the suggestion that TP53 mutations should be investigated before each therapy in CLL patients.19
Next-generation sequencing (NGS) technologies currently enable mutation analyses in cancer patients with previously unattainable sensitivity, reaching as far as fractions of percentages. The clinical significance of minor-clone TP53 mutations has recently been demonstrated by Rossi et al.18 Therefore, we utilized this powerful tool to study the clonal evolution of TP53 mutations in detail. We used an amplicon ultra-deep NGS approach with a high coverage to reach maximum sensitivity, and we used a highly accurate proof-reading polymerase to minimize the sequencing errors. The aims of this NGS-based study were to disclose (i) whether minor TP53-mutated clones had already been present before the preceding therapy, and if yes, (ii) whether some patients, who are TP53-wt after therapy, harbor minor TP53 mutations that are not selected. These two issues should address whether NGS is capable of identifying patients at risk of TP53 mutation selection by treatment.
Materials and methods
Patients’ cohort
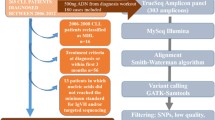
The study was performed on CLL patients’ peripheral blood samples at the University Hospital Brno (with written informed consent provided in accordance with the Declaration of Helsinki). Four common cytogenetic aberrations including 17p deletions were detected by Interphase fluorescent in situ hybridization using probes from MetaSystems (Altlussheim, Germany), and were classified according to the hierarchical cytogenetic model.20 TP53 mutations were identified by yeast functional analysis (FASAY) coupled to Sanger sequencing of DNA templates from red colonies bearing non-functional p53.16 Patient selection criteria for NGS analysis were: Cohort I: (i) TP53 mutational status change from wild-type (wt) to mutated documented using FASAY; (ii) only one therapy applied between the last TP53-wt examination and new TP53 mutation detection. This criterion was established to enable the tracking of clonal evolution during just one relapse; (iii) available DNA from the time when the sample was TP53-wt. Cohort II: (i) available results of consecutive FASAY analyses performed in relapse(s) with no TP53 mutational status change; (ii) DNA available from the period preceding therapy (Supplementary Figure 1).
Statistical analyses
Fisher’s exact test was used to assess the association between categorical variables. Mann–Whitney test was used to compare the continuous variables. Wilcoxon signed-rank test was used for paired comparison of mutation numbers. Survival analysis and time to mutation detection were calculated using the Kaplan–Meier survival estimator. Overall survival was assessed from the date of diagnosis; only disease-related death was considered as an event. Time to mutation detection was assessed from the date of diagnosis to the date of new TP53 mutation detection (event) or the last TP53-wt examination (censored).
Median survival, time to mutation detection, differences between the curves, and hazard ratios were evaluated by the log-rank test using the GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA, USA).
Ultra-deep NGS
NGS analysis was performed on MiSeq (Illumina, San Diego, CA, USA) using gDNA from cryopreserved peripheral blood separated CD19+ B-lymphocytes or mononuclear cells; the percentage of CLL cells (CD5+CD19+) was assessed using flow cytometry and was >80% in all cases. In all, 25 ng of patient DNA was amplified with highly accurate proof-reading Q5 Polymerase (New England Biolabs, Ipswich, MA, USA) using TP53 exon-specific primers (Supplementary Table 1). The experimental design and reaction conditions followed the manufacturer recommendations. Briefly, PCR products were pooled, purified with Agencourt AMPure XP (Beckman Coulter, Brea, CA, USA), and quantified using Qubit dsDNA HS Assay Kit (Life Technologies, Waltham, MA, USA). The purified amplicon mixes were diluted to a total amount of 1 ng. The indexed paired-end library was prepared with Nextera XT DNA Sample Preparation Kit (Illumina) and sequenced using MiSeq Reagent Kit v2 300 cycles (Illumina). To avoid cross-contamination, samples obtained from the same patient in different time periods were sequenced in separate runs. Amplicons and libraries for each run were prepared separately. The median coverage per base achieved was 31 599 reads (range 2601–177 021).
An in-house bioinformatics pipeline was established to call the sequencing variants. For read preprocessing and alignment, we used CLC Genomic Workbench (Qiagen, Hilden, Germany). Variant calling was performed using the deepSNV R-package21 with a statistical approach applying the shearwater algorithm to compute Bayes classifiers based on a betabinomial model.22, 23 By the reproducibility test, we disclosed that we were able to reliably distinguish point mismatches and ⩾2 nt insertions/deletions (indels) at the level of 0.2% of variant reads, and 1-nucleotide deletions at the level of 1% of variant reads as these may be artificially introduced during the sequencing and alignment process. For further details, see Supplementary material. Moreover, to evaluate the established pipeline, 20 control samples (TP53 exons 4–10) derived from healthy individuals were sequenced and no alteration in any sample was observed on the above-mentioned detection limits.
Results
Consecutive TP53 mutational analysis confirms the prominent impact of newly acquired TP53 mutations on survival
Consecutive TP53 mutation investigation using FASAY was performed in 330 patients in at least 2 serial samples. All patients harbored intact TP53 gene at the time of the first analysis (for patients’ characteristics, see Supplementary Table 2). Among 121 patients who did not receive any therapy during the follow-up, new TP53 mutations were observed in only one patient (median follow-up of the group 50 months). In contrast, analyses performed at the time of relapse after one or several therapy lines (n=209 patients; median follow-up of the group 61 months) identified new TP53 mutation(s) in 43 patients. Altogether, the risk of TP53 mutation acquisition at 5 years after diagnosis was 1% in untreated vs 17% in treated patients (hazard ratio 0.25 (95% CI 0.13–0.47; P<0.001)) (Figure 1).

Time from diagnosis to TP53 mutation acquisition. Patients with TP53-wt status at first investigation were repeatedly tested. Time to mutation detection was assessed from the date of diagnosis to the date of new TP53 mutation detection (event) or the last TP53-wt examination (censored). Patients treated during the follow-up acquired new TP53 mutation significantly more often than untreated patients.
To assess the importance of TP53 mutation status change from wt to mutated, we used our cohort analyzed using FASAY and compared the overall survival from diagnosis in patients who acquired a new dominant mutation in relapse with patients who remained TP53-wt in relapse, and patients who already harbored TP53 mutations at diagnosis (Figure 2). The overall survival was significantly reduced in the group of patients who had selected TP53 mutations compared with patients assessed as wt in relapse (P=0.03). The shortest survival was noted for patients with TP53 mutations already detected at diagnosis.

Overall survival according to the TP53 mutational status in relapse. Overall survival from diagnosis in patients who acquired a new TP53 mutation at relapse (n=18; median survival 64 months) in comparison with patients who remained TP53-wt at relapse (n=78; median survival undefined; pairwise comparison P=0.03), and patients who harbored TP53 mutation already at diagnosis (n=49; median survival 39 months; pairwise comparison P=0.02). Only patients with TP53 status examined at diagnosis or 12 months thereafter were included. All patients included in the analysis underwent treatment and patients having TP53-wt status at diagnosis were repeatedly tested for TP53 mutation presence at subsequent relapse(s).
NGS analysis reveals the presence of minor mutated clones before their therapy-driven selection
In the first part of our retrospective study, we focused on 20 patients who had acquired a new TP53 mutation in relapse, as assessed by FASAY coupled to Sanger sequencing (Cohort I; Sample 2). In these patients, we used ultra-deep NGS to examine samples taken before the preceding therapy, which showed TP53-wt status using Sanger sequencing and FASAY (Sample 1). In 10 patients, these retrospective samples were treatment naïve (Cohort IA), while the remaining 10 patients had already been pretreated at the time of NGS analysis (Cohort IB) (Table 1). The schematic visualization of samples’ inclusion criteria is shown in Supplementary Figure 1. The mutations analyzed consisted of 16 missense mutations, 2 non-sense mutations and 2 deletions and were hence representative of the p53 mutation profile in CLL.24 To decipher TP53 mutagenesis, we sequenced not only the affected regions but also all commonly mutated exons 4–9 (ref. 24) with a high median coverage for the positions containing mutations (25 709 reads; range 5245–64 979). We were able to detect minor-proportion mutations in 18/20 samples (90%), with a proportion of 0.20–3.71% of the reads showing mutations. In 2 of the 18 patients, we surprisingly detected other TP53 mutations that had not undergone expansion. The results are summarized in Table 1, for details see Supplementary table 3.
Interestingly, in addition to the presumed retrospective mutations, we also identified other minor-proportion TP53 mutations in both treatment-naïve and pretreated samples (10/20 patients; 2–6 mutations per patient) (Table 1). It indicates that in a proportion of patients, there is a pool of TP53 mutations available for therapy-driven selection.
We next intended to investigate whether the minor TP53-mutated subclones detectable by NGS in pretreated samples and undergoing selection in subsequent relapse (Cohort IB) had already been present before first therapy. Therefore, we used NGS in four available treatment-naïve samples (patients no. 149, 365, 542 and 1043) and confirmed the presence of respective mutation in one of them (patient no. 1043—mutation c.844C>G (p.R282G) detected in 0.2% of NGS reads). This observation suggests that preexisting mutations may expand after the first but also after subsequent therapies at least in some patients.
Minor TP53 mutations detectable before therapy are rare in patients remaining TP53-wt at relapse
As the next step, we analyzed 40 samples taken before first treatment in patients showing wt-TP53 status at relapse after one or several therapy lines (Cohort II). These cases were selected from the cohort of relapsing patients, and the inclusion criteria were chosen to collect the cohort with biological and clinical characteristics matching Cohort I (Table 2; Supplementary Figure 1). In this experiment, besides exons 4–9, exon 10 was also sequenced as it may occasionally harbor mutations.24
We found TP53 mutation in only 1 of the 40 patients (2.5%). Specifically, the mutation c.797G4A (p.G266E) was detected in 0.55% (148/32 973) of sequencing reads, and its presence was verified by an independent NGS run. This mutation did not undergo a clonal expansion during the disease course despite several treatment lines—the patient was treated consecutively with three distinct therapy lines (FCR, Alemtuzumab and Rituximab+Dexamethasone) and achieved two complete remissions. In the last available sample from the time of relapse after Rituximab+Dexamethasone treatment (follow-up 47 months) the same mutation was present in 1.4% of reads.
Clonal selection frequently affects multiple TP53 mutations simultaneously
As emerged from the previous analyses, multiple minor-clone TP53 mutations are commonly observed in CLL patients. To further explore this phenomenon in relation to the expansion of major mutations, we performed ultra-deep NGS of TP53 gene in samples taken at relapse(s) (Sample 2 in Supplementary Figure 1). For this analysis, we had chosen the following patients from Cohort I: (i) six patients with more than one mutation detected in sample 1; (ii) six patients with a single mutation detected in sample 1; and (iii) two patients with no mutation detected in sample 1. Furthermore, the patient with a single non-expanding mutation from Cohort II was also included. An increase in the number of mutations compared with the preceding samples was observed in 13/14 patients from Cohort I (Table 3; Figure 3a). In the paired analysis restricted to samples taken before the first therapy and in the first relapse (Cohort IA), a significant increase in the number of mutations per patient was observed (mean number of mutations per patient 2.1 vs 6.7; P=0.02). In the patient from Cohort II, only one mutation was found in both samples.

Kinetics of multiple mutations in subsequent samplings. (a) Increase in number of mutations detectable using NGS during the disease course. All patients form Cohort I with repeated NGS analysis are shown (n=14). (b) Dynamics of clonal evolution in patient no. 820. Clone bearing mutation p.249del that was detected at Relapse 2 using FASAY first expanded and was later outgrown by another mutation p.R175H. Splicing mutation c.375+2T>A slightly expanded and coexisted as a minor subclone. Subclonal dynamics of additional minor clones present below 2% is shown in detail. (c) Examples of patients with no prominent expansion of one mutation is shown. Proportion of variant reads in individual disease time points is illustrated. Mutation detected using FASAY in the second sampling is highlighted in bold.
Regarding the evolution of individual subclones, the most frequently observed event (7/14 patients) was the clear expansion of one mutation from minor to dominant clone accompanied with the occurrence of additional minor TP53-mutated clones. In addition to that, we also observed other specific situations: (i) in one case the consecutive selection of two different dominant TP53 mutations at the first and then the subsequent relapse was noted (one mutation replaced by the other) (patient no. 820; Figure 3b); (ii) in four patients there was not a prominent clonal expansion of one mutation, but multiple clones expanded simultaneously (patients no. 8, 178, 354 and 485; Figure 3c); (iii) one patient underwent only a very slight expansion of a single minor-clone mutation in the first relapse (from 0.2 to 1.46% reads), in the second relapse the proportion of the mutation also increased only slightly (to 2.82% reads) and two other minor-proportion mutations appeared (patient no. 503; Table 3). The results summarizing the rise in the number of mutations in all performed NGS analyses are recapitulated in Supplementary Table 3.
Since the analysis of mutated patients disclosed an increased occurrence of minor TP53 mutations after treatment, we further analyzed 15 randomly selected patients from Cohort II after 1–4 therapy lines using NGS. No TP53 mutations were observed in any patient.
Molecular features of mutations
In total, we identified 148 mutations in 21 patients (Supplementary Table 3) in all the NGS analyses performed. The mutation profile is shown in Supplementary Figures 2 and 3. Compared with the reference study on TP53 mutation profile in CLL24 our results showed the following: (i) a similar proportion of missense mutations (79 vs 74%; P=0.4) and non-sense mutations (both studies 4%); (ii) the same frequency of mutations at major hot spot codons (175, 179, 220, 248, 273 and 281) (20% of all mutations in both studies); (iii) a significantly higher proportion of splice-site mutations (9 vs 2%; P=0.005) and, on the other hand (iv) a significantly lower frequency of indel mutations (7 vs 20%; P=0.0003). Concerning point mutations, transitions represented 61% with only 29% of them (17% of all mutations) occurring at CpG sites. The G-A transitions at CpG predominated C-T transitions (G-A:C-T ratio 2:1). The lower proportion of CpG transitions and the prevalence of G-A exchanges coincided with the reference study.24
Comparison of TP53 mutation profiles in cases with unmutated immunoglobulin heavy chain gene (IGHV; U-CLL) vs mutated IGHV (M-CLL) showed no difference in mutation frequency within sequence motif (RGYW/WRCY) recognized by activation-induced cytidin deaminase in U-CLL vs M-CLL (20 vs 18% of point mutations P=0.8). In M-CLL, a significant prevalence of alterations in A:T pairs was found compared with U-CLL (56 vs 27% of point mutations; P=0.0008). The A:T alteration predominance was the most prominent in case of A:T>C:G transversions (12% in M-CLL vs 1% in U-CLL; P=0.009; Figure 4).

Comparison of TP53 mutation profile in patients with unmutated IGHV (U-CLL) vs mutated IGHV (M-CLL). Percentage from all point mutations shown.
As the number of mutations increased after therapy, we also compared the molecular profile of mutations detected in pretherapy samples only (n=24) with mutations that occurred exclusively after treatment (n=103) and we did not observe any significant differences regarding the proportion of hot spot mutations, transversion-to-transition ratio, proportion of transitions at CpG sites and G:C to A:T ratio (data not shown).
Discussion
The mechanisms leading to p53 mutation acquisition and accumulation in CLL are poorly understood. The direct induction of TP53 mutations by DNA-damaging chemotherapy, namely alkylating agents, has been suggested.25 In contrast, a large collaborative study involving 268 p53 mutations indirectly showed that the impact of therapy on de novo mutation induction is unlikely, as mutation spectra are similar in untreated and treated patients.24 This observation, however, may not serve as definitive proof of the neutral impact of therapy on TP53 mutagenesis, since similar mutations could evolve through different mechanisms. The current progress in highly sensitive techniques, specifically in ultra-deep NGS, allows the possibility to explore whether therapy merely selects TP53 mutations present in minor CLL clones before drug administration. Moreover, identifying TP53 defects as early as possible during their evolution may represent a significant achievement in the clinical management of high-risk CLL, since TP53-deffective patients could be offered alternative treatment.1 The clinical impact of minor-proportion TP53 defects is currently a matter of debate.18, 26, 27 Their relevance for relapse development is supported by the actual number of mutated cells. For instance, at common pretherapy leukocytosis achieving 100 × 10e9 per liter with 90% CLL cells, a patient harbors approximately 4.5 × 10e11 CLL cells in peripheral blood, not considering other organs like the spleen. In this case, a 1% TP53 mutation corresponds to ~4.5 × 10e9 cells. Moreover, the clinical significance of small TP53-mutated clones under the detection limit of Sanger sequencing have very recently been manifested by the study of Rossi et al.18 showing their similar unfavorable prognostic impact compared with clonal TP53 defects.
With this report, we focused on two principal issues: (i) exploration of TP53-mutated clone evolution and (ii) assessment of NGS utilization in TP53 mutation expansion prediction in clinical practice. Both these issues are important with respect to the clear negative impact of newly acquired TP53 mutations on patients’ prognosis, which was evidenced by Rossi et al.28 using time-dependent Cox regression analysis, and is also confirmed here by survival analysis of patients with new mutations.
Concerning the clonal evolution, we documented that the risk of new TP53 mutation acquisition at 5 years after diagnosis is 17% in patients requiring treatment, contrasting with 1% in untreated patients (P<0.001), and we confirmed that selection of preexisting mutated clones by therapy is the predominant mechanism for TP53 mutations’ accumulation. Moreover, we showed that mutations expanding during relapse are detectable before the preceding therapy in the majority of patients. Admittedly, based on our study we cannot entirely exclude that at least some TP53 mutations are the consequence of DNA damaging drugs25 since many minor-proportion mutations were undetectable before first therapy despite using ultra-deep NGS. Although we have not observed any profound difference in the mutation profile of these mutations compared with the mutations present before treatment, they could be induced by therapeutic agents or spontaneous mutagenesis during relapse. Alternatively, they may be present in a very low proportion of leukemic cells under the NGS detection limit.
Our study independently confirms two recent reports18, 29 showing that in a proportion of patients there are multiple minor-clone TP53 mutations (under the Sanger sequencing detection limit). These mutations may or may not accompany a major clonal mutation.29 We noted the presence of multiple TP53 mutations in patients with clonal selection of dominant TP53 mutations, and also in patients with dominant TP53 mutation detected at diagnosis (7/10 patients; data not shown). We further observed that selection may affect not only single TP53-mutated minor clone, but also in some patients multiple mutations simultaneously. In fact, at least some cases without prominent expansion of one mutation underwent a slight selection of a burden of different TP53-mutated clones that are not detectable by Sanger sequencing. Using FASAY, these patients were assessed as ‘mutation acquisition’ since with this methodology the overall percentage of red colonies equals the sum of all mutations present.
The striking aspect of our study is the actual number of multiple TP53 mutations, as according to our observation even tens of mutations may be present in individual patients. Our conclusion that these multiple alterations are true mutations and not NGS artifacts is supported by the following: (i) the point mutations present in ⩾0.2% of NGS reads were confirmed in a reproducibility test; (ii) the same variants were often observed in consecutive samples, (iii) some of the minor-proportion mutations were also noted in individual colonies during FASAY analysis (Supplementary Table 3); this also shows that the mutations are present on separate alleles as FASAY is based on subcloning template molecules; (iv) only one mutation was detected in 56 samples from patients remaining TP53-wt throughout disease (40 pretherapy and 16 relapsed samples from Cohort II), and no mutation was observed in any healthy control sample (n=20); and finally (v) the molecular profile of additional mutations was similar to that described for the reference cohort24 with the common hot spots being the most prevalent mutations.
Despite the similarities between the mutation profile of additional mutations and the reference TP53-mutated CLL cohort, we noticed several specificities. The low number of indel mutations among additional mutations can likely be accounted to the NGS methodology itself as it is generally difficult to distinguish minor-proportion 1-nucleotide deletions from background. An interesting observation is the high number of minor-proportion splice-site mutations, predominantly in intron 6. These mutations are often present at the subclonal level; however for a yet unknown reason they only rarely expand to dominant clone. Apart from this, we were not able to find any rule concerning a preferential selection of distinct mutation types. For instance, we recorded patients in which a truncating mutation outgrew the clone carrying hot spot mutation with documented dominant-negative and gain-of-function effect. Therefore, there should be other factors contributing to the preferential selection of particular TP53-mutated subclones, for example, mutations in other genes or distinct stimulation by the microenvironment. In addition, an obvious important factor represents deletion 17p, since the wt allele absence may contribute to the selection advantage of a particular subclone. The new 17p deletion accompanying the new TP53 mutation was found in 8/20 patients and in another two patients a new 17p copy-neutral loss of heterozygosity was noted. However, to determine the exact allele composition of minor subclones carrying different TP53 mutations would require single-cell analysis, which was beyond the scope of this study.
The surprisingly large number of mutations led us to explore the mechanisms of TP53 mutagenesis with regard to lymphoid-specific hypermutation machinery. No bias regarding mutations in sequence motifs recognized by activation-induced cytidin deaminase was found. Interestingly, we observed a prevalence of mutations in A:T pairs in patients with mutated IGHV compared with unmutated IGHV, which was most prominent in A:T>C:G transversions. A similar disproportion was found in a whole-genome sequencing study30 and is most likely to be attributed to the operation of error-prone polymerase eta.31
The observation that the majority of new dominant mutations are already present before therapy offers the opportunity to predict their expansion later during the disease course and change the patients’ care strategy. The obvious prerequisite for such clinical utilization is that persisting minor-proportion TP53 mutations’ existence is not a common phenomenon among patients who do not undergo massive mutation selection. Our long-term observation based on sensitive FASAY analyses in consecutive samples indicates that minor TP53-mutated clones may persist in occasional cases without significant expansion. Such a case was also documented here; in one patient we observed only a very slow increase of TP53-mutated subclone proportion in consecutive relapses. To explore the general incidence of non-selected mutations, we employed NGS and analyzed 40 pretherapy samples from patients remaining wt after treatment line(s) and observed that non-selected mutations are in fact rare since 39/40 patients were devoid of any mutation.
When considering the applicability of highly sensitive NGS in diagnostics, it is important to bear in mind that (i) the original clone size may be variable and under the detection limit of any method and (ii) the dynamics of the expansion process may vary among individual patients due to competition between CLL subclones and, potentially, also the type of therapy.
As we observed in our study, minor-clone mutations do not have to undergo selection after the first treatment. One may consider that the type of treatment could be critical for clonal selection, with the more intensive regimen being more likely to facilitate clonal selection as we indicated in the previous studies.32, 33 However, we document here that in some cases even administrating intensive chemo-immunotherapy resulting in complete remission does not necessarily result in clonal expansion. It is highly likely that there are other factors impacting the selection rate like other genomic defects present either in the TP53-mutated subclone itself or in the TP53-wt cells.
In conclusion, we show in our study that multiple TP53 clonal evolution scenarios are possible, with some of them being more likely to occur (Figure 5). In cases when a minor-proportion TP53-mutated clone(s) is detected, the patient is at high risk of mutation selection by therapy in the first or subsequent relapse, and the presence of the new dominant mutation should be considered as a clearly negative factor impacting the patient’s outcome. Moreover, our detailed analysis of TP53 mutations at the subclonal level at different time points suggests that some patients are intrinsically prone to acquire TP53 mutations and in the majority of these patients more than one clone carrying a different mutation with a different predisposition for expansion occur. Owing to deep sequencing, it is now technically possible to shift TP53 mutation identification to time preceding therapy administration. It seems now especially interesting to explore whether similar rules drive the clonal evolution of other recurrently mutated genes in CLL.

Schematic representation of different scenarios of TP53-mutated subclones clonal evolution.
References
Stilgenbauer S, Zenz T . Understanding and managing ultra high-risk chronic lymphocytic leukemia. Hematol Am Soc Hematol Educ Program 2010; 2010: 481–488.
Zenz T, Eichhorst B, Busch R, Denzel T, Häbe S, Winkler D et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol 2010; 28: 4473–4479.
Gonzalez D, Martinez P, Wade R, Hockley S, Oscier D, Matutes E et al. Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol 2011; 29: 2223–2229.
Byrd JC, Gribben JG, Peterson BL, Grever MR, Lozanski G, Lucas DM et al. Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol 2006; 24: 437–443.
Badoux XC, Keating MJ, Wang X, O'Brien SM, Ferrajoli A, Faderl S et al. Fludarabine, cyclophosphamide, and rituximab chemoimmunotherapy is highly effective treatment for relapsed patients with CLL. Blood 2011; 117: 3016–3024.
Stilgenbauer S, Zenz T, Winkler D, Bühler A, Schlenk R, Groner S et al. Subcutaneous alemtuzumab in fludarabine-refractory chronic lymphocytic leukemia: clinical results and prognostic marker analyses from the CLL2H study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2009; 27: 3994–4001.
Pettitt AR, Sherrington PD, Stewart G, Cawley JC, Taylor AM, Stankovic T . p53 dysfunction in B-cell chronic lymphocytic leukemia: inactivation of ATM as an alternative to TP53 mutation. Blood 2001; 98: 814–822.
Ouillette P, Fossum S, Parkin B, Ding L, Bockenstedt P, Al-Zoubi A et al. Aggressive chronic lymphocytic leukemia with elevated genomic complexity is associated with multiple gene defects in the response to DNA double-strand breaks. Clin Cancer Res 2010; 16: 835–847.
Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013; 152: 714–726.
Rossi D, Cerri M, Deambrogi C, Sozzi E, Cresta S, Rasi S et al. The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of Del17p13: implications for overall survival and chemorefractoriness. Clin Cancer Res 2009; 15: 995–1004.
Zainuddin N, Murray F, Kanduri M, Gunnarsson R, Smedby KE, Enblad G et al. TP53 mutations are infrequent in newly diagnosed chronic lymphocytic leukemia. Leuk Res 2011; 35: 272–274.
Zenz T, Häbe S, Denzel T, Mohr J, Winkler D, Bühler A et al. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood 2009; 114: 2589–2597.
Shanafelt TD, Witzig TE, Fink SR, Jenkins RB, Paternoster SF, Smoley SA et al. Prospective evaluation of clonal evolution during long-term follow-up of patients with untreated early-stage chronic lymphocytic leukemia. J Clin Oncol 2006; 24: 4634–4641.
Stilgenbauer S, Sander S, Bullinger L, Benner A, Leupolt E, Winkler D et al. Clonal evolution in chronic lymphocytic leukemia: acquisition of high-risk genomic aberrations associated with unmutated VH, resistance to therapy, and short survival. Haematologica 2007; 92: 1242–1245.
Zenz T, Krober A, Scherer K, Habe S, Buhler A, Benner A et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood 2008; 112: 3322–3329.
Malcikova J, Smardova J, Rocnova L, Tichy B, Kuglik P, Vranova V et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: selection, impact on survival, and response to DNA damage. Blood 2009; 114: 5307–5314.
Ouillette P, Saiya-Cork K, Seymour E, Li C, Shedden K, Malek SN . Clonal evolution, genomic drivers, and effects of therapy in chronic lymphocytic leukemia. Clin Cancer Res 2013; 19: 2893–2904.
Rossi D, Khiabanian H, Spina V, Ciardullo C, Bruscaggin A, Famà R et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 2014; 123: 2139–2147.
Pospisilova S, Gonzalez D, Malcikova J, Trbusek M, Rossi D, Kater AP et al. ERIC recommendations on TP53 mutation analysis in chronic lymphocytic leukemia. Leukemia 2012; 26: 1458–1461.
Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000; 343: 1910–1916.
R Core Team. A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria. http://www.R-project.org/2013.
Gerstung M, Papaemmanuil E, Campbell PJ . Subclonal variant calling with multiple samples and prior knowledge. Bioinformatics 2014; 30: 1198–1204.
Gerstung M, Beisel C, Rechsteiner M, Wild P, Schraml P, Moch H et al. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun 2012; 3: 811.
Zenz T, Vollmer D, Trbusek M, Smardova J, Benner A, Soussi T et al. TP53 mutation profile in chronic lymphocytic leukemia: evidence for a disease specific profile from a comprehensive analysis of 268 mutations. Leukemia 2010; 24: 2072–2079.
Sturm I, Bosanquet AG, Hermann S, Guner D, Dorken B, Daniel PT . Mutation of p53 and consecutive selective drug resistance in B-CLL occurs as a consequence of prior DNA-damaging chemotherapy. Cell Death Differ 2003; 10: 477–484.
Catovsky D, Richards S, Matutes E, Oscier D, Dyer MJ, Bezares RF et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 2007; 370: 230–239.
Tam C, Shanafelt T, Wierda W, Abruzzo L, Van Dyke D, O'Brien S et al. De novo deletion 17p13.1 chronic lymphocytic leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo Clinic experience. Blood 2009; 114: 957–964.
Rossi D, Rasi S, Spina V, Bruscaggin A, Monti S, Ciardullo C et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 2013; 121: 1403–1412.
Jethwa A, Hüllein J, Stolz T, Blume C, Sellner L, Jauch A et al. Targeted resequencing for analysis of clonal composition of recurrent gene mutations in chronic lymphocytic leukaemia. Br J Haematol 2013; 163: 496–500.
Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011; 475: 101–105.
Zhao Y, Gregory MT, Biertümpfel C, Hua YJ, Hanaoka F, Yang W . Mechanism of somatic hypermutation at the WA motif by human DNA polymerase η. Proc Natl Acad Sci USA 2013; 110: 8146–8151.
Panovská A, Smolej L, Lysák D, Brychtová Y, Šimkovič M, Motyčková M et al. The outcome of chronic lymphocytic leukemia patients who relapsed after fludarabine, cyclophosphamide, and rituximab. Eur J Haematol 2013; 90: 479–485.
Trbusek M, Smardova J, Malcikova J, Sebejova L, Dobes P, Svitakova M et al. Missense mutations located in structural p53 DNA-binding motifs are associated with extremely poor survival in chronic lymphocytic leukemia. J Clin Oncol 2011; 29: 2703–2708.
Acknowledgements
We thank Lenka Jurackova and Jitka Kabathova for their technical help with experiments and Matthew Smith for text editing. This study was supported by MZ CR grants NT13519-4 and NT13493-4, Central European Institute of Technology project CZ.1.05/1.1.00/02.0068 from the European Regional Development Fund, FP7-HEALTH-2012-INNOVATION-1 (NGS-PTL/2012–2015/no.306242), MSMT (2013–2015, no. 7E13008), MUNI/A/0830/2013, SoMoPro II Programme—no. 4SGA8684 (MM) co-financed by the EU and the South-Moravian Region), and EHA Research Fellowship award (MM), and the Czech Leukemia Study Group for Life (CELL).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Malcikova, J., Stano-Kozubik, K., Tichy, B. et al. Detailed analysis of therapy-driven clonal evolution of TP53 mutations in chronic lymphocytic leukemia. Leukemia 29, 877–885 (2015). https://doi.org/10.1038/leu.2014.297
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2014.297
- Springer Nature Limited
This article is cited by
-
Unresolved questions in selection of therapies for treatment-naïve chronic lymphocytic leukemia
Journal of Hematology & Oncology (2023)
-
Evolution of TP53 abnormalities during CLL disease course is associated with telomere length changes
BMC Cancer (2022)
-
Protease associated domain of RNF43 is not necessary for the suppression of Wnt/β-catenin signaling in human cells
Cell Communication and Signaling (2020)
-
Clinical significance of TP53, BIRC3, ATM and MAPK-ERK genes in chronic lymphocytic leukaemia: data from the randomised UK LRF CLL4 trial
Leukemia (2020)
-
ToTem: a tool for variant calling pipeline optimization
BMC Bioinformatics (2018)