
Abstract
Nothofagus cunninghamii is a long-lived, wind-pollinated tree species that dominates the cool temperate rainforests of southeastern Australia. The species’ distribution is more or less continuous in western Tasmania but is fragmented elsewhere. However, it is unknown whether this fragmentation has affected the species’ genetic architecture. Thus, we examined N. cunninghamii using 12 nuclear microsatellites and 633 individuals from 18 populations spanning the species’ natural range. Typical of wind-pollinated trees, there was low range-wide genetic structure (FST=0.04) consistent with significant gene flow across most of the species’ range. However, gene flow was not high enough to overcome the effects of drift across some disjunctions. Victorian populations (separated from Tasmania by the 240 km wide Bass Strait) formed a genetic group distinct from Tasmanian populations, had lower diversity (mean allelic richness (Ar)=5.4 in Victoria versus 6.9 in Tasmania) and were significantly more differentiated from one another than those in Tasmania (FST=0.045 in Victoria versus 0.012 in Tasmania). Evidence for bottlenecking was found in small populations that were at least 20 km from other populations. Interestingly, we found little divergence in microsatellite markers between the extremes of genetically based morphological and physiological altitudinal clines suggesting adaptive differentiation is strongly driven by selection because it is likely to be occurring in the presence of gene flow. Even though the cool temperate rainforests of Australia are highly relictual, the species is relatively robust to population fragmentation due to high levels of genetic diversity and gene flow, especially in Tasmania.
Similar content being viewed by others

Introduction
Understanding the processes affecting the distribution of genetic diversity of temperate trees is important because high genetic diversity can facilitate rapid adaptation to changing environments. In particular, populations that lack genetic diversity can have limited adaptive potential and relatively low fitness (Reed and Frankham, 2003). The cool temperate rainforests of southeastern Australia are ideal for considering how genetic diversity and gene flow affect the ways in which species respond to environmental change. These forests are remnants of closed forests that were more widespread and species rich until about 1.5 million years ago and subsequently underwent repeated periods of range contraction and expansion in response to the glacial–interglacial climate cycles (Hill, 2004). These processes would have inscribed patterns in the genetic structure and diversity of these tree populations that may still be visible today. Thus, areas of glacial survival can show elevated levels of diversity in both nuclear and plastid genomes (Anderson et al., 2006). In addition, patterns of dispersal can sometimes be identified using neutral markers. Unlike vegetation in many other parts of the world where signals of deeper time events have potentially been masked by anthropogenic disturbance, the ranges of the cool temperate rainforests of southeastern Australia have not been substantially altered by human activity.
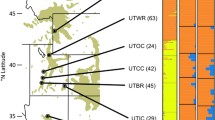
Nothofagus cunninghamii (Hook.) Oersted (Nothofagaceae) is the dominant tree species in most of southeastern Australia’s cool temperate rainforest. This species plays a central role in understanding Australian vegetation history because it is used as a marker of mesic environments in the palaeontological record and its biology and climatic tolerances are well understood. The species has survived for at least 1 million years (Jordan and Hill, 1999), exposing it to repeated glacial periods cold enough to induce glaciation in Tasmania, alternating with periods of warm and relatively mesic climates. Although very common in western Tasmania, it has a fragmented distribution elsewhere with several major disjunctions (Figure 1).

Location of the 18 Nothofagus cunninghamii populations sampled for this study (larger circles) with the known species’ distribution (small circles) underlain. Populations were selected to represent almost the full range of the species considering also major disjunctions; Bass Strait and dryland areas of Tasmania (the Midlands) and Victoria (Latrobe Valley, Western Plains and Yanakie Isthmus).
The strong geographic structuring of chloroplast haplotypes in N. cunninghamii provides evidence both that this species persisted in multiple glacial refugia across all major parts of its range and, assuming that the chloroplast genome is dispersed by seed, that seed-mediated gene flow has been restricted since the last glacial (Worth et al., 2009). Furthermore, the species shows significant morphological and physiological variation across its range for important functional traits such as leaf thickness, area, specific leaf area and stomatal density, and some of this variation is genetic (Hovenden and Vander Schoor, 2006). However, it is unknown how the fragmented range of the species has impacted the distribution of its nuclear genetic variation, the source of most adaptive potential. Indeed, the pollen of N. cunninghamii is likely to be much more mobile than its seed, which are gravity dispersed. This is because the species is wind pollinated and is strongly represented in modern pollen counts even at tens of kilometres distance from source trees (Fletcher and Thomas, 2007). Thus, it is unclear whether substantial geographical barriers to seed-mediated gene flow created by major areas of inhospitable habitat, such as Bass Strait and the dry Midlands region of Tasmania (Worth et al., 2009), are in fact barriers to gene flow via pollen.
We present results from a range-wide study of N. cunninghamii that uses 12 microsatellite markers to investigate whether the species’ fragmented range has impacted its genetic architecture (that is, the structure of the gene pool and genetic diversity of its populations) or whether the species’ mobile pollen has overcome these putative barriers to gene flow. Specifically, we assess whether: (1) the disjunctions across Bass Strait and dryland barriers have impacted the species’ genetic architecture; and (2) altitudinal gradients occur in spite of gene flow. This is the first study to use nuclear markers to better understand population genetic processes in a southern Australian cool temperate rainforest tree species.
Materials and methods
The study species and sampling strategy
N. cunninghamii is confined to cool, humid climates where rainfall exceeds ~1000 mm per year with at least 50 mm during the driest month and typically occurs in areas with low fire frequency (Jackson, 1965). In addition to dominating cool temperate rainforest, the species also commonly occurs in the understory of tall wet eucalypt forests and as a shrub in montane vegetation. The species’ distribution is more or less continuous in western Tasmania, but patchy in Victoria and parts of eastern Tasmania (Figure 1). The widest biogeographical barriers within its range are Bass Strait (an average of ~200 km of open water between the southern Victoria and mainland Tasmania) and the Western Plains in Victoria (a 100-km wide volcanic grassland plain). These barriers are likely to have varied through time. In particular, during glacial periods, Australia was connected to Tasmania and climates were colder and drier. The Latrobe Valley (30 km wide), the Yanakie Isthmus and numerous smaller barriers further fragment populations within Victoria. The dry Midlands of Tasmania (50 km wide) separates the largest group of populations of the species (in the west, south and southeast of the island) from the second largest group, which occur in the north-east highlands of Tasmania. The three smallest and most geographically isolated populations occur at Yarlington Tier in Tasmania, and Kinglake and Wilsons Promontory in Victoria. These populations are more than 24, 32 and 45 km from any other N. cunninghamii populations, respectively. The Yarlington Tier population also occurs in a drier climate than any other populations of the species.
A total of 18 populations of N. cunninghamii were sampled, representing almost the full geographic range of the species and spanning all major disjunctions (Figure 1). The sampling followed a nested design, with three subpopulations sampled within 13 of the populations, two subpopulations from the Tarra Bulga and Douglas Apsley populations and only one each at the small and disjunct Kinglake, Wilsons Promontory and Yarlington Tier populations. Each of the sampled populations was separated by at least 15 km from each other, while subpopulations were separated by 1.5–10 km. Individuals were sampled randomly within populations but at least 20 m from each other to reduce the chance of over-representing particular family groups, which would artificially inflate inbreeding measures. This spacing was not possible in the very small populations at Kinglake and Yarlington Tier, where an adequate sample size could only be achieved by sampling closer individuals, and at Wilsons Promontory, where samples were more widely spaced than in other populations because of the scattered nature of the species in this region. Between 10 and 15 trees were sampled from each subpopulation giving a total of 770 individuals. However, 137 individuals were excluded from the final data (n=633) due to missing data or failed amplification. Four of the populations sampled (Mt Barrow, Projection Bluff, Mt Field and Mt Dundas) were chosen for comparison of high and low altitude populations. In these cases, 20 samples were collected from each high and low altitude subpopulation. High altitude subpopulations occurred at the local altitudinal limit of the species. The low altitude subpopulations were ~400 m lower in altitude than, and between 1.5 and 5 km distant from high altitude populations. The low altitude plants were tall, erect trees with large, thin leaves, whereas the high altitude plants grew as shrubs and had small, thick leaves.
Molecular methods
Genomic DNA was extracted from the samples using the CTAB protocol of Doyle and Doyle (1990), with the following modifications: ~50 mg of fresh leaf tissue was frozen in liquid nitrogen and ground with a tissue disruptor (TissueLyser, Qiagen, Chadstone, VIC, Australia) using a tungsten carbide bead; the ground tissue was then mixed with CTAB buffer containing 4% polyvinylpyrrolidone (PVP40) and 0.7% of dithiothreitol (DTT) and incubated at 56 °C for 30 min. DNA concentration and quality was assessed using agarose gel electrophoresis with Goldview (Guangzhou Geneshun Biotech Ltd, Guangzhou, China) staining and compared with a λ HindIII molecular weight marker.
Nuclear microsatellites previously developed for N. cunninghamii by Jones et al. (2004) were used to identify patterns of genetic variation. These 14 microsatellites were amplified by PCR on a Bio-Rad C1000 thermal cycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA) using Qiagen multiplex master mixes. Loci were separated into two multiplex mixes (Table 1), with each reaction containing 2.5 μl of Qiagen multiplex PCR buffer, 0.5 μl of Q-solution, 5–10 ng of genomic DNA for each reaction and RNase-free water to a final volume of 5 μl. The following PCR programme was used: 95 °C for 15 min; 30 cycles of 94 °C for 30 s, annealing temperature (see Table 1) for 90 s and 72 °C for 60 s; an extension at 60 °C for 10 min; completing with a final holding temperature of 15 °C. PCR product quality was assessed using gel electrophoresis as explained previously. PCR products were diluted to a 1:10 ratio with RNase-free water and 1 μl of diluted PCR product was air dried at 55 °C for 30 min and sent to the Australian Genome Research Facility (Adelaide, Australia) for capillary separation with an AB3730 DNA analyser (Applied Biosystems, Foster City, CA, USA). Genotypes were scored using GeneMapper version 3.7 (Applied Biosystems) with the microsatellite default analysis method and a size standard of ‘GS500 (-250) LIZ’. All microsatellites used in this study have dinucleotide repeats and bin sets were established as either odd or even numbers at least two base pairs apart. Of the 14 microsatellite loci identified by Jones et al. (2004), two (ncutas10 and ncutas16) showed high levels of missing data or peaks with too much stutter, rendering them difficult to score. These two loci were therefore excluded from further analysis.
Data integrity
The frequencies of null alleles (alleles that are not detected) of each locus was estimated using Micro-Checker version 2.2.3 (Van Oosterhout et al., 2004) with a 99% confidence interval. The average frequency of null alleles was 7% across the whole data set, but two loci (ncutas02 and ncutas15) had particularly high frequencies (25 and 21%, respectively; Table 1). To determine the overall effect these two loci had on the results, all analyses were performed with these loci both present and absent. Although excluding these loci reduced overall average null frequency to only 3.7%, FST and diversity metrics did not change greatly.
The average percentage of missing data per population and locus was calculated using GenoDive version 2.0b23 (Meirmans and Van Tienderen, 2004). The total average missing data value was 2%, with highest incidence of 4.7% in the Otway Ranges population (Table 2). Repetition of PCR steps and blind scoring of 72 random samples gave an overall error rate across loci of only 3.1% (Table 1). Both total average missing data and error rate in this study were at levels considered acceptable for microsatellite data (Selkoe and Toonen, 2006). To test each locus for neutrality, we used an FST outlier method implemented by LOSITAN (Antao et al., 2008) with 10 runs of 50 000 simulations and 99.5% confidence interval under an infinite allele mutation mode. The results indicated that all loci in this study were likely to be neutral.
Data analysis
Observed number of alleles (A), observed heterozygosity (HO), expected heterozygosity (He) and Wright’s F statistics were calculated using GenoDive (Meirmans and Van Tienderen, 2004). Allelic richness (Ar) and private allelic richness (PAr) were both rarefied to the smallest population size of 11 using HP-Rare (Kalinowski, 2005). To geographically present spatial trends in Ar and PAr, interpolated surfaces were generated with the inverse distance weighted method incorporated in ArcGIS version 10.2 (ESRI, Redlands, CA, USA). The overall genetic structuring of microsatellite variation was investigated using analysis of molecular variance (AMOVA), Bayesian clustering and principal coordinate analysis.
To allow for the potential impact of null alleles, the inbreeding coefficient FIS was calculated using INEST 2.0 (Chybicki and Burczyk, 2009) with a Gibbs sampler of 500 000 iterations and a burn-in of 50 000 cycles and a Bayesian IIM model. FSTAT version 2.9.3.2 (Goudet, 1995) was used to calculate other inbreeding coefficients: FIT, inbreeding in individuals relative to all populations studied; and FST, the proportion of the total genetic variance contained in a population relative to the total genetic variance. To determine the contribution of stepwise mutation to population differentiation RST and FST were calculated using SPAGeDi (Hardy and Vekemans, 2002), and RST>FST was investigated by testing whether the observed RST was larger than the value obtained after permuting allele sizes among alleles within populations (Hardy et al., 2003). We assessed the partitioning of genetic variation by combining two 3-way AMOVAs with 999 permutations implemented in GenAlEx version 6.501 (Peakall and Smouse, 2006). The first AMOVA had states (Victoria versus Tasmania), populations and individuals as factors and the second AMOVA had populations, subpopulations and individuals. These two analyses were combined by partitioning the variance among individuals within populations (from the first analysis) into variance among individuals within subpopulations and variance among subpopulations within populations from the second analysis (Jones et al., 2006). Bayesian clustering of individuals, with no a priori hypothesis, was conducted using STRUCTURE version 2.3.4 (Pritchard et al., 2000). STRUCTURE employs an admixture ancestry model and an allele frequency model that assumes that allele frequencies are correlated among populations. Ten independent STRUCTURE runs were performed, each with K values allowed to range from 1 to 8, a burn-in period of 100 000 and 500 000 Markov chain Monte Carlo iterations. STRUCTURE HARVESTER (Earl and vonHoldt, 2012) was used to infer the most likely K. Principal coordinate analysis was conducted using GenAlEx (Peakall and Smouse, 2006) and based on a matrix of FST among populations.
To test for isolation by distance, we used the natural logarithm of geographic distance and a genetic relationship matrix between subpopulations calculated as FST/(1−FST) using SPAGeDi (Hardy and Vekemans, 2002). The strength of the correlation between these variables was tested in GenAlEx using a Mantel test with 999 permutations. To predict putative barriers to gene flow, we used BARRIER version 2.2 (Manni et al., 2004) to find the edges associated with the highest rate of genetic change according to Monmonier’s maximum difference algorithm. Allelic frequency histograms were constructed for each population to assess whether allele distributions followed patterns expected for bottlenecked populations (Luikart et al., 1998). To further examine evidence of recent bottlenecks, we used the M-ratio method of Garza and Williamson (2001), which is the mean ratio of the number of alleles to the range in allele size, M, calculated from a population sample of microsatellite loci. Populations that have experienced a recent bottleneck are considered to exhibit a low M-ratio of <0.68 (Garza and Williamson 2001).
Approximate Bayesian computation was implemented in DIYABC 2.0.4 (Cornuet et al., 2010) to investigate the time of genetic isolation of the two main STRUCTURE groups, Tasmania and Victoria. To do this, the geographically and genetically intermediate populations in southern Victoria (see Supplementary Figure S1) were excluded following the strategy of Kimura et al. (2014) and the small Kinglake and Yarlington Tier populations were also excluded because they may have inflated estimates of divergence times. Although coalescence analysis may not be as precise as direct evidence (for example, fossil data) for the timing of events, this method can provide information on the temporal hierarchy of intraspecific gene flow and demographic events caused by distributional changes (Kimura et al., 2014). We compared 12 alternative scenarios for the split between Tasmania and Victoria with and without bottlenecks: four that assumed that Victoria split from Tasmania, four that assumed that Tasmania split from Victoria and four that the populations in the two regions split from a common ancestral population (Supplementary Figure S2). Nine scenarios assumed a bottleneck for one or both of the populations. The priors for the historical models are shown in Supplementary Table S1. A total of 6.85 million simulated data sets (571 428 per scenario) of which the 1% closest to the observed data were used to estimate the relative posterior probabilities of each scenario via a logistic regression approach using 20 summary statistics including the number of alleles, heterozygosity, FST and dμ2 (a measure of pairwise distance between populations). Garza and Williamson’s (2001) M-ratio (within and between populations) was not used because values obtained under all scenarios were significantly different from the observed values. We also tested the effect of constraining the effective population sizes (while keeping all other priors the same) with Tasmanian having a minimum and maximum Ne of 800 and 1500 while Victoria was set at 200 and 400, respectively. These reflected the range in Ne for both populations estimated from the microsatellite data using the linkage disequilibrium method implemented in NeEstimator v.2 (Do et al., 2014)
Results
Genetic diversity
The 12 microsatellites used in this study were highly polymorphic with 8–31 alleles per locus (an average of 20 alleles per locus; Table 1). Observed heterozygosity (HO) per population varied from 0.51 to 0.64 (overall mean=0.59), which was considerably lower than expected heterozygosity (HE=0.58–0.76; overall mean=0.72; Table 2). Similarly, the inbreeding statistic FIS was very low except in the Wilsons Promontory population, in which the FIS value may be inflated by the sampling of more widely spread individuals than in other populations (Table 2). Thus, inbreeding appears to be low, except possibly at Wilsons Promontory. Expected heterozygosity of Tasmanian populations (HE=0.74) was significantly higher (P=0.015 after 1000 permutations) than that of Victorian populations (HE=0.69). Both mean rarefied allelic richness (Ar) and mean rarefied private allelic richness (PAr) were significantly higher for Tasmania than for Victoria (Ar=6.86 versus 5.41; PAr=0.26 versus 0.07; P=0.001 after 1000 permutations in both cases; Table 2; Figure 2). The populations with the highest allelic richness were Franklin-Gordon in western Tasmania and Adamson’s Peak in southeastern Tasmania (Ar=7.53 and 7.38, respectively). Only two Tasmanian populations, Yarlington Tier and Douglas Apsley, had low genetic diversity, with only Yarlington Tier comparable to Victorian populations.

Geographic patterns of genetic diversity in Nothofagus cunninghamii as indicated by (a) estimated allelic richness (rarefied to smallest populations size) and (b) estimated private allelic richness (alleles found in a population that are absent from all other population measured per individual per locus). Subpopulations are indicated by dark dots on the map. Barriers to gene flow predicted with BARRIER are indicated by grey lines; the thickness of the edge of a barrier is proportional to the extent of the barrier and the adjacent numbers relate to the proportion of times the barrier was observed across 100 bootstrap replicates. Populations with high levels of private alleles are labelled.
The genetic variation in Victoria was largely a subset of the variation found within Tasmania: only three alleles present in Victoria were not found in Tasmanian populations, but 87 alleles were present in Tasmania but not in Victoria. Among Victorian populations, the Otway Ranges had the highest allelic richness and number of private alleles, but these were still considerably lower than that for most Tasmanian populations. Kinglake in Victoria had the lowest allelic richness (Ar=3.65) of all populations.
Genetic structure
The species had relatively low level of differentiation with FST of only 0.039±0.005. Most of this was contributed by differentiation of Victorian populations (FST=0.045±0.006), which were considerably more differentiated than those in Tasmania (FST=0.012±0.002). A Z-test indicates that this difference was highly significant (Z=5.4; P<0.001). RST (mean=0.0337±0.0057) was not significantly different from FST (Table 1), indicating that drift was more important for differentiation among populations than mutation (Hardy et al., 2003). The partitioning of the variance using AMOVA based on all 12 loci or 10 loci (that is, excluding loci with high incidences of null alleles) indicated significant population substructure (Supplementary Figure S3). The 10-locus analysis is preferred for this analysis because null alleles can result in overestimation of genetic differentiation (Chybicki and Burczyk, 2009). In that analysis, most of the genetic variation (80%) was within individuals, but 11.6% occurred among individuals within populations, 1.3% occurred among subpopulations within populations, 2.0% was among populations within state and 4.7% between states (Supplementary Figure S3). All variance components were significantly greater than zero (P<0.05). The 12-locus analysis differed mainly in having higher variation among individuals within populations (Supplementary Figure S3).
The best estimate of the number of genetic groups identified by STRUCTURE was two (K=2), according to STRUCTURE HARVESTER. One genetic group was strongly characteristic of Tasmanian individuals and the other of Victorian individuals (Figure 3). However, the estimated memberships for individuals from the two most southern Victorian populations, the Otway Ranges and Wilsons Promontory, indicated more affinity with the Tasmanian population than any other Victorian region (Supplementary Figure S1). The next best level of structuring was K=5, but this was a much poorer fit according to the ΔK method (Earl and vonHoldt, 2012) and will not be discussed further.

Assignment of Nothofagus cunninghamii individuals to genetic clusters based on STRUCTURE analysis, assuming two clusters. Each vertical line represents an individual tree with colour codes according to the estimated proportion of ancestry derived from each cluster identified.
As in the STRUCTURE analysis, the principal coordinate analysis clearly segregated Victorian and Tasmanian populations, and again the most southerly populations in Victoria (Otway Range and Wilsons Promontory) were genetically more similar to populations from Tasmania than were the other Victorian populations (Figure 4). There was no clear geographic patterning within Tasmania but in Victoria, Kinglake was particularly divergent from all other populations. A Mantel test showed that within Tasmania, geographic distance was not significantly correlated (P=0.125) with genetic distance among subpopulations. In contrast, genetic distance between subpopulations was significantly correlated with geographic distance in Victoria, both overall (P=0.006) and also when the genetically and geographically isolated Kinglake population was excluded (P<0.001; Supplementary Figure S4).

Principal coordinates analysis for all populations of Nothofagus cunninghamii based on FST. Note the clear divergence between Tasmanian and Victorian populations, the significant divergence of Kinglake and that the two most southerly populations from Victoria are the closest to those from Tasmania.
The most significant barriers to gene flow (as inferred using BARRIER) were Bass Strait, and the dry regions surrounding the very small populations found at Yarlington Tier in Tasmania, Wilsons Promontory and Kinglake in Victoria (Figure 2). A minor barrier to gene flow was inferred around the Otway Ranges.
Pairwise and average FST comparisons reveal low levels of differentiation between high and low altitude populations (Projections Bluff FST=0.007; Mount Barrow FST=0.016; Mount Dundas FST=0.017; Mount Field FST=0.0016; average FST=0.007; all not significant). Furthermore, analysis of the relationship between altitude and allelic richness across the whole sample set (Supplementary Figure S5) found no statistical correlation (P=0.45) between them providing good evidence of gene flow over these short distances.
Population bottlenecks
Several small and isolated populations showed indications of bottlenecks. In particular, Yarlington Tier in southeastern Tasmania showed very low M-ratio; only 0.56 compared to the value of 0.68 considered indicative of a bottleneck, as well as low allelic diversity (Table 2). Under-representation of rare alleles in this population (Supplementary Figure S6) is further evidence for bottlenecking. Kinglake in Central Victoria also showed all these indicators of bottlenecking (Table 2), although the M value of 0.65 was only slightly below the critical value. This population had the most strongly distorted allele distribution and had HO that was only marginally lower than He giving further evidence for severe bottlenecking. Other populations from Victoria also showed some indications of possible bottlenecking with low allelic richness and under-representation of private alleles, and in the case of Otway Ranges and Tarra Bulga, low M values (M=0.65).
Timing of isolation across Bass Strait
By using the logistic regression method, the best supported scenario under the non-constrained analysis was scenario 3 with a posterior probability of 0.39 (95% confidence interval=0.36–0.43) where both Tasmanian and Victorian populations diverged from a common ancestor. Under scenario 3 the populations diverged 224 generations ago (T1, median value), the effective population size of Tasmania was nearly eight times larger than the central highlands Victoria (9500 versus 1230, respectively, median values; see Supplementary Tables S3 and S4 and Supplementary Figure S7 for detailed results of approximate Bayesian computation analysis). This implied a divergence time of 22 400 years ago assuming the generation time of 100 years estimated by Hickey (1994). Mean relative bias values were generally low (Supplementary Table S3) indicating that estimates are relatively accurate. The second best scenario was scenario 2 (with Tasmanian populations diverged from Victorian populations) with a posterior probability of 0.34 (0.30–0.38). Under this scenario the populations diverged 226 generations ago (T1, median value) or 22 600 years ago. However, scenario 2 is difficult to reconcile with the presence of much greater diversity of chloroplast haplotypes in Tasmania than in Victoria (Worth et al., 2009). When Ne was constrained scenario 1, where Victorian populations diverged from Tasmanian ones, had the highest posterior probability (0.54, 95% confidence interval=0.39–0.69). Under this scenario lower divergence times for Tasmanian and Victorian populations were obtained 31 generations ago (T1, median value) or 3100 years.
Discussion
Overall, the low genetic structure found in this study indicates relatively high gene flow; consistent with the fact that N. cunninghamii is wind pollinated. This gene flow is most likely occurring through pollen dispersal because chloroplast DNA evidence indicates low gene flow by seed in this species (Worth et al., 2009). Nonetheless, the presence of significant genetic patterns at the nuclear level across the species range can be explained by the combined effects of geographic isolation and small population size. Thus, the largest geographic barrier, Bass Strait, created the strongest differentiation in the species. Population size also played a role, with evidence for higher differentiation and lower genetic diversity within the small and relatively isolated Victorian populations compared to those from Tasmania (except for the tiny Yarlington Tier population). The effect of population size was expected because it has been shown to affect both rates of gene flow and rates of drift. This is because small source populations can be expected to produce less pollen than large populations and therefore contribute less to gene flow, and small populations are intrinsically more prone to drift and therefore more susceptible to genetic depletion (Dai and Fu, 2011). The relatively low differentiation of N. cunninghamii is consistent with the ‘moderate’ differentiation found in its close relative from ~700 km further north, N. moorei (Taylor et al., 2005). This latter species has a highly fragmented distribution and therefore may be expected to show greater differentiation.
Divergence across Bass Strait
The presence of a genetic barrier between Victorian and Tasmanian populations is not surprising because Bass Strait (>200 km wide) is wider than maximum reported distances of effective pollen dispersal, which range from 10 to 100 km in wind-pollinated tree species (Kremer et al., 2012). Chloroplast evidence implies long-term survival of the species on both sides of Bass Strait: in the Central Highlands of Victoria and in multiple locations in Tasmania (Worth et al., 2009). In contrast, the Bayesian coalescent analysis estimates that the mean time of genetic isolation across Bass Strait was <25 000 years ago and older than 3000 years ago, a time period that includes the Last Glacial Maximum when sea levels were sufficiently low for Victoria and Tasmania to be connected by land. However, we argue that it is unlikely that a continuous or near-continuous population of N. cunninghamii occurred at that time. This is because the Bass Strait region is modelled as having been extremely hostile for N. cunninghamii during the Last Glacial Maximum (Worth et al., 2014b). The coalescence age estimates also include much of the Holocene, and would include even more if true generation times were shorter than our estimate. Sea levels were sufficiently high (~72 m below current sea level) to form the Bass Strait seaway between Victoria and Tasmania by about 13 000 years ago, late in the Last Glacial (Lambeck et al., 2014). N. cunninghamii would therefore have little time to allow for migration out of its highly restricted refugia (Worth et al., 2009) and form a continuous population across a region in which none of the landmasses (islands) are currently within the bioclimatic range of the species (Worth et al., 2014a). The major pattern in nuclear microsatellites reported here therefore most likely reflects movement across Bass Strait of pollen that pollinated the descendants of N. cunninghamii individuals that survived in glacial refugia. The divergence times obtained using approximate Bayesian computation analysis may therefore be biased by low levels of pollen-mediated gene flow over tens of thousands of years, and the major split in the populations may be considerably older. Indeed it is plausible that the current genetic isolation of Victorian populations is only partial, with ongoing pollen-mediated gene flow occurring, but at rates that are too low to entirely overcome past or present drift. Consistent with the idea of recent or ongoing low level gene flow is that the most southern populations in Victoria at Wilsons Promontory and Otway Ranges are the most similar of all Victorian populations to those in Tasmania.
Contrasting genetic structuring within Tasmania and Victoria
The striking pattern of substantially lower genetic structure and greater genetic diversity in Tasmania compared to Victoria attests to the combined effects of low levels of genetic exchange and high levels of habitat fragmentation in Victoria compared to the more continuous distribution of N. cunninghamii in Tasmania. Thus, the FST in Tasmania (0.012) was low compared to the genetic divergence for trees in general of 0.05 (Petit and Hampe, 2006) but comparable to those observed in other ecologically dominant and widespread wind-pollinated trees with similar life histories, for example, Fagus crenata (0.027; Hiraoka and Tomaru, 2009a) and F. japonica (0.023; Hiraoka and Tomaru, 2009b), but was low compared to related Nothofagus species (0.049–0.14 for South American members of Nothofagus subgenus Lophozonia; Vergara et al., 2014). The lack of differentiation in N. cunninghamii is evident even between eastern and western Tasmanian populations, which means that the dry Midlands region of Tasmania, which is over 50 km wide, poses little barrier to gene flow. Our results therefore suggest that gene flow and/or population size is sufficient to largely override the effects of drift in almost all Tasmanian populations of N. cunninghamii. However, it is difficult to disentangle the impact of the two. Indeed, gene flow has not been enough to erase some patterns of genetic diversity in Tasmania that may be signatures of glacial refugia including ‘hotspots’ of genetic diversity (Ar and PAr) in the known area of refugial survival in the Franklin-Gordon area of southwest Tasmania (Macphail et al., 1993). Similarly, the high diversity of private alleles at Mt Victoria in northeastern Tasmania is consistent with a refugium in that local region as indicated by an endemic chloroplast haplotype (Worth et al., 2009). These private alleles have not dispersed to the west, possibly due to the general westerly wind direction in Tasmania.
In contrast to Tasmania, genetic divergence was higher in Victoria, as evidenced by an FST nearly four times higher than in Tasmania, principal coordinate analysis and significant genetic differentiation with increasing geographic distance. These results are consistent with the low effective population size—only 13% of that of Tasmania, and the more fragmented range of the species in Victoria.
Divergence, low diversity and bottlenecking in isolated populations
Genetic divergence and depauperate diversity shows that even the high rates of gene flow are not sufficient to overcome the genetic consequences of isolation in small populations of N. cunninghamii. Similar findings have been observed in small, fragmented populations of other wind-pollinated species, such as Juniperus communis (Provan et al., 2008). The strongest evidence for bottlenecking was in the two smallest populations, Kinglake in central Victoria and Yarlington Tier in southeastern Tasmania. These populations are both very small (less than 40 adult individuals) and more than 20 km from any other populations. Kinglake is the most significantly diverged of all individual populations and shows evidence of strong bottlenecking; the M value was low and allelic frequency histograms were strongly distorted. Yarlington Tier had low allelic richness and a value of M well below the critical level of 0.68. For both populations, it is unclear when bottlenecking occurred, particularly because of uncertainty about the origins of these populations. Fossil pollen evidence shows the continuous presence of the species at Yarlington Tier for at least 9000 years (Harle et al., 1993), but the age of the Kinglake population is unknown. If these populations survived the last glacial, the signals could not only be a legacy of extreme isolation and population reduction during this period, but also could be a consequence of small population size throughout the Holocene. In contrast, if the population are of Holocene origin via rare long-distance seed dispersal events the bottlenecks may represent founder effects combined with low population sizes.
Evidence for adaptation in N. cunninghamii
Worth et al. (2014b) proposed that adaptation may have contributed significantly to changes in the climatic ranges, in which N. cunninghamii has occurred over the past 25 000 years. This concept is consistent with evidence from common garden trials in controlled environments showing a partly genetically controlled altitudinal cline in N. cunninghamii in a suite of characteristics that are considered to be functionally significant (rates of photosynthesis, leaf thickness, leaf mass per unit area and degree of plasticity in these and other traits; Hovenden and Brodribb, 2000; Hovenden and Vander Schoor, 2006). The argument for a functional relationship of these traits to altitude is reinforced by similar patterns of clinal differentiation in a related species in South America (Premoli and Brewer, 2007; Premoli et al., 2007). Such functional variation could have arisen through drift or reflect adaptation. Given the low levels of differentiation in putatively neutral molecular markers between populations at the extreme of these clines and because high altitude populations did not suffer from low genetic diversity, one has to assume that the differentiation for functional traits has been driven by selection sufficiently strong to overcome gene flow along altitudinal gradients. Such causes of local adaptation are relatively common in forest trees (Aitken and Whitlock, 2013; Kremer et al., 2014).
Conclusion
Results from this study confirm that this wind-pollinated keystone rainforest tree, N. cunninghamii, shows significant gene flow across relatively long distances (at least 60 km). However, historical fragmentation has caused genetic drift in small populations. This study therefore shows that if gene flow is restricted to such an extent that it cannot overcome the impact of drift due to large barriers of unsuitable habitat or small population size, genetic drift can result in significant population divergence even in trees with effective dispersal methods. These data have conservation importance because understanding the genetic consequences of historical population dynamics sheds light on how trees with similar life history traits may respond to modern environmental stressors. If fragmentation of the range of N. cunninghamii increases under future climate change as predicted by Worth et al. (2014a), Victorian populations will probably be vulnerable to loss of genetic diversity. In contrast, western and northeastern Tasmania not only harbour most of the diversity in the species but are also predicted to contain the most suitable climates for the species under projected climate change (Worth et al. 2014a). This matching of projected environment to genetics may reflect the long-term roles of these regions as refugia for N. cunninghamii and species dependant on it.
Data Archiving
Data were available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.dn175.
References
Aitken SN, Whitlock MC . (2013). Assisted gene flow to facilitate local adaptation to climate change. Annu Rev Ecol Evol Syst 44: 367–388.
Anderson LL, Hu FS, Nelson DM, Petit RJ, Paige KN . (2006). Ice-age endurance: DNA evidence of a white spruce refugium in Alaska. Proc Natl Acad Sci USA 103: 12447–12450.
Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G . (2008). LOSITAN: a workbench to detect molecular adaptation based on a FST-outlier method. BMC Bioinformatics 9: 323–327.
Chybicki IJ, Burczyk J . (2009). Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100: 106–113.
Cornuet J-M, Ravigné V, Estoup A . (2010). Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v1. 0). BMC Bioinformatics 11: 401.
Dai Q, Fu J . (2011). When central populations exhibit more genetic diversity than peripheral populations: a simulation study. Chin Sci Bull 56: 2531–2540.
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR . (2014). NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14: 209–214.
Doyle J, Doyle J . (1990). Isolation of plant DNA from fresh tissue. Focus 12: 13–15.
Earl DA, vonHoldt BM . (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4: 359–361.
Fletcher M-S, Thomas I . (2007). Modern pollen–vegetation relationships in western Tasmania, Australia. Rev Palaeobot Palynol 146: 146–168.
Garza J, Williamson E . (2001). Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10: 305–318.
Goudet J . (1995). FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86: 485–486.
Hardy OJ, Vekemans X . (2002). SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2: 618–620.
Hardy OJ, Charbonnel N, Fréville H, Heuertz M . (2003). Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163: 1467–1482.
Harle K, Kershaw A, Macphail M, Neyland M . (1993). Palaeoecological analysis of an isolated stand of Nothofagus cunninghamii (Hook.) Oerst. in eastern Tasmania. Aust J Ecol 18: 161–170.
Hickey JE . (1994). A floristic comparison of vascular species in Tasmanian oldgrowth mixed forest with regeneration resulting from logging and wildfire. Aust J Bot 42: 383–404.
Hill RS . (2004). Origins of the southeastern Australian vegetation. Philos Trans R Soc Lond B Biol Sci 359: 1537–1549.
Hiraoka K, Tomaru N . (2009a). Genetic divergence in nuclear genomes between populations of Fagus crenata along the Japan Sea and Pacific sides of Japan. J Plant Res 122: 269–282.
Hiraoka K, Tomaru N . (2009b). Population genetic structure of Fagus japonica revealed by nuclear microsatellite markers. Int J Plant Sci 170: 748–758.
Hovenden MJ, Brodribb T . (2000). Altitude of origin influences stomatal conductance and therefore maximum assimilation rate in Southern Beech, Nothofagus cunninghamii. Aust J Plant Physiol 27: 451–456.
Hovenden MJ, Vander Schoor JK . (2006). The response of leaf morphology to irradiance depends on altitude of origin in Nothofagus cunninghamii. New Phytologist 169: 291–297.
Jackson WD. : Vegetation. In: Davies J (ed) Atlas of Tasmania. 1965 Department of Lands and Surveys: Hobart, pp 30–35.
Jones RC, Vaillancourt RE, Jordan GJ . (2004). Microsatellites for use in Nothofagus cunninghamii (Nothofagaceae) and related species. Mol Ecol Notes 4: 14–16.
Jones TH, Steane DA, Jones RC, Pilbeam D, Vaillancourt RE, Potts BM . (2006). Effects of domestication on genetic diversity in Eucalyptus globulus. Forest Ecol Manag 234: 78–84.
Jordan GJ, Hill RS . (1999). The phylogenetic affinities of Nothofagus (Nothofagaceae) leaf fossils based on combined molecular and morphological data. Int J Plant Sci 160: 1177–1188.
Kalinowski ST . (2005). HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5: 187–189.
Kimura MK, Uchiyama K, Nakao K, Moriguchi Y, San Jose-Maldia L, Tsumura Y . (2014). Evidence for cryptic northern refugia in the last glacial period in Cryptomeria japonica. Ann Bot 114: 1687–1700.
Kremer A, Potts BM, Delzon S . (2014). Genetic divergence in forest trees: understanding the consequences of climate change. Functional Ecology 28: 22–36.
Kremer A, Ronce O, Robledo-Arnuncio JJ, Guillaume F, Bohrer G, Nathan R et al. (2012). Long-distance gene flow and adaptation of forest trees to rapid climate change. Ecology Letters 15: 378–392.
Lambeck K, Rouby H, Purcell A, Sun Y, Sambridge M . (2014). Sea level and global ice volumes from the Last Glacial Maximum to the Holocene. Proc Natl Acad Sci USA 111: 15296–15303.
Luikart G, Allendorf FW, Cornuet J-M, Sherwin WB . (1998). Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89: 238–247.
Macphail MK, Jordan GJ, Hill RS . (1993). Key periods in the evolution of the flora and vegetation in western Tasmania. I. The early-middle Pleistocene. Aust J Bot 41: 673–707.
Manni F, Guerard E, Heyer E . (2004). Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier's algorithm. Hum Biol 76: 173–190.
Meirmans PG, Van Tienderen PH . (2004). GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4: 792–794.
Peakall R, Smouse PE . (2006). GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295.
Petit RJ, Hampe A . (2006). Some evolutionary consequences of being a tree. Annu Rev Ecol Evol Syst 37: 187–214.
Premoli AC, Brewer CA . (2007). Environmental v. genetically driven variation in ecophysiological traits of Nothofagus pumilio from contrasting elevations. Aust J Bot 55: 585–591.
Premoli AC, Raffaele E, Mathiasen P . (2007). Morphological and phenological differences in Nothofagus pumilio from contrasting elevations: evidence from a common garden. Austral Ecol 32: 515–523.
Pritchard J, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Provan J, Beatty GE, Hunter AM, McDonald RA, McLaughlin E, Preston SJ et al. (2008). Restricted gene flow in fragmented populations of a wind-pollinated tree. Conserv Genet 9: 1521–1532.
Reed DH, Frankham R . (2003). Correlation between fitness and genetic diversity. Conserv Biol 17: 230–237.
Selkoe KA, Toonen RJ . (2006). Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett 9: 615–629.
Taylor KJ, Lowe AJ, Hunter RJ, Ridgway T, Gresshoff PM, Rossetto M. . (2005). Genetic diversity and regional identity in the Australian remnant Nothofagus moorei. Aust J Bot 53: 437–444.
Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P . (2004). MICRO‐CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Vergara R, Gitzendanner MA, Soltis DE, Soltis PS . (2014). Population genetic structure, genetic diversity, and natural history of the South American species of Nothofagus subgenus Lophozonia (Nothofagaceae) inferred from nuclear microsatellite data. Ecol Evol 4: 2450–2471.
Worth JRP, Harrison PA, Williamson GJ, Jordan GJ . (2014a). Whole range and regional-based ecological niche models predict differing exposure to 21st century climate change in the key cool temperate rainforest tree southern beech (Nothofagus cunninghamii. Austral Ecol 40: 126–138.
Worth JRP, Jordan GJ, McKinnon GE, Vaillancourt RE . (2009). The major Australian cool temperate rainforest tree Nothofagus cunninghamii withstood Pleistocene glacial aridity within multiple regions: evidence from the chloroplast. New Phytol 182: 519–532.
Worth JRP, Williamson GJ, Sakaguchi S, Nevill PG, Jordan GJ . (2014b). Environmental niche modelling fails to predict Last Glacial Maximum refugia: niche shifts, microrefugia or incorrect palaeoclimate estimates? Global Ecol Biogeogr 23: 1186–1197.
Acknowledgements
We thank Sascha Wise for molecular laboratory support; Adam Smolenski and Peter Harrison for discussion and advice; and Forestry Tasmania, the Department of Primary Industries, Parks, Water and Environment, Tasmania, the Department of Sustainability and Environment, Victoria, and Ion Maher from Parks and Wildlife Victoria for help with access and permission to collect samples. This research was supported by the Australian Research Council (DP120100501).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Duncan, C., Worth, J., Jordan, G. et al. Genetic differentiation in spite of high gene flow in the dominant rainforest tree of southeastern Australia, Nothofagus cunninghamii. Heredity 116, 99–106 (2016). https://doi.org/10.1038/hdy.2015.77
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2015.77
- Springer Nature Switzerland AG
This article is cited by
-
Climate-driven adaptive responses to drought of dominant tree species from Patagonia
New Forests (2022)
-
Population genetic divergence in Lycium chinense Mill. (Solanaceae) is driven by both hybridization and geo-environmental factors
Genetic Resources and Crop Evolution (2020)
-
Development of EST-SSR markers and their application in an analysis of the genetic diversity of the endangered species Magnolia sinostellata
Molecular Genetics and Genomics (2019)
-
Historical range contraction, and not taxonomy, explains the contemporary genetic structure of the Australian tree Acacia dealbata Link
Tree Genetics & Genomes (2018)
-
Characterization of novel microsatellite markers of the Emei Shan Liocichla using restriction site-associated DNA sequencing
Avian Research (2017)