
Abstract
The oncogenic exogenous Jaagsiekte sheep retrovirus (JSRV), responsible for ovine pulmonary adenocarcinoma, has several endogenous counterparts termed enJSRVs. Although many of these elements have been inactivated over time by the accumulation of deleterious mutations or internal recombination leading to solo long terminal repeat (LTR) formation, several members of enJSRVs have been identified as nearly intact and probably represent recent integration events. To determine the level of enJSRV polymorphism in the sheep population and related species, we have undertaken a study by characterizing enJSRVs copies and independent integration sites in six domestic sheep and two wild species of the sheep lineage. enJSRVs copies were detected by amplifying the env-LTR region by PCR, and for the detection of the insertion sites, we used two approaches: (1) an in silico approach based on the recently published Sheep Reference Genome Assembly (OARv3.0) and (2) an experimental approach based on PCR suppression and inverse PCR techniques. In total, 103 enJSRV sequences were generated across 10 individuals and enJSRV integrations were found on 11 of the 28 sheep chromosomes. These findings suggest that there are still uncharacterized enJSRVs, and that some of the integration sites are variable among the different species, breeds of the same species, subspecies and geographic locations.
Similar content being viewed by others


Introduction
An essential step in the replication cycle of retroviruses is the integration of their genome into the host genomic DNA. During evolution, exogenous retroviruses occasionally infected the germ cells of their hosts, resulting in stably integrated endogenous retroviruses (ERVs) that are passed to subsequent generations following Mendelian rules (Gifford and Tristem, 2003). The continuous accumulation of new retroviral integrations over millions of years resulted in the genomes of all vertebrates being heavily colonized by ERVs. Most ERVs have accumulated genetic defects that render them unable to express infectious virus or proteins. However, some ERVs are transcriptionally active and have maintained intact open reading frames for some of their genes, raising the possibility that some of these elements may be beneficial to their hosts (Stoye, 2001). One of the possible explanations for the selection of some ERVs in vertebrates is their ability to provide protection against infection of related exogenous pathogenic retroviruses. Actually, some enJSRVs appear to block JSRV at two levels. The first block acts at the level of virus entry by receptor interference, whereas the second step blocks most likely viral particle transport or exit. The first step is provided by expression of the enJSRVs env. The cellular receptor for JSRV, the product of the hyaluronidase-2 gene, can interfere with the exogenous and pathogenic JSRV at the level of virus entry (Spencer et al., 2003), but also interact with exogenous env both at the membrane or cytoplasm levels. The env-hyaluronidase-2 interaction decreases the JSRV receptor availability at the cell surface, inhibiting this way virus entry. The second block is provided by Gag expression of some enJSRVs loci (for example, enJS56A1). In 2004, Mura et al. showed that the ERVs JS56A1 particles (with a dominant Gag protein that interferes with its exogenous counterpart as mentioned above) had a regular organization and demonstrated the presence in these groupings of incomplete cores, suggesting that assembled particles were not able to traffic properly to the cell membrane.
Sheep (Ovis aries) provide an interesting animal model system to study retroelements. One of the major infectious diseases of sheep, ovine pulmonary adenocarcinoma (OPA), is caused by a retrovirus called Jaagsiekte sheep retrovirus (JSRV) that has highly related ERV counterparts called enJSRVs (York et al., 1992). OPA has been classically compared with the human bronchioloalveolar carcinoma, and thus studies on Jaagsiekte retrovirus in domestic sheep could provide insights into the genetic basis governing lung carcinogenesis (Archer et al., 2007).
Recently, it has been demonstrated that the invasion of the sheep genome by ERVs of the JSRV/enJSRVs group is still in progress (Arnaud et al., 2007). Moreover, some newly inserted proviruses acquired a defective Gag polyprotein resulting in a transdominant phenotype able to block late replication steps of related exogenous retroviruses (Mura et al., 2004; Arnaud et al., 2007; Murcia et al., 2007; Armezzani et al., 2011).
To date, <30 copies related to Jaagsiekte retrovirus have been identified in 65 different sheep breeds and in 8 related species: Capra hircus, C. falconeri, O. aries musimon, O. ammon, O. canadensis, O. dalli, Bos taurus and Budorcas taxicolor (Arnaud et al., 2007; Mozorov et al., 2007; Wang et al., 2008; Chessa et al., 2009). Furthermore, many of these copies have demonstrated to be polymorphic and their distribution among the Caprinae genome is not yet known totally.
Traditional methods can only analyze a small sample of the total genetic variability of polymorphic retroelements (Boissinot et al., 2004). Inverse PCR (iPCR) technique has been widely used to identify virus insertion points into the genome or sequences flanking transposable elements; (Ochman et al., 1988) but in 2000, a new technique called PCR suppression was used to detect polymorphic insertions/deletions in the genome of different and unsequenced species, and facilitated the creation of a specific library of these flanking regions (Siebert et al., 1995; Lebedev et al., 2000; Mamedov et al., 2002). In addition, the International Sheep Genomics Consortium recently announced the release of the third build of the sheep reference genome assembly called OARv3.0 (Archibald et al., 2010). Thanks to this reference genome assembly, it is now also possible to analyze the chromosomal distribution of enJSRV sequences in silico.
The aim of this study was to characterize enJSRV copies and to map their integration sites by both experimental and in silico methods. In total, we were able to map enJSRV copies in 13 sheep chromosomes and we found at least 32 different enJSRV copies. Owing to the high variability found, this data suggest that there could be more uncharacterized enJSRVs, and that some of the integration sites are variable among different species.
Materials and methods
Samples and breeds
Samples were collected from individuals from a diverse set of species, subspecies and geographic locations (Table 1). DNA extractions were attempted from three species of the subfamily Caprinae: three domestic sheep breeds (Latxa, Assaf and Rasa aragonesa), two Mouflon individuals (O. aries musimon) and two Pyrenean chamois individuals (Rupicapra pyrenaica), some of them known to carry enJSRVs (Arnaud et al., 2007; Chessa et al., 2009). Sheep samples were obtained from experimental flocks that have been monitored for chronic respiratory diseases and also from several flocks from which animals were sampled at culling. The lung samples were macroscopically and microscopically evaluated to confirm the presence/absence of OPA tumors. Tissue samples were fixed in 10% buffered formalin, processed for paraffin inclusion, sectioned at 4–5 μm and stained with hematoxylin and eosin for light microscopy examination. All sheep samples used in this study were negative for OPA to avoid the amplification of the exogenous Jaagsiekte virus whose long terminal repeat (LTR) is homologous to recently inserted enJSRVs.
Preparation of DNA
Briefly, lung tissues were lysed by the addition of TES (0.01 M Tris, 0.1 M EDTA and 0.5% sodium dodecyl sulfate), digested with RNase (100 μg ml−1) and proteinase K (100 μg ml−1) and extracted by traditional phenol/chloroform extraction. DNA from Pyrenean chamois and Mouflons was extracted from muscular tissue.
Experimental detection of enJSRVs
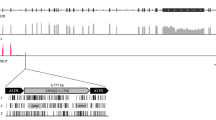
To determine the sequence diversity of enJSRVs among the species and subspecies examined, a primer pair, designed env-LTR was used, which has a forward primer based on enJSRV env sequence and a reverse primer based on the enJSRV LTR region, and thus would only amplify 3′-LTRs in proviruses with a coding region adjacent. These LTR primers were designed using conserved regions of previously sequenced enJSRV LTRs in sheep (GenBank accession numbers: AF136224, AF136225, AF153615 and EF680296-319) (Palmarini et al., 2000; Arnaud et al., 2007). Primer sequences and locations are shown in Figure 1.

Endogenous JSRV structure showing sequenced regions and primer sequences. (a) enJSRV proviral genome structure showing the positions of targeted by three sets of primers amplicons (iPCR and env-LTR primers). R: restriction-site. (b) Oligonucleotide primers used to carry out the iPCR technique. (c) Forward and reverse oligonucleotide primer sequences used for sequencing of enJSRVs.
All PCRs were performed in a total volume of 25 μl including 1 × PCR buffer for Advantage 2 (BD Biosciences, Clontech, Palo Alto, CA, USA), 0.2 mM dNTPs, 1.5 mM MgCl2, 0.4 μM of each primer and 0.5 U per reaction of Advantage 2 Polymerase Mix. PCR method consisted of an initial denaturation at 95 °C for 3 min, followed by a total of 35 cycles of 95 °C for 30 s, 64 °C for 30 s and 72 °C for 30 s, with a final elongation step of 5 min at 72 °C. The PCRs were performed using high-fidelity Advantage 2 Polymerase Mix to minimize polymerase errors. PCR products were analyzed by electrophoresis, purified and directly sequenced in order to verify the identity of the region amplified as the target region and to detect as much variation as possible.
Some enJSRV copies apparently monophyletic to R. pyrenaica were found. To test whether this clade of enJSRVs was also present in O. aries, but its relative abundance was too low to be detected, we designed a pair of primers based on the chamois enJSRV sequences obtained in the current study (GenBank accession number KC407957). These primers were named ‘Chamois-like enJSRV’: 5′-TTTGTTTAGCTCCTTGCCTTAT-TCG_F-3′ and 5′-GTCCTGGAGCCTTAAGGGTAATAAA_R-3′.
enJSRV nomenclature and classification
We followed the nomenclature used by Palmarini et al. (2000) and Arnaud et al. (2007) for the previously characterized enJSRVs. The two major clades previously identified by Palmarini et al. (2000) and designed enJSRV-A and enJSRV-B were used to describe the two main phylogenetic clusters . The most ancient enJSRV copies belong to the enJSRV-A group and recent enJSRV copies (including polymorphic enJSRVs) are clustered into the enJSRV-B group. Finally, we found some new and previously undescribed enJSRVs that are referred as ‘New enJSRVs’ and named with local references.
Genomic neighborhood of sheep enJSRVs
Two different approaches were used to amplify the neighboring regions of enJSRVs: suppression PCR and iPCR.
Suppression PCR
A fraction of endogenous JSRV flanking 5′-LTR sequence was amplified using the selective PCR suppression technique (Siebert et al., 1995; Lebedev et al., 2000; Mamedov et al., 2002). This technique has four main steps: (1) targeted sequences cleaved by a restriction enzyme, (2) ligation of restricted sites with an adapter pair of complementary oligonucleotides of unequal length, (3) amplification of the adapter-containing restricted fragments, and (4) preparation of libraries of amplicons. The PCR amplification of adapter-containing cleaved fragments takes place with the simultaneous use of two primers: A-primer, complementary to the adapter sequence, and primer complementary to a single-stranded part of the stem–loop structure of the fragment, called T primer. Thus, this technique allows the amplification of the neighboring region of a known sequence.
As the adapters are useful as long as they do not have homologous sequences in the analyzed genome (I. Mamedov, personal communication), we did a BLAST search using the adapter sequence as a query to ensure that there was no similar sequence on the sheep genome.
We chose restriction enzymes that did not cut enJSRV LTRs but at the same time had quite common restriction sites to cleave both the inner part of the enJSRV and the neighboring sequence. To do this, known nucleotide sequences of enJSRVs obtained from databases of the National Centre for Biotechnology Information (GenBank accession numbers: AF136224, AF136225, AF153615 and EF680296-319) were analyzed with the software WebCutter version 2.0 (Heiman, 1997). In total, the following five restriction enzymes were selected: VspI, DraI, MspI, TspRI and BsmAI.
A and T primers (Mamedov et al., 2002; Table 2) were used in the PCR suppression technique. LTR primer (designed and named LTRR by Chessa et al. (2009)) amplifies the region surrounding the 5′-LTRs and the 5′-LTR itself, respectively (including the flanking genomic DNA sequences of the host). The PCR mixture contained 10 ng of the ligate in 25 ml of 1 × PCR buffer for Advantage 2 (BD Biosciences, Clontech) containing 200 mM each of dNTPs, 0.4 mM each of primers and 0.5 ml of 50 × Advantage 2 polymerase mix (BD Biosciences, Clontech). The PCR was carried out as follows: 72 °C for 4 min, 23 cycles at 95 °C for 25 s, 65 °C for 25 s, 72 °C for 25 s and a final step of 72 °C for 30 min to ensure the presence of 3′-A-overhangs in the PCR product. The PCR products obtained were gel purified by PureLink extraction kit (Invitrogen, Burlington, ON, Canada) in order to separate different bands by size, and then these were precipitated and dissolved in sterile water.
Inverse PCR
iPCR technique is based on the digestion of DNA with restriction enzymes and circularization of cleavaged products before amplification using primers synthesized in the opposite orientations to those normally employed for PCR. By using inverted primers in a circularized strand, it is possible to amplify in vitro the flanking region of a known sequence (Tsuei et al., 1994).
To carry out the iPCR technique, primers previously described (Chessa et al., 2009) were used but synthesized in the opposite orientation (that is, the forward primer is synthesized as the reverse and the reverse primer as the forward), except for the pair of primers complementary to the LTR (Figure 1b). ProvR_R and ProvF_R (designed and named ProvR and ProvF by Chessa et al. (2009) amplify the inner structural region surrounding the 5′- and 3′-LTRs, respectively; and LTRR and LTRF primers amplify the LTR itself. Finally, a set of primers designed Flank were used as a positive control of the iPCR and suppression PCR techniques, as there were complementary to the host’s sequences flanking enJSRVs (Figure 1b). All the primers were designed to amplify enJSRV-18 that is insertionally polymorphic but appears to be almost fixed in the Latxa sheep breed (Chessa et al., 2009). The restriction enzymes were the same used for the suppression PCR.
The PCR mixture contained 10 ng of the ligate in 8 μl of 1 × PCR buffer for Advantage 2 (BD Biosciences, Clontech) containing 200 mM each of dNTPs, 0.4 mM each of primers and 0.5 ml of 50 × Advantage 2 polymerase mix (BD Biosciences, Clontech). As a positive control for the iPCR, the primers were combined as follows: ProvR_R and 5′-FlankF_R to amplify region flanking 5′-LTR; and ProvF_R and 3′-FlankR_R to amplify 3′-LTR surrounding gene. The PCR was carried out as follows: 95 °C for 1 min, 35 cycles at 95 °C for 15 s, 65 °C (in the case of 5′-LTR region) or 60 °C (for 3′-LTR region) for 15 s and 72 °C for 30 s, and finally a last step of 72 °C for 30 min.
Cloning and sequencing
To examine the degree of polymorphism within samples, purified products were cloned into a TOPO-TA vector and grown in One Shot TOP10 chemically competent Escherichia coli cells (Invitrogen). To test the correct insertion of the transformants, a PCR analysis was performed and M13 Forward (−20) and M13 Reverse primers were used. PCR products were analyzed by gel electrophoresis. Sequences were generated using both sense and antisense primers by the sequencing-kit BigDye v.3.1 (Applied Biosystems, Foster City, CA, USA), or for difficult sequences kit GTPBigDye (Applied Biosystems) was used.
Bioinformatic analyses
Sequence verification and alignment
The sequences obtained were initially verified as enJSRVs using the NCBI BLAST algorithm. All sequences were aligned with Mafft version 5.531 (Katoh et al., 2002, 2005).
Phylogenetic analyses
Within-individual clones that produced identical sequences may have been due to repeated amplification of the same locus within an individual sample. Thus, when two within-individual clones had identical sequences, duplicate sequences were removed, with only one of the sequences chosen for the phylogeny as representative of other duplicate sequences in that individual.
We investigated the phylogenetic history of enJSRV sequences using maximum likelihood (ML) inference and Bayesian inference. ML analyses were conducted with RAxML VIHPC (Stamatakis, 2006) with 1000 bootstrap replicates and for the Bayesian inference, we used the MrBayes 3 (Huelsenbeck and Ronquist, 2001). To assess the appropriate number of runs for the Metropolis-coupled Marcov chain Monte Carlo methods, we used the default value of 106 generations. As the split deviation of both runs was <0.1, we considered to perform the simulation in two runs for 106 generations, with trees sampled every 100 generations under the GTR+I+G model of sequence evolution. The first 2500 trees were discarded in the burn-in and a 50% majority-rule consensus tree was computed from the remaining trees. Finally, to assess the burn-in length, we used the default value of 25% of trees, as the plato of ML values of the trees reached in few generations.
The ovine enJSRV copies that we detected were defined based on the support of phylogenetic trees. A cluster was considered a copy of a previously described enJSRV when the clustering was significant in at least one of the phylogenetic methods (bootstrap values of 70 for ML and Bayesian posterior probability of 95 for Bayesian inference).
Population genetic analyses
Genetic diversity of enJSRVs (based mainly in enJSRVs previously found in sheep) was calculated for each host taxon in MEGA4 (Tamura et al., 2007), using the 118 sequences for env-LTR data. The mean diversity (d) was calculated for each taxon using the Kimura 2-parameter substitution model (Kimura, 1980) such that:

where xi is the frequency of ith sequence in the sample from taxon i; q is the number of different sequences from each taxon; and dij is the pairwise distance between sequences i and j (Tamura et al., 2007). Finally, Kimura 2-parameter distance was calculated for the three pairs of taxa based on enJSRV sequences of each taxon.
In silico detection of enJSRVs and their integration sites
Owing to the short length of the enJSRV copies amplified experimentally in this work, we decided to map them by using whole nucleotide sequences of previously sequenced enJSRVs (GenBank accession numbers: AF136224, AF136225, AF153615 and EF680296-319) (Palmarini et al., 2000; Arnaud et al., 2007). This strategy was based on the similarity of sequences, and previously sequenced enJSRVs were used as a search query. Homologous gene segments were searched in each sheep chromosome (GenBank accession numbers: CM001582-CM001608) using the BLAST alignment tool (http://blast.ncbi.nlm.nih.gov). We did not use the BLAST functionality of the Sheep Reference Genome, as the sequence length of the query is limited to 2000 bps and we only took into account homologous sequences of the same length to the provirus used as query. The resulting homologous sequences were then used as input sequences in the Sheep Genome Browser to analyze their flanking sites. We analyzed 5 kb upstream and downstream of each enJSRV, and to observe whether there was any consensus or common sequence flanking enJSRVs, we compared them on the BLAST alignment tool.
Results
Sequence diversity of enJSRVs
One pair of conserved primers (designed ‘env-LTR’) was used to amplify and sequence a region of enJSRV including the env and 3′-LTR. PCR was attempted on 10 individuals and sequences were successfully generated for all the individuals (Table 1). A total of 118 sequences were generated for enJSRV clones, but among the clones 10 within-individual sequences were identical. These duplicate sequences within the same individual were removed from most analyses, except for the determination of the sequence diversity (Π). Eight sequences were excluded from the data set due to inconsistencies. Thus, subsequent analyses of enJSRVs included a total of 103 sequences of the env-LTR region.
Alignments of the two sets of sequences indicated that there was considerable diversity among species, among individuals within a species and within each individual. This diversity included point mutations and indels. Among the species examined, the Mouflon had the highest level of enJSRV diversity (d: 0.06589 and s.e.: 0.00689), whereas the Pyrenean chamois was found to have the lowest genetic diversity values (d: 0.03309 and s.e.: 0.00412). We also observed significant differences among the three sheep breeds. Latxa sheep breed showed the highest diversity level (d: 0.0644 and s.e.: 0.00815), whereas the diversity of Rasa Aragonesa breed was quite low (d: 0.04905 and s.e.: 0.00435) compared with the mean diversity of O. aries (d: 0.0569 and s.e.: 0.00673) (Figure 2).

Summary by taxon of the genetic diversity present among enJSRVs.
Phylogenetic analyses of experimentally detected enJSRVs
Phylogenetic analyses were performed using the 103 final sequences (after removal of within-individual duplicates) generated using primer pair env-LTR. The analysis included previously published enJSRV sequences from O. aries that are available in GenBank and that overlapped completely the relevant region of enJSRV (GenBank accession numbers: AF136224, AF136225, AF153615 and EF680296-319) (Palmarini et al., 2000; Arnaud et al., 2007). Phylogenetic analyses of the resulting provirus sequences revealed two major clades (Figure 3) previously identified by Palmarini et al. (2000) and designed enJSRV-A and enJSRV-B, which cluster ancient and recent insertions, respectively (Arnaud et al., 2007).

Phylogenetic relationships inferred for 105 sequences across species within the sheep lineage using a 727-bp alignment spanning env to the 3′-LTR region of enJSRV. The sequences shown were generated from cloned amplicons following PCR using the primer set env-LTR (Figure 1; Table 1) and include 27 sequences from GenBank (white labeling) (GenBank accession numbers: AF136224, AF136225, AF153615 and EF680296-319). Sequence labels contain the species and sample number followed by the clone number (L for Latxa, R for Rasa aragonesa, M for Mouflon, A for Assaf and S for chamois), and are colored by taxon: O. aries (dark green), O. aries musimon (light green) and R. pyrenaica (orange). The phylogeny was inferred from partitioned ML and Bayesian analyses. Above and below the branches, the ML bootstrap values are shown and the Bayesian posterior probability is shown (red color for best scores and blue for low scores). The most ancient enJSRV copies belong to the enJSRV-A group and recent enJSRV copies (including polymorphic enJSRVs) are clustered in the enJSRV-B group (Palmarini et al., 2000).
In this work, at least five new enJSRV copies were detected. Clustering of sequences by taxon was only evident for R. pyrenaica, and thus this cluster was designed ‘Chamois-like’. The Chamois-like cluster appeared to have a single retroviral copy. No amplification was detected in sheep with the chamois-specific primers, thus this was the only copy to be demonstrated experimentally in this work to be species specific. All these new enJSRVs seem to be ancient insertions according to the phylogenetic analyses (Figure 3). However, many of the enJSRVs from the enJSRV-B cluster (recent insertions) were too similar between them to determine whether they were new proviruses or not (at least, based only on the analyzed env-LTR sequence).
Comparisons of enJSRVs within and across species
Within species, R. pyrenaica was found to have the lowest genetic diversity values indicating a relatively high homogeneity of enJSRV sequences (Figure 4). This is consistent with the clustering evident for this species in the env-LTR phylogeny (Figure 3) in which 93% of R. pyrenaica sequences fell into a single clade.

The proportion of env-LTR sequences for each taxon that were in each of the clades or groups. All enJSRV types were named according to the groups established by Arnaud et al. (2007), in which the different enJSRV copies were clustered depending on their presence or absence in different species of the sheep lineage. The proportion of enJSRV-A and enJSRV-B copies for each species is also shown. A highly dissimilar copy found in chamois is tagged with a question mark.
The enJSRV sequence diversity observed within species and breeds was also consistent with the proportion of enJSRV types for each taxon and reflected the degree of clustering within the tree (Figure 3). The Mouflon was found to have almost the same enJSRV copies of those of sheep, although the proportion of enJSRV-A copies on O. aries musimon was slightly higher. In the Pyrenean chamois, only three different enJSRV copies were found, and 12 of the sequences appear to be a copy of a previously undescribed enJSRV (Figure 3). Finally, the Kimura 2-parameter distance was estimated to compare enJSRV sequences of different species and subspecies. The highest distance corresponds to comparison involving R. pyrenaica and O. aries (0.1039±0.01328), and the lowest involving O. aries and O. aries musimon (0.06633±0.01195).
Genomic environment of sequences flanking enJSRVs
We used three independent methodologies to analyze the genomic neighborhood of sheep enJSRVs: two experimental methods (suppression PCR and iPCR) and a computational method based on the Sheep Reference Genome Assembly OARv3.0 (see Materials and Methods).
Among the 98 sequences generated for suppression PCR and iPCR clones, only 32 were long enough or contain information of the sites flanking the enJSRVs. However, some of the flanking sequences we obtained were also described previously (Carlson et al., 2003) validating our results. By computational analyses, 19 flanking sequences were generated, thus subsequent analyses included a total of 51 sequences of the enJSRVs flanking sites. Collected sequences ranged from 97 bp to 5 kb (in the case of computationally obtained sequences).
All the sequences were aligned in the BLAST tool: the longest ones (obtained computationally) were selected, and we then assessed if the experimentally obtained sequences overlapped and extended the longest ones. When aligning the 5-kb sequences upstream and downstream of each enJSRV copy, we observed that most of the copies were located in noncoding regions, whereas there was a small proportion of copies inserted into gene introns (Gastrokine-1 in chromosome 3, Neuropolin and Toll-like protein 2 in chromosome 14, and Myotubularin-related protein 13 in chromosome 15). However, in chromosome 20, an enJSRV copy appeared to be inserted between TRIM39 (60 kbp downstream) and HLA-B (85 kbp upstream) (Figure 5).

Chromosomic distribution of enJSRV copies. Bands are colored according to the enJSRV type. Bands marked with an asterisk represent enJSRV copies found in this work; C (Chessa et al., 2009); A (Armezzani et al., 2011). Note that only 2 polymorphic copies (enJSRV-14 and enJSRV-15) were found in the OARv3.0 and on the contrary, copies of enJSRV-20 appear to be distributed along five different chromosomes. Genes neighboring enJSRV copies are also shown.
Integration landscape
Using the Sheep Reference Genome Assembly OARv3.0, we mapped previously described enJSRV copies on sheep chromosomes and we found them on 11 of the 28 sheep chromosomes. When comparing the insertion sites of those enJSRV copies, we found that many of recently inserted and thus, probably polymorphic enJSRVs had a different insertion site or there were not present in the OARv3.0 (Figure 5). The most common enJSRV copy found in this study was enJSRV-20, with copies distributed among five different chromosomes. enJS56A1 appeared to be as well widely distributed, with copies in three different chromosomes. Finally, we found a single copy of enJSRV-15 in chromosome 6, a single copy of enJSRV-3 and a copy of enJSRV-25 in chromosome 14, a single copy of enJSRV-19 in chromosome 6, a single copy of enJSRV-4 in chromosome 15, and a single copy of enJSRV-14 in chromosome 19 (Figure 5). enJSRV-14 and enJSRV-15 were the only polymorphic enJSRV copies found.
Discussion
Diversity of enJSRVs across the species of the sheep lineage
The current study is the first work studying enJSRV diversity quantitatively and also the first analyzing enJSRV diversity in R. pyrenaica populations to date. We observed that enJSRV diversity was quite different among the three species, among individuals within a species and within each individual examined.
The greatest differences detected among enJSRV sequences between taxa were found in comparisons that involved both sheep or Mouflon and chamois. We observed that R. pyrenaica showed the lowest diversity patterns and this was consistent with the low number of enJSRV copies that were found in this species. It seems that R. pyrenaica suffered mainly a single enJSRV infection and that the colonization of its genome by other enJSRV copies was anecdotal. This could be due to the isolation of this species in comparison with sheep populations that usually live in herds. This is in agreement with the previous suggestion that herd size could be a risk factor for certain diseases (Gardner et al., 2002).
In comparison, O. aries and O. aries musimon appeared to have almost the same enJSRV copies but the proportion of ancient copies is higher in O. aries musimon. Interestingly, it has been suggested that Mouflon could be the remnant of the first domesticated sheep re-adapted to feral life (Hiendler et al., 2002). Thus, Mouflon could have suffered from enJSRV infections when they first became domesticated ∼9000 years ago (Harris, 1996).
Finally, O. aries was the species with the higher percentages of recent enJSRV copies that suggests that the sheep has experienced recently a higher proliferation of this type of enJSRVs, reinforcing the results obtained by Arnaud et al. (2007). Indeed, the main provirus that is under positive selection, enJS56A1 provirus (and its transdominant copy, enJSRV-20 with a W21 Gag variant) represents a recent insertion event (Armezzani et al., 2011). Owing to the difficulty to define different enJSRV copies among recent insertions, it is possible that many of the recent copies are in fact enJS56A1 copies, and thus are under positive selection too. Actually, it has been proposed that the domestic sheep has acquired, by genome amplification, several copies of this provirus (Armezzani et al., 2011). In consequence, these data could explain the high proportion of enJSRV copies found in the enJSRV-B group. However, to test this hypothesis complete sequences (including the transdominant W21 Gag) would be needed.
These results were also consistent with the clustering and/or distinctiveness of the enJSRVs evident for these three species in the env-LTR phylogeny that supports the subdivision of enJSRVs into two major clades, designed enJSRV-A and enJSRV-B (Palmarini et al., 2000) (Figure 3): all but two R. pyrenaica enJSRV sequences were clustered in the Chamois-like clade (enJSRV-A), whereas O. aries and O. aries musimon sequences were present in both enJSRV-A and enJSRV-B clades.
Although the current study is the first quantitative analysis of the proportion of all enJSRV copies in the Caprinae lineage, some presence/absence analyses for each enJSRV copy have been made (Palmarini et al., 2000; Arnaud et al., 2007). Studies on endogenous feline leukemia viruses in the germ lines of the domestic cat and related wild species of the genus Felis revealed similar results to those describe in the current study, suggesting a complex history of interactions between FeLV and the germ lines of the species that comprise the domestic cat lineage (Polani et al., 2010). These findings are also analogous to the different patterns seen across species of primates in studies involving ERVs also found in humans (Bannert and Kurth, 2006). In particular, the different enJSRV patterns detected across species and breeds of the sheep lineage likely resulted from invasion, proliferation or homogenization of enJSRVs that varied among these hosts.
Genomic neighborhood of sheep enJSRVs
Our analysis of enJSRV copies in the Caprinae genome revealed common elements flanking retroviral sequences, resembling some sheep genes and domains. Furthermore, a portion of these sequences were previously described by Carlson et al. (2003) validating our results. As ERVs are sequences able to ‘jump’ into new locations within genomes, they can induce various types of rearrangements by transposition and recombination, and thus it has been demonstrated that mobiles elements in general and ERVs in particular are a source of evolution and functional diversification of gene families (Deininger et al., 2003).
It is possible that during the transposition and the reintegration procedure of these ERVs, short sequences of the previous target sites turn out to be excised and inserted into the new locations. The ability of enJSRVs to encode a reverse transcriptase activity and move by retrotransposition could also explain why these flanking sequences appear in both the direct and inverse directions.
Finally, when we analyzed enJSRV locations within the chromosomes, we found that the most common enJSRV copies in the sheep genome OARv3.0 were enJSRV-20 and enJS56A1 (the same as in diversity studies), the latter containing the defective Gag polyprotein that blocks the late replication steps of the Jaagsiekte exogenous sheep retrovirus (exJSRV). The fact that many of the polymorphic enJSRV copies are not present in the OARv3.0 could have two possible explanations: (1) it could be that the two only animals analyzed in this version of the sheep genome did not have, in fact, any polymorphic copy apart from enJSRV-14 and enJSRV-15; or (2) as the sequencing of this reference genome is still under development, it is possible that there are still more uncharacterized virus. If so, the insertion sites of the various copies of enJSRVs would be those described in this work. However, we cannot predict which enJSRV copy would be inserted in a specific location.
Taken together, these results suggest a complex co-evolutionary interactions of JSRVs and the germ lines of the species that comprise sheep lineage. Owing to the high proportion of insertionally polymorphic enJSRV copies found, there is evidence to suggest that the invasion of the sheep genome by enJSRVs is still in progress.
Data archiving
Sequence data have been submitted to GenBank (accession numbers KC407956-93 and KC414197). Only sequences clustered in different branches and thus, considered new enJSRVs were deposited.
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
Archer F, Jacquier E, Lyon M, Chastang J, Cottin V, Mornex JF et al. (2007). Alveolar type II cells isolated from pulmonary adenocarcinoma: a model for JSRV expression in vitro. Am J Respir Cell Mol Biol 36: 534–540.
Archibald AL, Cockett NE, Dalrymple BP, Faraut T, Kijas JW, Maddox JF et alInternational Sheep Genomics Consortium. (2010). The sheep genome reference sequence: a work in progress. Anim Genet 41: 449–453.
Armezzani A, Arnaud F, Caporale M, Pia Di Meo G, Iannuzzi L, Murgia C et al. (2011). The signal peptide of a recently integrated endogenous sheep betaretrovirus envelope plays a major role in eluding Gag-mediated late restriction. J Virol 85: 7118–7128.
Arnaud F, Caporale M, Varela M, Biek M, Chessa B, Alberti A et al. (2007). A paradigm for virus–host coevolution: sequential counter-adaptations between endogenous and exogenous retroviruses. PLoS Pathog 3: 1716–1729.
Bannert N, Kurth R . (2006). The evolutionary dynamics of human endogenous retroviral families. Annu Rev Genomics Hum Genet 7: 149–173.
Boissinot S, Entezam A, Young L, Munson PJ . (2004). The insertional history of an active family of L1 retrotransposons in humans. Genome Res 14: 1221–1231.
Carlson J, Lyon M, Bishop J, Vaiman A, Cribiu E, Mornex JF et al. (2003). Chromosomal distribution of endogenous Jaagsiekte sheep retrovirus proviral sequences in the sheep genome. J Virol 77: 9662–9668.
Chessa B, Pereira F, Arnaud F, Amorim A, Goyache F, Mainland I et al. (2009). Revealing the history of sheep domestication using retrovirus integrations. Science 324: 532–536.
Deininger PL, Moran JV, Batzer MA, Kazazian HHJ . (2003). Mobile elements and mammalian genome evolution. Curr Opin Genet Dev 13: 651–658.
Gardner A, Willeberg P, Mousing J . (2002). Empirical and theoretical evidence for herd size as a risk factor for swine diseases. Anim Health Res Rev 3: 43–55.
Gifford R, Tristem M . (2003). The evolution, distribution and diversity of endogenous retroviruses. Virus Genes 26: 291–315.
Harris DR . (1996) The Origins and Spread Of Agriculture and Pastoralism in Eurasia In: Harris DR (ed.). Smithsonian Institution Press: Washington, DC, USA Vol. 5, pp 552–573.
Heiman M . (1997) Webcutter 2: 0http://rna.lundberg.gu.se/cutter2/.
Hernández-Fernàndez M, Vrba ES . (2005). A complete estimate of the phylogenetic relationships in Ruminantia: a dated species-level supertree of the extant ruminants. Biol Rev Camb Philos Soc 80: 269–302.
Hiendleder S, Kaupe B, Wassmuth R, Janke A . (2002). Molecular analysis of wild and domestic sheep questions current nomenclature and provides evidence for domestication from two different subspecies. Proc Biol Sci 269: 893–904.
Huelsenbeck J, Ronquist F . (2001). MrBayes: Bayesian inference of phylogeny. Bioinformatics 17: 754–755.
Katoh K, Misawa K, Kuma K-I, Miyata T . (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30: 3059–3066.
Katoh K, Kuma K, Toh H, Miyata T . (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33: 511–518.
Kimura M . (1980). A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16: 111–120.
Lebedev YB, Belonovitch OS, Zybrova NV, Khil PP, Kurdyukov SG, Vinogradova TV et al. (2000). Differences in HERV-K LTR insertions in orthologous loci of humans and great apes. Gene 247: 265–277.
Mamedov I, Batrak A, Buzdin A, Arzumanyan E, Lebedev Y, Sverdlov ED . (2002). Genome-wide comparison of differences in the integration sites of interspersed repeats between closely related genomes. Nucl Acids Res 30: e71.
Mozorov VA, Mozorov AV, Lagaye S . (2007). Endogenous JSRV-like proviruses in domestic cattle: analysis of sequences and transcripts. Virology 367: 59–70.
Mura M, Murcia P, Caporale M, Spencer TE, Nagashima K, Rein A et al. (2004). Late viral interference induced by transdominant Gag of an endogenous retrovirus. Proc Natl Acad Sci 101: 11117–11122.
Murcia PR, Arnaud F, Palmarini M . (2007). The transdominant endogenous retrovirus enJS56A1 associates with and blocks intracellular trafficking of the JSRV Gag. J Virol 81: 1762–1772.
Ochman H, Gerber AS, Hartl DL . (1988). Genetic applications of an inverse polymerase chain reaction. Genetics Soc America 120: 621–623.
Palmarini M, Hallwirth C, York D, Murgia C, De Oliveira T, Spencer T et al. (2000). Molecular cloning and functional analysis of three type D endogenous retroviruses of sheep reveal a different cell tropism from that of the highly related exogenous Jaagsiekte sheep retrovirus. J Virol 74: 8065–8076.
Polani S, Roca AL, Rosensteel BB, Kolokotronis S, Bar-Gar GK . (2010). Evolutionary dynamics of endogenous feline leukemia virus proliferation among species of the domestic cat lineage. Virology 405: 397–407.
Siebert PD, Chenchik A, Kellogg DE, Lukyanov KA, Lukyanov SA . (1995). An improved PCR method for walking in uncloned genomic DNA. Nucleic Acids Res 23: 1087–1088.
Spencer TE, Mura M, Gray CA, Griebel PJ, Palmarini M . (2003). Receptor usage and fetal expression of ovine endogenous betaretroviruses: implications for coevolution of endogenous and exogenous retroviruses. J Virol 77: 749–753.
Stamatakis A . (2006). RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690.
Stoye JP . (2001). Endogenous retroviruses: still active after all these years? Curr Biol 11: 914–916.
Tamura K, Dudley J, Nei M, Kumar S . (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599.
Tsuei DJ, Chen PJ, Lai MY, Chen DS, Yang CS, Chen JY et al. (1994). Inverse polymerase chain reaction for cloning cellular sequences adjacent to integrated hepatitis B virus DNA in hepatocellular carcinomas. J Virol Methods 49: 269–284.
Wang Y, Liu S, Li J, Han M, Wang Z . (2008). Cloning and sequence analysis of genome from inner Mongolia strain of the endogenous betaretroviruses (enJSRV). Virol Sinica 23: 15–24.
York DF, Vigne R, Verwoerd DW, Querat G . (1992). Nucleotide sequence of the Jaagsiekte retrovirus, an exogenous and endogenous type D and B retrovirus of sheep and goats. J Virol 66: 4930–4939.
Acknowledgements
We thank Dr JA Rodriguez for helping on the experimental techniques; Dr K García-Etxebarria for advice on bioinformatics analysis and Dr A Larruskain for comments on the article. We also thank I Montes for advice on the population genetic analyses. This work was supported by the University of the Basque Country UPV/EHU (grant GIU10/22 given to BMJ and UFI11/20). MS-P was supported by a fellowship from the Department of Education of the Basque Government (RBFI-2012-63).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Sistiaga-Poveda, M., Jugo, B. Evolutionary dynamics of endogenous Jaagsiekte sheep retroviruses proliferation in the domestic sheep, mouflon and Pyrenean chamois. Heredity 112, 571–578 (2014). https://doi.org/10.1038/hdy.2013.136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2013.136
- Springer Nature Switzerland AG