
Abstract
Deletion of HAX1 in mice causes a severe reduction in the numbers of lymphocytes in the bone marrow and in the spleen. Additionally, B220+ B progenitor cells in the bone marrow are reduced, suggesting an important function of HAX1 in B cell development. HAX1 is thought to play a protective role in apoptotic processes; therefore, we investigated the role of HAX1 in bone marrow B progenitor cells and splenic B cells. We did not observe an effect on the survival of Hax1−/− bone marrow cells but detected enhanced survival of splenic Hax1−/− B cells upon in vitro starvation/growth-factor withdrawal. To explain this apparent inconsistency with previous reports of HAX1 function, we also studied the B cell receptor (BCR)-induced apoptosis of IgM-stimulated splenic naïve B cells and found that apoptosis decreased in these cells. We further found impaired internalization of the BCR from Hax1−/− splenic B cells after IgM crosslinking; this impaired internalization may result in decreased BCR signaling and, consequently, decreased BCR-mediated apoptosis. We measured HAX1 binding to the cytoplasmic domains of different Ig subtypes and identified KVKWI(V)F as the putative binding motif for HAX1 within the cytoplasmic domains. Because this motif can be found in almost all Ig subtypes, it is likely that HAX1 plays a general role in BCR-mediated internalization events and BCR-mediated apoptosis.
Similar content being viewed by others


Introduction
HAX1 was originally identified as an interaction partner of the hematopoietic lineage cell-specific protein 1 (HS1)1 and was shown to affect receptor-mediated apoptotic and proliferative responses of lymphocytes.2,3,4 Generally, HAX1 is closely associated with not only the mitochondrial membrane but also the endoplasmic reticulum, nuclear envelope, and plasma membrane.1,4,5,6 The fact that HAX1 functions as an interaction partner of many different proteins, including caspase-9,7 Omi/HtrA2,8,9 cortactin/EMS1,5 the α subunit of G13 heterotrimeric G protein,10 polycystic kidney disease protein 2 (PKD2),5 ILK (integrin-linked kinase),11 and the cytoplasmic tail of ανβ6 integrin,12 to intracellular pre-IL1α13 and to some virus-associated proteins,14,15,16,17 suggests that HAX1 has complex functionality.
Because of the structural similarity of HAX1 to the anti-apoptotic protein Bcl-2,1 HAX1 was thought to play an important role in the regulation of programmed cell death and in the promotion of cell survival. This hypothesis was confirmed by several studies that demonstrated a protective function of HAX1 against apoptotic cell death.7,8,18,19 In humans, HAX1 deficiency causes autosomal recessive severe congenital neutropenia (SCN),20,21 and HAX1 was suggested to be a major regulator of myeloid homeostasis, thereby emphasizing its effect on apoptosis during neutrophil development. In psoriasis, disturbed epidermal differentiation correlates with increased HAX1 expression, which may result in prolonged resistance to terminal differentiation.22
In addition to its anti-apoptotic role, HAX1 has been shown to have proapoptotic properties that may be mediated by the upregulation of HAX1 splice variant II23 (shown in rat). This change in function could be mediated partly by the ability of the HAX1 splice variants to form homotypic and heterotypic dimers. The human and rat HAX1 genes are heavily spliced.24,25,26 In patients, HAX1 mutations affecting splice variant I lead to SCN, whereas mutations affecting splice variants I and II lead to CNS symptoms.21,26,27 In mouse, only three different mRNAs encoded by two genes on chromosomes 2 and 3 have been described thus far.19,25 Chromosome 3 encodes an mRNA of 1121 base pairs (HAX-1s) and a splice variant (HAX-1xs) with an internal deletion before the ends of exons 2 and 3 that keeps the reading frame intact.19 In addition, a third species for the long HAX1, called Silg111, which does not contain introns and which is believed to be a pseudogene, originates from chromosome 2.28
We generated a mouse model in which we deleted exons 2 and 3 of HAX1 on chromosome 3 and abolished the production of both splice variants; this model was used to demonstrate the crucial function of HAX1 in B cell development and in hematopoietic stem cell homeostasis maintenance.29 Additionally, as described in detail by Chao et al.,8 HAX1 deletion led to death at the age of 12 weeks; this premature death was caused by a loss of motor coordination, leading to a failure to drink and eat. Here, we aimed to explain the reduced number of B cells in more detail, and we asked whether the general viability of the bone marrow B progenitor cells and of the splenic B cells might be affected by the proposed anti-apoptotic function of HAX1. We first investigated the viability of the bone marrow cells and splenocytes upon starvation using eFluor staining and further investigated the influence of HAX1 deletion on apoptotic processes of anti-IgM-stimulated immature and mature B cells (using live/dead and apoptosis staining). Additionally, we addressed the question of whether HAX1 affects the internalization kinetics of the IgM-B cell receptor (BCR) using FACS. Because HAX1 is an IgE tail-interacting protein,30 we further analyzed the binding of HAX1 to different cytoplasmic immunoglobulin domains by co-immunoprecipitation (co-IP) experiments and by surface plasmon resonance analysis (SPRA).
Materials and methods
Mice
The BALB/c Hax1−/− mouse strain was generated by Peckl-Schmid et al.29 Breeding and maintenance were conducted in the animal facility at the University of Salzburg according to the institutional and national guidelines for animal care and use.
Cell lines and media
The murine B lymphoma cell line A20 (TIB-208) was maintained in RPMI 1640 supplemented with 10% FBS, 50 μM β-mercaptoethanol, 20 mM L-glutamine, 1x penicillin and streptomycin, 1 mM sodium pyruvate (PAA Laboratories), and 25 mM HEPES (Gibco). A20 cells that were stably transfected with chimeric constructs expressing human IgE/mouse IgM, IgG1, IgG2a, IgE, or IgA receptors were cultured in supplemented RPMI enriched with 400 μg/ml G418 (Gibco). The original human IgE plasmid (dEMPD) was kindly provided by Facundo Batista.29,31
Antibodies
For the in vitro stimulation of splenic B cells and bone marrow cells, we used goat anti-mouse IgM (Southern Biotechnology, 1020-01) antibody. For BCR internalization experiments, we used rat anti-mouse IgM-FITC (BD Pharmingen, R6-60-2) and goat anti-mouse IgM-pHrodo (Southern Biotechnology, 1020-01) antibodies. Apoptosis assays were performed with annexin V-FITC and the corresponding binding buffer (eBioscience) with eFluor780 and eFluor450 (eBioscience). For FACS analysis, the cells were further stained with CD45R/B220-APC (BD Pharmingen, clone RA3-6B2) antibody. For co-IP, we used anti-HAX1 (BD Transduction Lab) and anti-human IgE-HRP (KPL) antibodies.
Viability and apoptosis assays
Bone marrow cells and splenocytes were isolated from 8-week-old wild type (WT) and Hax1−/− mice, and red blood cells were lysed using 1x BD Pharm Lyse buffer (BD Biosciences). In total, 1 × 106 washed and pelletized cells were resuspended in 1 ml supplemented RPMI medium (described above), seeded in 48-well plates and incubated in a humidified atmosphere at 37°C.
Viability assay. The cells received no additional stimuli. Duplicates (1 × 106 per well) were analyzed for their viability from days 0 to 4. The cells were harvested, washed with 1 ml 1x PBS, and stained with eFluor780 for 30 minutes at 4°C. The stained cells were washed two times with 1x PBS.
Apoptosis assay. In total, 5 × 106 cells were stimulated with 2 μg goat anti-IgM (Southern Biotechnology, clone 1020-01). Then, 1 × 106 cells were analyzed at days 0, 1, 2, 3, and 4. The cells were washed with PBS and stained with eFluor450 and CD45R (B220) for 30 minutes at 4°C. After the cells were washed with 1 ml FACS buffer (1x PBS and 3% FCS) and 1 ml 1x binding buffer (eBioscience), they were stained with 5 μl annexin V-FITC (eBioscience), which was diluted in 100 μl 1x binding buffer, for 15 minutes in the dark at room temperature (RT). Then, the cells were washed with 1x binding buffer and apoptosis analysis was performed using a FACSCantoII flow cytometer and FlowJo 9.7.6 software.
Internalization assays
Single cell suspensions of splenocytes were prepared with 1x PBS, and red blood cells were lysed using 1x BD Pharm Lyse buffer (BD Biosciences).
Internalization of IgM-FITC. After the cells were washed, they were stained on ice with anti-IgM-FITC for 15 minutes and washed with ice-cold PBS. The cells were resuspended in 37°C pre-warmed 1x PBS/3% FCS and incubated at 37°C. At the time points 0, 5, 10, 15, 20, 25, and 30 minutes, an aliquot was stopped with ice-cold PBS. The cells were additionally stained with B220-APC (BD Pharmingen) and eFluor450 (eBioscience) on ice for 15 minutes and washed again with ice-cold PBS.
Internalization of IgM-pHrodo. In total, 100 μg goat anti-mouse IgM (clone 1020-01) was labeled using a pHrodo Red Microscale Labeling Kit (Molecular Probes by Life Technologies) according to the manufacturer’s instructions. Then, 1 × 106 cells were resuspended in 3 ml PBS/30 μl IgM-pHrodo and incubated at 37°C. At the time points 0, 15, 30, 45, 60, 75, and 90 minutes, 300 μl was stopped with ice-cold PBS, centrifuged and stained with B220-APC and eFluor450.
Co-immunoprecipitation of HAX1 with the hIgE antigen receptor
In total, 1 × 106 transfected A20 cells were centrifuged and resuspended in 1 ml lysis buffer (25 mM Tris (pH 7.5), 140 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, and 1 mM SDS) containing a protease inhibitor (Complete Mini; Roche) and rotated at 4°C for 1 hour. The mixtures were centrifuged thereafter, and the supernatant was transferred into fresh tubes. Then, 5 μg HAX1 antibody was added, and the mixtures were rotated for an additional 3 hours at 4°C. Next, 50 μl Protein G Sepharose 4 Fast Flow (GE Healthcare) was added and incubated overnight by rotating at 4°C. On the next day, Protein G Sepharose was centrifuged for 1 minute at 14 000 rpm and washed two times with lysis buffer and two times with wash buffer (100 mM Tris (pH 8.1) and 0.5 M LiCl). After the pellets were washed, 40 μl 4x XT Sample buffer/20x XT Reducing Agent (1:5) (Bio-Rad) was added to the pelletized Protein G Sepharose and heated at 95°C for 5 minutes. Then, the samples were loaded onto a 10% polyacrylamide gel. The separated proteins and the Bio-Rad Kaleidoscope marker were transferred onto a methanol-activated Immobilon-P PVDF membrane (Millipore Corporation, Bedford, MA, USA) by electroblotting at 250 mA. Then, the membrane was blocked (blocking buffer: 10 mM Tris (pH 7.5), 100 mM NaCl, 0.1% Tween 20, and 5% nonfat dry milk) for 1 hour at RT under gentle agitation and incubated overnight with 5 μg anti-human IgE-HRP antibody. Detection was performed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Production of recombinant HAX1 protein
Complete HAX1 cloned into the pHIS-parallel 2 vector29,30 was used for the production of recombinant HAX1 protein. Protein expression was induced in the E.coli strain BL21 at a density of OD600 = 0.8 by adding 0.75 M IPTG (stock: 1 M) and incubating at 26°C overnight. Then, the overnight culture was disrupted by sonification. Recombinant His-tagged HAX1 protein was purified from the supernatant using nickel-nitrilotriacetic acid agarose (Qiagen) according to the manufacturer’s instructions, i.e., washing the column with binding buffer (8 M urea, 50 mM NaH2PO4, 300 mM NaCl, and 20 mM imidazole), followed by protein elution using elution buffer (8 M urea, 50 mM NaH2PO4, 300 mM NaCl, and 250 mM imidazole). The purified protein was analyzed by gel electrophoresis.
Surface plasmon resonance analysis with Biacore X
Recombinant His-tagged HAX1 protein (1 mg/ml in 10 mM NaH2PO4 (pH 7.5)) was diluted in 10 mM Na-acetate (pH 4) to a final concentration of 12 ng/μl and coupled to a CM5 chip (GE Healthcare) according to the manufacturer’s instructions. The empty flow cell 2 was used as a reference. Synthetic peptides of the corresponding M2 regions (cytoplasmic domains) were injected at different concentrations (1–1000 μM).
Data analysis
The data are shown as mean ± SD. The statistical significance (n.s., p > 0.05; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001) was calculated using the unpaired Student’s t-test.
Results
Hax1−/− bone marrow cells show a decreased number of B220+ cells
HAX1 deletion dramatically decreased the number of bone marrow cells,8,29 among which the overall cell type distribution was only marginally affected (i.e., a trend toward a decreased percentage of lymphocytes) (Figure 1a). However, within the lymphocyte population, we found that the percentage of B220+ cells decreased significantly (Figure 1b), which suggests impaired lymphopoiesis throughout the development of B220+cells. Surface expression of the B220 marker is a characteristic of B cell development, and the decreased number of B220+ cells within the lymphocytes reflects impaired B cell development, resulting in a low number of B cells released from the bone marrow into the peripheral circulation of Hax1−/− mice.8,29 Representative FACS plots are shown in Figure 1c. The gating history is shown in Supplementary Figure 1.

Frequencies of bone marrow lymphocytes and B220+ cells. (a) Percentages of cells gated from live lymphocytes from four Hax1−/− and four WT mice. (b) The lymphocytes were further analyzed to determine the number of B220+ cells. (c) One representative comparison is shown. The means ± SD are shown; the significances were calculated using the unpaired Student’s t-test.
HAX1 deletion enhances the survival of splenic B cells upon starvation/growth-factor withdrawal
Hax1−/− mice display a severely reduced number of lymphocytes in the spleen, with an almost 70% reduction in the number of B220+ cells.29 Given the proposed anti-apoptotic function of HAX1, we investigated whether the survival of bone marrow cells, from which B cells are derived, and the survival of splenic B cells might be affected. Hence, we cultivated bone marrow cells and splenocytes from 8-week-old Hax1−/− and WT mice over a period of 4 days upon starvation, without adding stimulatory factors. We determined the rate of cell death of bone marrow cells and splenocytes by staining with the eFluor780 viability dye. During the observation period of 4 days, the number of dead Hax1−/− bone marrow cells was comparable to the number observed in the WT bone marrow cells (Figure 2a), whereas the Hax1−/− splenocytes surprisingly showed significantly enhanced survival on days 1 and 2 of cultivation (Figure 2b). The same trend could also be observed on days three and four.

Rate of cell death of Hax1−/− and WT bone marrow cells and splenocytes. (a) Bone marrow cells and (b) splenocytes from five Hax1−/− and five WT mice were cultured over a period of 4 days without further stimuli. The percentage of dead cells was determined every day using eFluor staining. The means ± SD are shown; the significances were calculated using the unpaired Student’s t-test.
HAX1 deletion affects the survival of immature and mature B cells upon BCR ligation
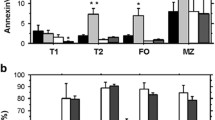
Furthermore, we investigated the apoptotic influences of HAX1 deletion on B220+ cells upon BCR ligation using anti-IgM antibody. Bone marrow cells and splenocytes from 8-week-old Hax1−/− and WT mice were cultured and then stimulated with anti-IgM antibody over a period of 4 days. B220+cells were analyzed for their eFluor450 (eF) and annexin V-FITC (A) staining. The cells were classified as follows: A−/eF− (living cells), A+/eF− (early apoptotic cells), A+/eF+ (late apoptotic cells), and A−/eF+ (dead cells).
Because only a small percentage of bone marrow cells, i.e., the immature B cells, carry the IgM-BCR, we expected a minor effect. Nevertheless, we detected a difference in the percentage of A−/eF− B220+ living cells on day 4 of our investigations, with Hax1−/− mice showing increased percentages of these cells. Additionally, we observed decreased percentages of dead Hax1−/− B220+ cells from day 2 onwards (Figure 3a). Increased percentages of A+/eF+ (late apoptotic) cells beginning on days 3 and 4 indicated a slower apoptotic rate in the Hax1−/− cells. For a better overview, a comparison of living and dead B220+ bone marrow cells is shown in Figure 3c (top panel (a, b)).

Effect of HAX1 on BCR-induced apoptosis. Bone marrow cells and splenocytes from four Hax1−/− and four WT mice were stimulated with anti-IgM and cultivated over a period of 4 days. B220/eFluor/annexin V staining was performed every day. (a) B220+ bone marrow cells and (b) B220+ splenic B cells were analyzed for annexin V and eFluor staining. (c) Comparison of the percentages of living and dead cells; (i) living and (ii) dead bone marrow cells; (iii) living and (iv) dead splenic B cells. Abbreviations: A, annexin V; eF, eFluor; A−/eF−, living cells; A+/eF−, early apoptotic cells, A+/eF+, late apoptotic, and A−/eF+, dead cells. The means ± SD are shown; the significances were calculated using the unpaired Student’s t-test.
This trend of increased survival observed in Hax1−/−-deficient immature B cells is even more pronounced in anti-IgM-stimulated B220+ splenocytes, with enhanced percentages of living cells and decreased percentages of dead cells on days 1 and 2 (Figure 3b and c, bottom panel (c, d)). On days 3 and 4, nearly all cells were dead, and the differences disappeared. The gating strategy is shown in Supplementary Figures 2 and 3. The means of Figure 3a and b are listed in Table 1 with significant differences indicated by a gray background.
HAX1 deletion affects the internalization of the IgM-BCR
We incubated splenocytes that had been treated with BCR-stimulating IgM-FITC at 37°C to investigate the internalization dynamics by measuring the mean fluorescent intensity (MFI) of FITC, which ceases to fluoresce in an acidic environment such as that of endosomes. The MFI of the Hax1−/− B cells remained at a higher level compared to WT, indicating decreased internalization dynamics (Figure 4a). This difference became significant after 30 minutes at 37°C, indicating that HAX1 plays an important role in BCR internalization. Because the FITC fluorescence seemed to be preserved to a certain extent, we sought to confirm the observed difference in internalization dynamics using pHrodo-labeled IgM antibodies as a second approach. pHrodo fluorescence increases in acidic environments; therefore, an increase in fluorescence reflects the internalization. The comparison of the IgM-pHrodo internalization dynamics confirmed the difference we observed between Hax1−/− and WT B cells using the FITC approach. Compared to WT, Hax1−/− B cells showed significantly lower fluorescence starting after 15 minutes (Figure 4b). One representative FACS comparison showing time points 0 and 90 minutes is shown in Figure 4c. Slower internalization was also confirmed by confocal microscopy (Supplementary Figure 4).

Internalization kinetics of splenic B cells. (a) The decrease in mean fluorescent intensities (MFI) upon IgM-FITC internalization at 37°C from three Hax1−/− and three WT mice is shown. The MFI of time point 0 was set at 100. The numbers of the following time points were calculated accordingly. (b) The increase in the MFI upon IgM-pHrodo internalization at 37°C from 3 Hax1−/− and 3 WT is shown. The MFI value of time point 0 was subtracted from the MFI of the following time points. (c) One representative Hax1−/− and WT IgM-pHrodo FACS comparison is shown and indicates internalization after 90 minutes. The means ± SD are shown; the significances were calculated using the unpaired Student’s t-test.
HAX1 can bind to the cytoplasmic tails of different immunoglobulin subtypes
We initially identified HAX1 as an IgE tail-interacting protein that affected IgE-BCR endocytosis upon stimulation.30 Here, we expanded upon these initial studies and tested the ability of HAX1 to bind to the cytoplasmic domains of IgA, IgG1, IgG2a, IgE, and IgM using a different approach. For that purpose, we exchanged the transmembrane and the cytoplasmic domains of the human IgE receptor with different mouse isotypes, as illustrated in Figure 5a, and stably transfected these constructs into A20 mouse B cell lymphoma cells. The MFIs of surface expression from the analyzed clones are shown in Figure 5b. The MFIs of the chimeric IgG1, IgE clone 1, and IgA receptors were equal; the MFIs of the chimeric IgG2a and IgE clone 2 receptors were slightly higher, whereas the chimeric IgM receptor showed the lowest MFI. A FACS histogram comparison is shown in Figure 5c. We precipitated HAX1 from the different stably transfected A20 cell lines with protein G Sepharose and determined its ability to bind to the chimeric human IgE receptors (Figure 5d). We detected the strongest interaction (strongest band) between HAX1 and the IgE tail (clones 1 and 2), followed by a strong interaction with the IgG1 and the IgG2a tails, whereas only a faint band was visible with the IgA tail. Because the expression level (i.e., MFI) of the IgA construct was comparable to those of the IgG1 and IgE clone 1 (Figure 5b and c), but the co-precipitation was much weaker with IgA, we concluded that the HAX1 interaction with the IgA tail is of low affinity. The MFI of the IgM tail construct was the lowest; nevertheless, a detectable interaction (faint band) that was stronger than the IgA band was visible. This somewhat weak band could be a result of the low surface expression of the chimeric IgM construct.

HAX1 interaction with the cytoplasmic domains of Ig subtypes. (a) The transmembrane (M1) and the cytoplasmic domains (M2) of human IgE were exchanged with the mouse IgM, IgG1, IgG2a, IgE, or IgA isotype, and the resulting chimeric plasmids were stably expressed in the A20 B lymphoma cell line. (b) The mean fluorescent intensities (MFI) reflecting the surface expression of the chimeric proteins are shown as bar graph, and (c) one representative FACS histogram is shown. (d) HAX1 was enriched from the stably transfected A20 cell lines using protein G Sepharose, and the binding of the chimeric human IgE/mouse tail receptors was determined using the anti-human IgE antibody. (e) The protein lysates used for co-IP were controlled for the presence of the HAX1 protein.
Identification of HAX1 as an N-terminal interaction partner of IgG1, IgG2a, and IgE by SPRA
To support the above data, we additionally performed SPRA to measure the affinity of synthetic peptides of the corresponding cytoplasmic domains (M2 regions) to the immobilized recombinant HAX1. The peptides were injected at different concentrations (1–1000 μM), and the determined KD values were calculated from the obtained curves (Table 2). In addition to the expected IgE-M2 binding (KD∼10−6), the cytoplasmic M2 tail regions of IgG1 and IgG2a showed high capacities (KD∼10−7) to bind to HAX1. Interestingly, we could not detect binding with the synthetic IgA peptide. This result supports the co-IP data and indicates a specific and relevant interaction of HAX1 with the immunoglobulin tails of IgE, IgG1, and IgG2a but not with that of IgA. To determine the possible binding motif of HAX1, we analyzed various truncated peptide versions. While the C-terminal deletions affected the binding behavior moderately, the N-terminal deletions of IgG1, IgG2a, and IgE led to a complete loss of interaction (Table 2). Based on these data, we propose that the N-terminal part KVKWI(V)F is the most likely binding motif for HAX1. To confirm this hypothesis, we fused the putative binding motif KVKWIF to the nonbinding IgA tail, which resulted in detectable binding (KD∼10−6). The short cytoplasmic tail motif of IgM, which consists of only three amino acids (KVK), is too short for meaningful SPRA; however, because KVK is part of the putative binding motif, a specific HAX1/IgM tail interaction is likely.
To define which chemical features within the cytoplasmic tail of membrane-bound IgA likely abolished HAX1binding, we compared the charge and polarity of the first 6 N-terminal amino acids of the different cytoplasmic immunoglobulin domains. This analysis revealed high similarity between IgG1, IgG2a, and IgE (Table 3). The greatest differences between the HAX1 binding tails (IgG1, IgG2a, and IgE) and the nonbinding IgA tail were visible at the first amino acid (IgG1, IgG2a, IgE, and IgM: K = polar/basic; IgA: T = polar/uncharged) and the fourth amino acid (IgG1, IgG2a, and IgE: W = nonpolar/hydrophobic; IgA: G = polar/uncharged), which could explain the altered binding. Glycine (G), the fourth amino acid within the IgA tail, primarily destroyed the nonpolar and hydrophobic properties of the putative binding motif. Because glycine (G) is a small amino acid (MW = 75) compared to tryptophan (W) (MW = 204), the size could additionally disturb HAX1 binding to the IgA tail through the reduced accessibility of this site. Taking together the co-IP data and the SPRA results, we conclude that HAX1 is not essential—or even is dispensable—for IgA-BCR functions such as internalization and/or signal transduction.
Discussion
The results of the present study demonstrate the effect of HAX1 on BCR internalization dynamics. HAX1 deficiency resulted in decelerated internalization of the BCR, which correlated with the enhanced survival of premature and mature B cells.
Deletion of HAX1 affected the total cell number of all stages of B and T cell development in the bone marrow and in the secondary lymphatic organs.8,29 HAX1 deletion also led to up to a 50% decrease in B220+ progenitor cells in the bone marrow, indicating that lymphopoiesis stays impaired throughout the developmental stages of B cells. This impairment increases with the developmental stage of the B cells; therefore, only an extremely small amount of progenitor cells reach the immature IgM-positive B cell stage, explaining the dramatic decrease in cell number in the periphery. Chao et al.8 determined similar cell numbers in the pre-pro B cell stage (B220+, CD19−), representing very early B progenitors; a significant and high reduction in the pro-B cell stage (B220+, CD43+); and the highest reduction in the pre-B cell stage (B220+, CD25+). In patients with SCN, marginal B cell abnormalities have been described, with the exception of one patient who developed acute lymphoblastic leukemia (ALL).32,33 The HAX1 mutation described in this patient affected splice variants I and II, but no further mutation was found. In ALL disease, many premature lymphocytes are produced that cannot mature into healthy blood cells. Generally, HAX1 deficiency in humans constitutes pre-leukemic conditions.32 Because of the short lifetime of Hax1−/− mice, the establishment of leukemic diseases could not be observed. We observed a slight reduction in the early B progenitor stage and a strong reduction in the late B progenitor stage in the bone marrow. Based on our current knowledge, we hypothesize that HAX1 plays a role in B cell development in mice and that B cell abnormalities in humans may depend on the site of the mutation within the HAX1 gene and on whether splice variants I and II are affected.
Several research groups have demonstrated the anti-apoptotic functions of HAX1.7,8,17,29,34 Thus, we investigated the general survival rate of total bone marrow cells and expected altered cell death rates, which we did not observe. We did not detect altered survival of Hax1−/− bone marrow cells, which confirmed our previousfinding.29 Trebinska et al.35 observed similar results for bone marrow-derived mast cells.HAX1 failed to protect against cytokine withdrawal-induced apoptosis, and these authors argued that the anti-apoptotic functions of HAX1 are cell type-specific and/or dependent on different stimuli. When we determined the survival of Hax1−/− splenocytes in the same manner, we surprisingly detected lowered death rates. This observation was even more prominent after stimulating splenic B cells with anti-IgM and could be observed from anti-IgM-stimulated bone marrow B progenitor cells, where Hax1−/− B220+ cells showed enhanced numbers of living cells and decreased numbers of dead cells in comparison to WT. In general, Hax1−/− bone marrow cells showed a decreased number of lymphocytes, with a lowered amount of B220+ cells. Only low numbers of immature B cells were released to the periphery, and we suggest that these cells survive as a result of decreased pre-BCR response.
Additionally, mature Hax1−/− B cells survived BCR-stimulation better than WT cells, which, as we hypothesized, was also due to decelerated BCR-mediated dynamics. Generally, BCR-mediated apoptosis is an important mechanism for the negative selection of self-reactive B cells.36,37 Triggering the BCR without T cell-derived survival signals induces apoptosis38,39 and prevents the survival of self-reactive B cells. Decelerated BCR internalization dynamics could facilitate the establishment of self-reactive B cells, leading to autoimmune diseases. However, this hypothesis needs to be studied in patients. In addition to our findings, the importance of HAX1 in internalization processes and cytoskeleton dynamics has been shown in previous publications.1,2,3,5,10,30,40 Thus, we are the first group to link this function to activation-induced B cell death.
Because HAX1 has been identified as an IgE tail-interacting protein30 that is involved in IgE-mediated internalization processes, we further investigated the interaction of HAX1 with different immunoglobulin tails and confirmed that HAX1 interacts with the IgG1- and IgG2a tails. HAX1 has a relatively high number of known interaction partners and is suspected to have “sticky” properties. We failed to detect binding between HAX1 and the IgA tail using both co-IP and SPRA, which suggests that the binding to IgG1, IgG2a, and IgE was specific. The short cytoplasmic domain of IgM (KVK) composes only a part of the interaction structure, which may offer an explanation for the relatively weak yet detectable interaction we found in our co-IP (Figure 5d). This weak interaction seems to be sufficient to mediate accelerated IgM-BCR internalization in the presence of HAX1. Using peptide sequence shuffling in SPRA, we identified the HAX1 interaction structure KVKWI(V)F in the binding tails of IgE, IgG1, and IgG2a. However, this sequence was completely absent in the IgA tail, whose sequence (TVRGPF) differs in charge and polarity, explaining the absent binding of the IgA tail to HAX1. As a final demonstration that KVKWI(V)F is sufficient for HAX1 binding, we restored the binding of HAX1 to the IgA tail by fusing the IgA tail to the KVKWIF motif. HAX1 was shown to be part of the pre-IL1α/IL1RII complex,13 where the putative binding site of HAX1 at pre-IL1α was determined to be KVLKKRR. The first amino acids of KVKWI(V)F and KVLKKRR are chemically similar; therefore, we suggest that the motif KVK is the important core structure for HAX1 binding at the immunoglobulin tails.
We showed the direct interaction of HAX1 with immunoglobulin tails and the effect of HAX1 on BCR-mediated internalization. However, the function of HAX1 at immunoglobulin tails remains unknown. The BCR controls the activity of α4β1 integrin (VLA-4),41 which mediates the adhesion of B progenitor cells to vascular cell adhesion molecule-1 (VCAM-1), which is present on bone marrow stromal cells.42,43,44,45 This interaction ensures B cell development in defined microenvironments. Additionally, integrins mediate trans-endothelial migration, which is required for the recirculation and homing of mature B cells46,47,48,49 and which is important for antigen-specific B cell differentiation.50,51,52,53,54 Ramsay et al.12 determined an important function of HAX1 in cell migration and clathrin-mediated endocytosis and identified HAX1 as a binding partner of ανβ6 integrin. The function of HAX1 at the tails is unknown and must be proven by further studies; however, HAX1 could play a role in transmitting the BCR signal to distinct integrins and/or in the regulation of integrin activity to coordinate the development, migration, and differentiation of B cells.
Based on the above-described results, we hypothesize that HAX1 plays a role in the development and maturation of B progenitor cells in the bone marrow. Additionally, we hypothesize that inhibited IgM-BCR response leads to decelerated apoptosis in Hax1−/− mice. Based on the presence of the KVKWI(V)F motif as the putative interaction structure at the cytoplasmic immunoglobulin domains, we suggest that HAX1 affects BCR internalization and apoptosis for the IgE-, IgG1-, and IgG2a isotypes but not for the IgA-BCR. The exact function of HAX1 at the BCR and how HAX1 affects the internalization dynamics must be evaluated in further studies. However, the intrinsic effects of HAX1 on B cells, i.e., BCR internalization and apoptosis, cannot be the sole explanation for the severe defects in B cell development observed in Hax1−/− mice in vivo, as described by Peckl-Schmid et al.29 Additionally, a defective developmental environment could contribute to the decreased cell number in the bone marrow of Hax1−/− mice; this possibility must evaluated by further experiments.
References
Suzuki Y, Demoliere C, Kitamura D, Takeshita H, Deuschle U, Watanabe T . HAX-1, a novel intracellular protein, localized on mitochondria, directly associates with HS1, a substrate of Src family tyrosine kinases. J Immunol 1997; 158: 2736–2744.
Fukuda T, Kitamura D, Taniuchi I, Maekawa Y, Benhamou LE, Sarthou P et al. Restoration of surface IgM-mediated apoptosis in an anti-IgM-resistant variant of WEHI-231 lymphoma cells by HS1, a protein-tyrosine kinase substrate. Proc Nat Acad Sci USA 1995; 92: 7302–7306.
Taniuchi I, Kitamura D, Maekawa Y, Fukuda T, Kishi H, Watanabe T . Antigen-receptor induced clonal expansion and deletion of lymphocytes are impaired in mice lacking HS1 protein, a substrate of the antigen-receptor-coupled tyrosine kinases. EMBO J 1995; 14: 3664–3678.
Yamanashi Y, Fukuda T, Nishizumi H, Inazu T, Higashi K, Kitamura D et al. Role of tyrosine phosphorylation of HS1 in B cell antigen receptor-mediated apoptosis. J Exp Med 1997; 185: 1387–1392.
Gallagher AR, Cedzich A, Gretz N, Somlo S, Witzgall R . The polycystic kidney disease protein PKD2 interacts with Hax-1, a protein associated with the actin cytoskeleton. Proc Nat Acad Sci USA 2000; 97: 4017–4022.
Ortiz DF, Moseley J, Calderon G, Swift AL, Li S, Arias IM . Identification of HAX-1 as a protein that binds bile salt export protein and regulates its abundance in the apical membrane of Madin-Darby canine kidney cells. J Biol Chem 2004; 279: 32761–32770.
Han Y, Chen YS, Liu Z, Bodyak N, Rigor D, Bisping E et al. Overexpression of HAX-1 protects cardiac myocytes from apoptosis through caspase-9 inhibition. Circul Res 2006; 99: 415–423.
Chao JR, Parganas E, Boyd K, Hong CY, Opferman JT, Ihle JN . Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature 2008; 452: 98–102.
Cilenti L, Soundarapandian MM, Kyriazis GA, Stratico V, Singh S, Gupta S et al. Regulation of HAX-1 anti-apoptotic protein by Omi/HtrA2 protease during cell death. J Biol Chem 2004; 279: 50295–50301.
Radhika V, Onesime D, Ha JH, Dhanasekaran N . Galpha13 stimulates cell migration through cortactin-interacting protein Hax-1. J Biol Chem 2004; 279: 49406–49413.
Dobreva I, Fielding A, Foster LJ, Dedhar S . Mapping the integrin-linked kinase interactome using SILAC. J Prot Res 2008; 7: 1740–1749.
Ramsay AG, Keppler MD, Jazayeri M, Thomas GJ, Parsons M, Violette S et al. HS1-associated protein X-1 regulates carcinoma cell migration and invasion via clathrin-mediated endocytosis of integrin alphavbeta6. Can Res 2007; 67: 5275–5284.
Kawaguchi Y, Nishimagi E, Tochimoto A, Kawamoto M, Katsumata Y, Soejima M et al. Intracellular IL-1alpha-binding proteins contribute to biological functions of endogenous IL-1alpha in systemic sclerosis fibroblasts. Proc Nat Acad Sci USA 2006; 103: 14501–14506.
Dufva M, Olsson M, Rymo L . Epstein-Barr virus nuclear antigen 5 interacts with HAX-1, a possible component of the B-cell receptor signalling pathway. J Gen Virol 2001; 82: 1581–1587.
Johns HL, Doceul V, Everett H, Crooke H, Charleston B, Seago J . The classical swine fever virus N-terminal protease N(pro) binds to cellular HAX-1. J Gen Virol 2010; 91: 2677–2686.
Kawaguchi Y, Nakajima K, Igarashi M, Morita T, Tanaka M, Suzuki M et al. Interaction of Epstein-Barr virus nuclear antigen leader protein (EBNA-LP) with HS1-associated protein X-1: implication of cytoplasmic function of EBNA-LP. J Virol 2000; 74: 10104–10111.
Sharp TV, Wang HW, Koumi A, Hollyman D, Endo Y, Ye H et al. K15 protein of Kaposi’s Sarcoma-associated herpesvirus is latently expressed and binds to HAX-1, a protein with antiapoptotic function. J Virol 2002; 76: 802–816.
Cassese G, Arce S, Hauser AE, Lehnert K, Moewes B, Mostarac M et al. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J Immunol 2003; 171: 1684–1690.
Cho K, Adamson L, Park JH, Zipkin R, Greenhalgh D . Identification of truncated form of mouse HAX-1s gene (HAX-1xs) and characterization of its expression in small intestine and thymus of mice after burn injury. Shock 2002; 18: 223–229.
Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schaffer AA et al. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Gen 2007; 39: 86–92.
Carlsson G, Melin M, Dahl N, Ramme KG, Nordenskjold M, Palmblad J et al. Kostmann syndrome or infantile genetic agranulocytosis, part two: understanding the underlying genetic defects in severe congenital neutropenia. Acta Paediatrica 2007; 96: 813–819.
Mirmohammadsadegh A, Tartler U, Michel G, Baer A, Walz M, Wolf R et al. HAX-1, identified by differential display reverse transcription polymerase chain reaction, is overexpressed in lesional psoriasis. J Investigat Dermatol 2003; 120: 1045–1051.
Koontz J, Kontrogianni-Konstantopoulos A . Competition through dimerization between anti-apoptotic and pro-apoptotic Hax-1 proteins. J Biol Chem 2013; doi: 10.1074/jbc.M113.536151 .
Grzybowska EA, Sarnowska E, Konopinski R, Wilczynska A, Sarnowski TJ, Siedlecki JA . Identification and expression analysis of alternative splice variants of the rat Hax-1 gene. Gene 2006; 371: 84–92.
Lees DM, Hart IR, Marshall JF . Existence of multiple isoforms of HS1-associated protein X-1 in murine and human tissues. J Mol Biol 2008; 379: 645–655.
Fadeel B, Grzybowska E . HAX-1: a multifunctional protein with emerging roles in human disease. Biochimica et Biophysica Acta 2009; 1790: 1139–1148.
Carlsson G, Fasth A . Infantile genetic agranulocytosis, morbus Kostmann: presentation of six cases from the original “Kostmann family” and a review. Acta Paediatrica 2001; 90: 757–764.
Segade F, Claudio E, Wrobel K, Ramos S, Lazo PS . Isolation of nine gene sequences induced by silica in murine macrophages. J Immunol 1995; 154: 2384–2392.
Peckl-Schmid D, Wolkerstorfer S, Konigsberger S, Achatz-Straussberger G, Feichtner S, Schwaiger E et al. HAX1 deficiency: impact on lymphopoiesis and B-cell development. Eur J Immunol 2010; 40: 3161–3172.
Oberndorfer I, Schmid D, Geisberger R, Achatz-Straussberger G, Crameri R, Lamers M et al. HS1-associated protein X-1 interacts with membrane-bound IgE: impact on receptor-mediated internalization. J Immunol 2006; 177: 1139–1145.
Batista FD, Anand S, Presani G, Efremov DG, Burrone OR . The two membrane isoforms of human IgE assemble into functionally distinct B cell antigen receptors. J Exp Med 1996; 184: 2197–2205.
Germeshausen M, Grudzien M, Zeidler C, Abdollahpour H, Yetgin S, Rezaei N et al. Novel HAX1 mutations in patients with severe congenital neutropenia reveal isoform-dependent genotype-phenotype associations. Blood 2008; 111: 4954–4957.
Yetgin S, Germeshausen M, Touw I, Koc A, Olcay L . Acute lymphoblastic leukemia in a patient with congenital neutropenia without G-CSF-R and ELA2 mutations. Leukemia 2005; 19: 1710–1711.
Shaw J, Kirshenbaum LA . HAX-1 represses postmitochondrial caspase-9 activation and cell death during hypoxia-reoxygenation. Circul Res 2006; 99: 336–338.
Trebinska A, Hogstrand K, Grandien A, Grzybowska EA, Fadeel B . Exploring the anti-apoptotic role of HAX-1 versus BCL-XL in cytokine-dependent bone marrow-derived cells from mice. FEBS Lett 2014; 588: 2921–2927.
Billian G, Mondiere P, Berard M, Bella C, Defrance T . Antigen receptor-induced apoptosis of human germinal center B cells is targeted to a centrocytic subset. Eur J Immunol 1997; 27: 405–414.
Eray M, Postila V, Eeva J, Ripatti A, Karjalainen-Lindsberg ML, Knuutila S et al. Follicular lymphoma cell lines, an in vitro model for antigenic selection and cytokine-mediated growth regulation of germinal centre B cells. Scand J Immunol 2003; 57: 545–555.
Norvell A, Mandik L, Monroe JG . Engagement of the antigen-receptor on immature murine B lymphocytes results in death by apoptosis. J Immunol 1995; 154: 4404–4413.
Tsubata T, Wu J, Honjo T . B-cell apoptosis induced by antigen receptor crosslinking is blocked by a T-cell signal through CD40. Nature 1993; 364: 645–648.
Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J et al. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature 1996; 379: 91–96.
Spaargaren M, Beuling EA, Rurup ML, Meijer HP, Klok MD, Middendorp S et al. The B cell antigen receptor controls integrin activity through Btk and PLCgamma2. J Exp Med 2003; 198: 1539–1550.
Arroyo AG, Yang JT, Rayburn H, Hynes RO . Differential requirements for alpha4 integrins during fetal and adult hematopoiesis. Cell 1996; 85: 997–1008.
Arroyo AG, Yang JT, Rayburn H, Hynes RO . Alpha4 integrins regulate the proliferation/differentiation balance of multilineage hematopoietic progenitors in vivo. Immunity 1999; 11: 555–566.
Leuker CE, Labow M, Muller W, Wagner N . Neonatally induced inactivation of the vascular cell adhesion molecule 1 gene impairs B cell localization and T cell-dependent humoral immune response. J Exp Med 2001; 193: 755–768.
Miyake K, Weissman IL, Greenberger JS, Kincade PW . Evidence for a role of the integrin VLA-4 in lympho-hemopoiesis. J Exp Med 1991; 173: 599–607.
Berlin-Rufenach C, Otto F, Mathies M, Westermann J, Owen MJ, Hamann A et al. Lymphocyte migration in lymphocyte function-associated antigen (LFA)-1-deficient mice. J Exp Med 1999; 189: 1467–1478.
Butcher EC, Picker LJ . Lymphocyte homing and homeostasis. Science 1996; 272: 60–66.
Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA . Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med 2001; 193: 741–754.
Kunkel EJ, Butcher EC . Chemokines and the tissue-specific migration of lymphocytes. Immunity 2002; 16: 1–4.
Freedman AS, Munro JM, Rice GE, Bevilacqua MP, Morimoto C, McIntyre BW et al. Adhesion of human B cells to germinal centers in vitro involves VLA-4 and INCAM-110. Science 1990; 249: 1030–1033.
Koopman G, Keehnen RM, Lindhout E, Newman W, Shimizu Y, van Seventer GA et al. Adhesion through the LFA-1 (CD11a/CD18)-ICAM-1 (CD54) and the VLA-4 (CD49d)-VCAM-1 (CD106) pathways prevents apoptosis of germinal center B cells. J Immunol 1994; 152: 3760–3767.
Koopman G, Keehnen RM, Lindhout E, Zhou DF, de Groot C, Pals ST . Germinal center B cells rescued from apoptosis by CD40 ligation or attachment to follicular dendritic cells, but not by engagement of surface immunoglobulin or adhesion receptors, become resistant to CD95-induced apoptosis. Eur J Immunol 1997; 27: 1–7.
Koopman G, Parmentier HK, Schuurman HJ, Newman W, Meijer CJ, Pals ST . Adhesion of human B cells to follicular dendritic cells involves both the lymphocyte function-associated antigen 1/intercellular adhesion molecule 1 and very late antigen 4/vascular cell adhesion molecule 1 pathways. J Exp Med 1991; 173: 1297–1304.
Kosco M H, Pflugfelder E, Gray D . Follicular dendritic cell-dependent adhesion and proliferation of B cells in vitro. J Immunol 1992; 148: 2331–2339.
Acknowledgements
We dedicate this work to the late Prof. Gernot Achatz. This work was supported by the FWF projects P-24543, P-22419, and W1213.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no financial or commercial conflict of interest.
Additional information
Supplementary Information accompanies the paper on Cellular & Molecular Immunology’s website. (http://www.nature.com/cmi).
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permissing from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Wolkerstorfer, S., Schwaiger, E., Rinnerthaler, M. et al. HAX1 deletion impairs BCR internalization and leads to delayed BCR-mediated apoptosis. Cell Mol Immunol 13, 451–461 (2016). https://doi.org/10.1038/cmi.2015.18
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2015.18
- Springer Nature Limited