
Abstract
Non-receptor tyrosine kinase activated cdc42 kinase was reported to participate in several types of cancers in mammals. It is also believed to have an anti-apoptotic function in Drosophila. Here, we report the identification of Drosophila activated cdc42 kinase as a growth promoter and a novel Hippo signaling pathway regulator. We find that activated cdc42 kinase promotes tissue growth through modulating Yorkie activity. Furthermore, we demonstrate that activated cdc42 kinase interacts with Expanded and induces tyrosine phosphorylation of Expanded on multiple sites. We propose a model that activated cdc42 kinase negatively regulates Expanded by changing its phosphorylation status to promote tissue growth. Moreover, we show that ack genetically interacts with merlin and expanded. Thus, we identify Drosophila activated cdc42 kinase as a Hippo pathway regulator.
Similar content being viewed by others

Introduction
Activated cdc42 kinase (Ack) belongs to a conserved family of non-receptor tyrosine kinase [1]. Several studies suggested that human ACK1 has important roles during the progression of several cancers. It is known that ACK1 activation is associated with poor prognosis and metastatic phenotypes in human tumors [2]. Advances in genome sequencing have revealed several ACK1 somatic mutations; however, gene amplification is a more important mechanism of ACK1 activation, which has been observed in many types of cancers (COSMIC and cBioPortal databases) [3]. During prostate cancer progression, ACK1 can phosphorylate tumor suppressor Wwox to promote Wwox’s degradation. It also phosphorylates androgen receptor that modulates the radiation resistance of cancer cells [4–6]. Despite above findings, Ack might function in a more complicated way rather than a simple participation in the development of spontaneous tumors [7]. In the genetically tractable animal organism Drosophila, Cdc42 signaling that leads to dorsal closure of the epidermis during embryogenesis has been shown to regulate Ack protein level [8]. Moreover, Ack has been reported to regulate Dock localization in male germ cells during spermatogenesis [9]. Notwithstanding these findings, the function of Drosophila Ack during tumorigenesis remains elusive. Not until recently, a study reported that Ack has an anti-apoptotic function, and it genetically interacts with Yorkie (Yki) to regulate cell proliferation and apoptosis [10].
The Hippo pathway controls organ size by orchestrating proliferation and apoptosis in diverse cells and tissues [11–13]. Most, if not all, of the Hippo pathway’s functions are exhibited through its downstream transcriptional coactivator Yki [14]. The activity of Yki is closely related with its localization. The localization of Yki can be fine-tuned by the Hippo pathway either through modulation of its phosphorylation status or through physical interactions [15, 16]. Central to the Hippo signaling pathway is a kinase cascade, the best demonstration of phosphorylation-dependent Yki regulation. The STE20 family serine/threonine kinase Hippo (Hpo) [17–21], activated by autophosphorylation and dimerization [22, 23], phosphorylates and activates the NDR family kinase Warts (Wts) [22, 24, 25], leading to Wts-mediated Yki phosphorylation. The adaptor proteins Salvador (Sav) [26, 27] and Mob as tumor suppressor (Mats) [28] facilitate the activities of Hpo and Wts. Phosphorylated Yki is captured by 14-3-3 protein and retained in cytosol; thus, its activity is inhibited [24, 25, 29, 30]. Expanded (Ex) is an important upstream component in the Hippo pathway. Ex, together with Merlin (Mer) and Kibra, transduces signals to the Hpo-Wts kinase cascade through multiple protein interactions [31]. In addition, Ex interacts with Yki through WW domain and PPxY motif to restrict Yki in cytosol [32, 33]. WW domain and PPXY/PY motif interactions participate in the regulation of Yki [34, 35]. For instance, WBP2 interacts with Yki’s WW domains through its PY motifs to promote Yki’s activity [36, 37].
In this study, we report the identification of Drosophila Ack as a growth promoter and a novel Hippo signaling pathway regulator. We show that Ack promotes tissue growth through enhancing Yki activity. Furthermore, we demonstrate that Ack interacts with Ex and negatively regulates Ex activity in a kinase-dependent manner by changing its phosphorylation status on multiple tyrosine residues. In addition, we determine that ack genetically interacts with merlin and ex. Taken together, our findings support that Drosophila Ack is a Hippo pathway regulator and promotes tissue growth via suppressing Ex-mediated Yki regulation.
Results
Ack overexpression upregulates Yki activity to promote growth
To identify novel regulators of the Hippo pathway, we performed mass spectrometry (MS) analysis using GST fused Yki tandem WW domains (refer to as GST-WW) as bait to identify its binding partners. As expected, some known Yki binding partners such as Wts, WWBP, Mop and Ack were found in the result (Supplementary Figure S1). An interaction of Ack and Yki was reported recently [10, 38], which was confirmed using co-immunoprecipitation (co-IP) experiment (Figure 1a).

Ack interacts with Yki and promotes tissue growth. (a) Ack interacts with Yki. S2 cells expressing Myc-Yki or blank vector were immunoprecipitated and probed with indicated antibodies. Experiment was repeated three times; and representative blots are shown. (b) A schematic of Hippo signaling pathway. (c) Ack promotes the transcriptional activity of the Yki–Sd complex in a kinase-dependent manner. S2 cells were transfected with the indicated constructs; and the cell lysates were subjected to dual luciferase assay. Quantitative data are expressed as mean±s.d. (triplicate wells). ***P<0.001. (d) Ack overexpression promotes tissue growth in vivo. Wild-type male wing and wings expressing MS1096-Gal4 with UAS-V5-Ack or UAS-V5-Ack-KD were shown. Ack and Ack-KD were inserted at the same attp site to ensure equal expression level. Overexpression of Ack induces wing overgrowth compared with control, whereas overexpression of Ack-KD suppresses wing growth. Quantification of wing area of each group is expressed as mean±s.d. (N=10). ***P<0.001. Scale bar is 500 μm. (e–j) Ack promotes eye growth and enhances Yki activity. Wild-type adult eye (e) or eyes expressing the indicated constructs (f–j) were shown. Dashed line indicated normal eye size. Scale bar is 300 μm. Experiments were repeated; and representative eyes are shown. (k) Yki RNAi suppresses Ack-induced wing growth. Wild-type male wings and wings expressing indicated constructs were shown. Quantification of wing area of each group is expressed as mean±s.d. (N=10). ***P<0.001. n.s., not significant. Scale bar is 500 μm.
Yki is the downstream Hippo pathway effector, and its activity is tightly regulated by upstream Hippo pathway components via inducing Yki Ser168 phosphorylation or physical interaction (Figure 1b) [39]. We then sought to determine whether Ack affects Yki activity. A dual luciferase assay was established to monitor the transcriptional activity of Scalloped (Sd)–Yki complex [40]. Coexpression of Yki and Sd in S2 cells activated the reporter gene (3×Sd2-Luc), which was promoted by Ack expression (Figure 1c). Coexpression of a kinase-dead form of Ack (Ack-KD, which contains a lysine to arginine substitution at amino acid 156) [8] could not promote Sd–Yki complex activity as wild-type Ack (Figure 1c), indicating that the kinase activity of Ack is essential for its function on promoting Sd–Yki transcriptional activity in vitro.
To verify whether Ack promotes Yki activity in vivo, we generated Ack transgenic flies using Gal4/UAS system [41]. Overexpression of Ack (but not Ack-KD) under the control of MS1096-Gal4 or GMR-Gal4 promoted growth in wings and eyes, respectively (Figure 1d–g). In addition, coexpression of Ack (but not Ack-KD) further promoted Yki-induced eye overgrowth (Figure 1h–j). Knockdown of Yki by RNAi suppressed Ack overexpression-induced overgrowth phenotype, generating phenotypes similar to that induced by depletion of Yki (Figure 1k). On the basis of these results, we speculated that Ack promoted tissue overgrowth through upregulating Yki’s activity.
Ack overexpression upregulates the Hippo pathway targets
To confirm our speculation, Ack expression was driven in posterior (P) compartment of wing discs using hedgehog-Gal4 (hh-Gal4); and the expression level of Yki target genes was evaluated. ex is one of the Yki target genes and encodes an upstream Hippo pathway component that suppresses Yki’s function [42]. Its transcription level can be reflected by Ex-LacZ (Ex-Z). Another well-known Yki target gene is diap1, whose transcriptional level can be monitored using a reporter gene diap1-GFP4.3 [40]. Our results showed that overexpression of Ack using hh-Gal4 enhanced Ex-Z signal and induced a significant upregulation of diap1-GFP signal in the P compartment (Figure 2a–b′ and d–e′). Furthermore, microRNA bantam, a previously reported Yki target, has been shown to control cell proliferation and apoptosis [43, 44]. As expected, the expression of its reporter, Bantam sensor GFP, that reversely correlates with the microRNA bantam expression, was downregulated in the P compartment when Ack was overexpressed using hh-Gal4 (Figure 2g–h′), indicating an elevation of bantam transcription. In addition, the P compartment of wing discs was enlarged in size when Yki target gene expression was elevated by Ack overexpression (Figure 2b–b′, e–e′ and h–h′). In contrast, overexpression of Ack-KD could not promote Yki target gene expression (Figure 2c–c′, f–f′ and i–i′). Together, these findings indicate that the regulation of Yki activity by Ack is kinase dependent in vivo.

Ack overexpression upregulates Hippo pathway targets. (a–i′) Ack overexpression upregulates Yki target genes. Wild-type wing discs (a, a′, d, d′ and g, g′) or wing discs expressing UAS-V5-Ack (b, b′, e, e′ and h, h′) or UAS-V5-Ack-KD (c, c′, f, f′ and i, i′) by hh-Gal4 were immunostained to show the expression of Ci and Hippo pathway markers, including Ex-lacZ (a–c′), diap1-GFP4.3 (d–f′) and bantam sensor (BS) GFP. Note that, in the P compartment, Ack overexpression upregulates the expression of Ex-lacZ and diap1-GFP and downregulates BS GFP, while Ack-KD overexpression cannot (g–i′). Arrows indicated the P compartment. White dashed lines indicated the A-P compartment boundary based on Ci staining. Scale bar is 100 μm. (j–k′′′) Drosophila wing discs expressing UAS-V5-Ack (j–j′′′) or UAS-V5-Ack-KD (k–k′′′) by hh-Gal4 were immunostained with Yki and V5 antibody. Compared with the A compartment, in the P compartment, Ack overexpression promotes Yki nuclear localization whereas Ack-KD overexpression cannot. l–l′′′ and m–m′′′ are magnified pictures of the dashed line indicated P compartment area in j–j′′′ and k–k′′′, respectively. DAPI was stained to show nuclei. White dashed line indicated the A-P compartment boundary. Scale bar is 20 μm.
Notably, cytoplasmic-nuclear translocation of endogenous Yki was affected when Ack (but not Ack-KD) was overexpressed (Figure 2j–k′′′). Yki was increasingly accumulated in the nucleus upon Ack overexpression (but not Ack-KD expression) (Figure 2l–m′′′), and this increase is correlated with the upregulation of its activity. Collectively, these results indicate that Ack overexpression enhances Yki activity in a kinase activity-dependent manner in vitro and in vivo.
Ack modulates Yki activity via regulating Ex activity
To shed some light on possible mechanisms by which Ack promotes growth, we performed western blot and co-IP analyses. As we noticed that Ack interacted with Yki in S2 cells and overexpression of Ack induced Yki nuclear translocation in vivo (Figures 1a and 2j–j′′′), we speculated that Ack promotes Yki’s activity directly. However, in S2 cells, Ack overexpression could not induce tyrosine phosphorylation of Yki (described later in Figure 3b). Ack expression slightly suppressed Hippo signaling-induced upregulation of Yki Ser168 phosphorylation (pS168) (Figure 3a), implying that Ack-induced Yki pS168 change might be indirect and through upstream components of Hippo signaling.

Ack interacts with and phosphorylates Ex. (a) Ack overexpression suppresses the upregulation of Yki S168 phosphorylation induced by Hpo/Sav/Wts. S2 cells expressing the indicated constructs were probed with the indicated antibody. Experiment was repeated three times, and representative blots are shown. (b) Ex interacts with Ack, and is phosphorylated by Ack. S2 cells expressing the indicated constructs were immunoprecipitated and probed with the indicated antibodies. Experiment was repeated three times, and representative blots are shown. (c) Ack inhibits Ex-induced Yki S168 phosphorylation. S2 cells expressing indicated constructs were probed with the indicated antibody. Note that Ex-induced upregulation of pS168 is inhibited by Ack but not by Ack-KD. Experiment was repeated three times, and representative blots are shown. (d) Ex interacts with Ack, while Mer does not interact with Ack. S2 cells expressing the indicated constructs were immunoprecipitated and probed with the indicated antibodies. Experiment was repeated three times, and representative blots are shown.
To verify our hypothesis, we checked whether Ack interacts with other components of Hippo signaling and analyzed whether Ack affects their activity in S2 cells. By co-IP experiments, we confirmed that Ack has no interaction with Hpo or Wts. We further tested upstream components such as Mer and Ex. We found that Ack and Ex interacted with each other in both directions (Figure 3b, lane 3 and 8). In addition, overexpressed Ex can form protein complex with endogenous Ack (Supplementary Figure S2A). As Yki interacted with both Ex and Ack, we asked whether Yki mediated Ack–Ex interaction. By co-IP experiment, we found that knockdown of Yki expression by double-strand RNA (dsRNA) treatment did not affect Ack–Ex interaction and Ack-induced Ex phosphorylation (Supplementary Figure S2B). Furthermore, overexpression of Ex induced Yki S168 phosphorylation significantly, while such increase was partially suppressed by Ack in a kinase-dependent manner (Figure 3c). In addition, we did not observe any association between Ack and Mer in S2 cells (Figure 3d), supporting that Ack modulates Yki’s activity by regulating Ex function.
To explore how Ack regulates Ex function, we tested whether Ack modulates Ex phosphorylation status as its function on promoting Yki activity was kinase activity dependent. A phosphor-tyrosine (p-Y) antibody was used to detect Ex phosphorylation status when Ack was coexpressed. We confirmed the specificity of the antibody by checking the autophosphorylation status of Ack (unpublished observations). As shown in Figure 3b, Ack induced tyrosine phosphorylation of Ex specifically (compare lane 1 and 4 with lane 3). Notably, Ack did not affect Yki tyrosine phosphorylation status (Figure 3b, lane 5), further suggesting that regulation of Yki by Ack is indirect.
Ack phosphorylates Ex on multiple tyrosine residues
To identify Ex tyrosine phosphorylation sites induced by Ack, we overexpressed Myc-tagged Ex with or without Ack in S2 cells. Ex-Myc protein was then immunoprecipitated and prepared for MS analysis. Five potential tyrosine phosphorylation sites of Ex were identified, including Y227, Y423, Y679, Y766 and Y1103 (Supplementary Figure S3). To verify whether these sites are actual Ex tyrosine phosphorylation sites by Ack, we first generated an Ex mutant that contains tyrosine to alanine substitution of all potential tyrosine phosphorylation sites (Ex-5YA) and checked Ex tyrosine phosphorylation status using the p-Y antibody. We found that Ex-5YA had a much lower tyrosine phosphorylation level than wild-type Ex when cotransfected with Ack (Figure 4a). As Ex-5YA did not lose tyrosine phosphorylation signal completely, and the decrement of p-Y signal is not because of the weaker interaction between Ack and Ex-5YA, we speculate that additional tyrosine phosphorylation site(s) might exist. To identify whether these five potential tyrosine phosphorylation sites are important for Ex’s function, we coexpressed Yki with Ex or Ex-5YA in S2 cells to detect Yki pS168 change. As predicted, Ex-5YA exhibited a stronger effect on Yki activity as it almost abolished Ack-induced Ex inhibition. Ex-5YA also induced a higher Yki pS168 level than wild-type Ex (Figure 4b, compare lane 5 with lane 2). However, we noticed that Ex-5YA activity was still responsive to Ack coexpression, which may be due to the fact that the other unidentified tyrosine phosphorylation sites exist for Ack.

Ack suppresses Ex activity via tyrosine phosphorylation. (a) Ex-5YA mutation blocks majority of the phosphorylation signal induced by Ack. S2 cells expressing the indicated constructs were immunoprecipitated and probed with the indicated antibodies. Experiment was repeated three times and representative blots are shown. (b) 5YA mutation partially blocks Ack-induced suppression of Ex activity. S2 cells expressing the indicated constructs were probed with the indicated antibody. Yki total protein level was used as a control to examine the change of Yki phosphorylation level. Quantification of relative Yki S168 phosphorylation level is expressed as mean±s.d. (N=3) **P<0.01. *P<0.05. n.s., not significant. (c) Ex-5YA induces stronger growth defects than wild-type Ex and does not response to Ack overexpression. Drosophila female adult wings expressing the indicated constructs were shown. Quantification of wing area of each group is expressed as mean±s.d. (N=10). ***P<0.001. *P<0.05. Scale bar is 500 μm.
To verify these findings in vivo, we generated Ex and Ex-5YA transgenic flies using the phiC31 integration system to ensure that all of the transgenes were expressed from the same chromosomal location (attp at 25C6) so that position effects on gene expression were eliminated [45]. Expression of Ex-5YA by MS1096-Gal4 on wing discs showed stronger activity compared with the wild-type Ex in regards to the reduction in wing size (Figure 4c). Whereas Ack coexpression partially suppressed Ex-induced inhibition of wing growth, it barely had any influence on wing growth defect induced by Ex-5YA expression (Figure 4c). We believe that this phenomenon is a result from disruption of Ack regulation caused by tyrosine to alanine mutations of Ex. These findings are consistent with our observations in cultured cells and show that Ack modulates the tyrosine phosphorylation status of Ex to regulate both Yki’s activity and tissue growth.
Y679 and Y766 are bona fide phosphorylation sites of Ack-induced phosphorylation
To further explore whether a change of any of these phosphorylation sites has a critical role for Ex during growth, we generated transgenic flies expressing Ex mutants with individual tyrosine to alanine substitution. We found that flies carrying several Ex single tyrosine mutation (Y423, Y679, Y766 and Y1103) exhibited stronger growth inhibition effect than those carrying wild-type Ex. We reasoned that, by abolishing Ack-induced Ex phosphorylation, Ex’s function on growth inhibition is enhanced. However, all of the Ex mutants bearing single mutation were partially suppressed by Ack overexpression in varying degrees (Figure 5a). Notably, Ex mutants, Y679A and Y766A, exhibited stronger repressive activity on wing growth compared with the wild-type Ex and other single mutations (Figure 5a). Thus, we predicted that Y679 and Y766 double mutants would have stronger effects on Ex’s activity. To test this, we generated Ex attp flies containing a combination of Y679A and Y766A mutations (Ex-2YA). We found that Ex-2YA exhibited more severe growth defects than any of single mutants and in a level similar to Ex-5YA (Figure 5b). Consistently, in S2 cells, Ex-2YA overexpression-induced increase of Yki S168 phosphorylation was barely affected by Ack coexpression, whereas Ex-Y679A- and Ex-Y766A-induced Yki S168 phosphorylation is still responsive to Ack coexpression (Figure 5c). These results suggest that Y679 and Y766 sites have major roles in the regulation of Ack-mediated Ex’s function. To confirm Y679 and Y766 are bona fide phosphorylation sites of Ack-induced phosphorylation, we generated phospho-specific antibodies against Y679 and Y766. We found that these antibodies specifically recognize phosphorylated wild-type Ex by Ack coexpression in S2 cells, and the signal was abolished by coexpression of Ack-KD or corresponding Ex-Y679A and Ex-Y766A mutations (Figure 5d). Of note, although Y679 and Y766 are the critical sites that respond to Ack-induced regulation, the tyrosine phosphorylation level of Ex-2YA was still higher than Ex-5YA (Figure 5e), suggesting that the rest three sites are still phosphorylated by Ack. Taken these results together, we conclude that Ack suppresses Ex, regarding its functions on growth restriction, through regulating multiple sites of phosphorylation, especially Ex Y679 and Y766 residues.

Ack phosphorylates Ex on multiple tyrosine sites to suppress Ex activity. (a) Ack suppressed the activity of wild-type Ex as well as Ex single site mutants. Quantification of wing area of each group is expressed as mean±s.d. (N=10). ***P<0.001. **P<0.01. *P<0.05. n.s., not significant. (b) Mutations of potential Ack-induced tyrosine phosphorylation sites affect Ex activity on inhibiting wing growth. Quantification of wing area of each group is expressed as mean±s.d. (N=10). **P<0.01. n.s. no significant. Scale bar is 500 μm. (c) S2 cells expressing the indicated constructs were probed with the indicated antibody. Yki total protein level was used as a control to examine the change of Yki phosphorylation level. Quantification of relative Yki S168 phosphorylation level is expressed as mean±s.d. (N=3). *P<0.05. n.s., not significant. (d) Ack induces Ex phosphorylation on Y679 and Y766. S2 cells expressing the indicated constructs were immunoprecipitated with Myc antibody and probed with the indicated antibody. (e) Ack-induced tyrosine phosphorylation level of Ex-2YA was lower than Ex wild type but higher than Ex-5YA. S2 cells expressing the indicated constructs were immunoprecipitated and probed with the indicated antibodies. Ex total protein level was used as a control to examine the change of Ex phosphorylation level. Quantification of relative p-Y/Myc is expressed as mean±s.d. (N=3).
Genetic interaction between Ack and Ex/Mer
To verify the genetic epistatic relationship of ack and ex in vivo, we used ack mutant flies generated by excision of a P-element KG00869 [46]. Surprisingly, although two Ack RNAi lines targeting different sites strongly suppress tissue growth (unpublished observations), homozygous ack mutant flies are viable without obvious growth defect. In addition, expression of Hippo pathway target genes did not change in ack null imaginal discs. Such a striking discrepancy implies that there may be potential problems for the RNAi lines. The viable property of ack mutant flies made it a suitable tool for examining epistatic relationships with other components in double mutant analysis. In our analysis, ex mutant mosaic induced massive overgrowth not only in eyes but also at the base of antenna (Figure 6, compare 6c with 6a), whereas mer mutant caused mild phenotype. Double mutant of ack and ex changed neither growth phenotype (Figure 6, compare 6d with 6b and c) nor fj-lacZ elevation in both wing and eye discs (Figure 6e–f′ and Supplementary Figure S4A–B′′) induced by loss of ex [47]. On the contrary, mer mutant-induced Ex upregulation [42, 48] was suppressed by ack mutant (Figure 6g–h′′). We also found that in S2 cells knockdown of Ack expression by dsRNA treatment promoted Mer expression-induced Yki pS168 level (Supplementary Figure S4C). It is interesting that loss of ack suppresses mer but not ex phenotype. Consistently, several previous reports also pointed out there are functional differences between Ex and Mer in Hippo signaling [49–51].

Genetic interaction between Ack and Ex/Mer. (a–d) Homozygous loss of ack cannot suppress ex mutant-induced tissue overgrowth. Scan electronic micrographs of adult fly head of the indicated genotype. Arrows indicated overgrowth in ex mutant. The genotypes were the following: eyflp; exe1 FRT40A/ubi-GFP FRT40A (c), eyflp; exe1 FRT40A/ubi-GFP FRT40A; Ack10b/Ack10b (d). Scale bar is 100 μm. (e–f′) Wing discs containing exe1 clones or exe1;Ack10b/10b clones were immunostained to show fj-lacZ (fj-Z) and GFP expression. exe1 clones were marked by the loss of GFP signal. Arrows indicated upregulation of fj-Z expression in exe1 clones. Note that homozygous loss of ack cannot suppress ex mutant-induced upregulation of fj-Z. The genotypes were the following: hsflp; exe1 fj-Z FRT40A/ubi-GFP FRT40A (e–e′), hsflp; exe1 fj-Z FRT40A/ubi-GFP FRT40A; Ack10b/Ack10b (f–f’). Scale bar is 50 μm. (g–h′′) Eye discs containing mer4 clones or mer4; Ack10b/10b clones were immunostained to show Ex and Mer expression. Note that homozygous loss of ack suppresses mer mutant-induced upregulation of Ex. Arrows indicated the clone boundary. The genotypes were the following: eyflp, mer4 FRT19A/FRT19A (g–g′′), eyflp, mer4 FRT19A/FRT19A;;Ack10b/Ack10b (h–h′′). Scale bar is 50 μm.
Ack may disrupt Ex–Yki binding to promote Yki activity
We have studied the regulation of Ex by Ack extensively but the significance of Ack–Yki interaction is not shown. It has been reported that Ex interacts with Yki directly through WW–PPxY association to inhibit Yki activity [32, 33], which bypasses the Hippo pathway core kinase cascade (i.e., a phosphorylation-independent Yki regulation). We then asked whether Ack-induced tissue growth is partially resulted from a modulation of Ex–Yki association. We found that the interaction between Ex and Yki was weakened by Ack partially in a kinase activity-dependent manner (Supplementary Figure S5A, compare lane 3 and 4 with lane 2). To verify this regulation in vivo, we expressed Ack and Ack-KD in hpo clones generated by Mosaic Analysis with A Repressible Cell Marker [52, 53] system. It blocks the regulation path via the Hippo pathway core kinase cascade. As reported, induction of hpo mutant clones leads to an overgrowth phenotype (Supplementary Figure S5B). Overexpression of Ack (but not Ack-KD) in hpo clones further promotes the overgrowth phenotype compared with hpo clones alone (Supplementary Figure S5, compare S5C and D with S5B). Yki-S168A mutation also blocked the kinase-dependent function of the Hippo pathway. We found that Ack (but not Ack-KD) further promoted the overgrowth phenotype induced by Yki-S168A (Supplementary Figure S5E–G). In toto, we speculated that Ack may disrupt Ex–Yki association, thus weaken Ex-induced retention of Yki in cytosol to modulate growth as well.
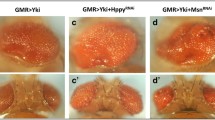
Taken together, our data lead to a model in which Ack promotes growth via suppressing Ex. To further determine whether this is true in vivo, we overexpressed Ack in ex, mer or kibra mutants using the Mosaic Analysis with A Repressible Cell Marker system. In control group, flies overexpressing Ack promoted adult eye overgrowth compared with wild-type flies (Figure 7a and b). Ack overexpression in ex mutant clones failed to cause dramatic change (Figure 7c and d), while Ack further promoted growth in the absence of mer or kibra (Figure 7e–h). These results suggest that Ack modulates growth mainly through negatively regulating Ex.

Ack promotes tissue growth mainly through regulating Ex. (a–h) Scan electronic micrograph of the adult eye of the indicated genotype was shown. Note that Ack overexpression in both mer (e and f) and kibra (g and h) mutant eye induces further growth effect but in ex (c and d) mutant eye did not cause dramatic change. The genotypes were the following: eyflp, ubi-Gal4, UAS-GFP; exe1 FRT40A/Gal80 FRT40A (c), eyflp, ubi-Gal4, UAS-GFP; exe1 FRT40A/Gal80 FRT40A; UAS-V5-Ack (d), eyflp, mer4 FRT19A/Gal80 FRT19A; ubi-Gal4, UAS-GFP (e), eyflp, mer4 FRT19A/Gal80 FRT19A; ubi-Gal4, UAS-GFP; UAS-V5-Ack (f), eyflp, ubi-Gal4, UAS-GFP; FRT82B kibra4/FRT82B Gal80 (g) and eyflp, ubi-Gal4, UAS-GFP; UAS-V5-Ack; FRT82B kibra4/FRT82B Gal80 (h). Scale bar is 100 μm.
Discussion
In this study, we report the identification of non-receptor tyrosine kinase Ack as a novel regulator of the Hippo pathway. Using Yki WW domain as a bait, we found that Ack interacts with Yki from our MS analysis, consistent with results from two earlier studies [10, 38]. We confirmed the interaction between Ack and Yki, and found that Ack modulates Yki activity (Figures 1 and 2) in a kinase activity-dependent manner to promote tissue growth.
Schoenherr et al. [10] reported an anti-apoptotic function of Ack, which is regulated by Yki. They proposed that Ack cannot promote Yki activity and nuclear localization, as they found that overexpression of Ack by GMR-Gal4 failed to promote Ex-Z expression. Consistently, we also found no change of Ex-Z in this condition. However, we noticed that the protein levels of other two Yki targets, Diap1 and cyclin E, were increased in Ack-overexpressing eye discs (unpublished observations). Taken together with the adult phenotype, we concluded that Ack modulates Yki activity in eye discs. The anti-apoptotic function of Ack may be partially explained by increased Yki activity since increased Diap1 protein level may inhibit cell death [54, 55]. The reason why Ex-Z shows no change may be due to varied sensitivities of the reporter in different tissues.
Our results imply that Yki might not be a substrate for Ack tyrosine phosphorylation (Figure 3b, lane 5). To elucidate the mechanism of how Ack promotes growth, we examined Hippo pathway upstream components and found that Ack interacts with Ex and induces Ex tyrosine phosphorylation, indicating that Ack might function through Ex. We further confirmed that Ack mediates growth via Ex phosphorylation on Tyr679 and Tyr766 (Figures 4 and 5). Interestingly, Ex overexpression induces Yki Ser168 phosphorylation dramatically. We suppose Ex phosphorylation induced by Ack reduces its ability to efficiently promote the Hpo/Wts core kinase cascade, thereby Yki is less phosphorylated. However, Yki may not be the only target that mediates Ack activity during tissue growth. Ack may have ex-dependent but Hippo pathway-independent function. One reason is that coexpression of Ack with YAP-S94A/S127A mutation form still could promote cell proliferation [10]. Another one is there is Hippo pathway-independent function of Ex, such as regulating receptor endocytosis [56]. Based upon our findings, it is intriguing to speculate that Ack regulates Hippo signaling in a similar way in mammals. Like most of other Hippo pathway components, Ack is highly conserved from Drosophila to human [1]. So far, no comparable Ex homologs have been found in mammals. Willin/FRMD6 was considered as the human Ex ortholog and its expression has been shown to activate Hippo signaling [57], while it lacks the C-terminus, where the most important phosphorylated tyrosine residues Y679 and Y766 are located. Therefore, it will be interesting to determine whether Ack inhibits Willin/FRMD6 in mammals.
It was previously reported that ACK1 transduces extracellular signals to cytosolic and nuclear effectors to promote cell survival and growth, and several somatic ACK1 mutations discovered in tumors could increase the ability of tumor cells to proliferate and migrate [58]. Whether Hippo signaling is affected in these tumor cells is an interesting question to explore. Recently, several small molecules that act as potential Ack inhibitors have been reported [59–63], and they may lead to effective drugs targeting Ack in the future [64, 65]. Our study thus provides the basis of the mechanism underlying these therapeutic approaches for curbing tumorigenesis.
Materials and Methods
Clones, mutants, Drosophila stocks and genetics
The full-length Ack DNA fragment (3 222 bp) was amplified from a BDGP DGC clone (clone ID: GH10777) and was inserted into pUAST-V5 or pUAST-Flag vector. Ack and Ex mutants that contain point mutation were generated by PCR-based site-directed mutagenesis.
Ex transgenic flies were generated by site-specific integration into the fly genome at 25C6 attp locus. ack null allele (Ack10b) was a kind gift from Nicholas Harden, Simon Fraser University. kibra null allele (kibra4) was a kind gift from Hugo Stocker, Institute of Molecular Systems Biology. fj-lacZ was a kind gift from Kenneth D. Irvine, The State University of New Jersey. Other RNAi or null alleles used in this study were as following: UAS-AckRNAi (NIG 14992R1 and VDRC 39857), UAS-YkiRNAi (VDRC 40497), exe1 [66] and mer4 [67]. MS1096-Gal4, GMR-Gal4, hh-Gal4, hpoBF33, Ex-lacZ, diap1-GFP4.3 and bantam sensor mic32-GFP were described previously [40, 68]. Flies and crosses were cultured at 25 °C unless otherwise indicated.
Protein pull-down and MS screen
For pull-down experiment, Drosophila S2 cells were cultured for 3 days (10 dishes) and then collected and lysed in immunoprecipitation (IP) buffer (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1% NP-40, 10% Glycerol, 1.5 mm EDTA, 10 mm NaF, 1 mm Na3VO4) with protease inhibitor cocktail (Sigma, St Louis, MO, USA). Cell lysates were pre-cleared using Glutathione sepharose (GE Healthcare, Little chalfont, UK) and then incubated with 200 μg immobilized GST-WW protein, which was expressed in BL21 E. coli and was purified with Glutathione Sepharose. GST protein was used as a negative control. Proteins in pull-down samples were separated by SDS-PAGE, and were stained using Colloidal Blue staining (Invitrogen, Carlsbad, CA, USA). Compared with control sample, specific bands were selected for liquid chromatography coupled with tandem MS analysis in Protein Centre, SIBCB.
Cell culture, transfection and co-IP
S2 cells were cultured in Drosophila Schneider’s Medium (Invitrogen) with 10% fetal bovine serum, 100 U ml−1 of penicillin and 100 mg ml−1 of Streptomycin. Plasmid transfection was carried out using LipofectAMINE (Invitrogen) according to the manufacturer’s instructions. The constructs transfected in S2 cells were all pUAST expression vectors unless otherwise indicated. A construct of ubiquitin-Gal4 was cotransfected with pUAST expression vectors for all transfection experiments.
For co-IP experiment, S2 cells expressing the indicated constructs were collected and lysed in IP buffer (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1% NP-40, 10% Glycerol, 1.5 mm EDTA, 10 mm NaF, 1 mm Na3VO4) with protease inhibitor cocktail (Sigma). Cell lysates were then divided into two parts, and 2 μg antibody was used to do co-IP in each part. Proteins in IP samples were then washed with IP buffer and subjected for western blot analysis.
Western blot, immunostaining and luciferase reporter assay
Western blot analysis and immunostaining were performed as previously described [40]. Primary antibodies used in western blot and immunostaining were as follows: mouse anti-Myc (1:5 000, Sigma), mouse anti-Flag (1:5 000, Sigma), mouse anti-V5 (1:5 000, Invitrogen), mouse anti-HA (1:5 000, Sigma), mouse anti-phospho tyrosine (1:1 000, Cell Signaling Technology, Danvers, MA, USA), rabbit anti-Ack (1:500), rat anti-Ci (1:500, Developmental Studies Hybridoma Bank, DSHB), rabbit anti-Ex (a gift from Allen Lauhgon, University of Wisconsin), guinea pig anti-Mer (a gift from Richard G. Fehon, University of Chicago) and rabbit anti-lacZ (1:1 000, Invitrogen). Rabbit anti-Ack (556-1073AA) was generated by Abclonal (Wuhan, China). Rabbit anti-phospho Ex (Y679 and Y766) was generated by Abclonal, with peptide as antigen (Y679, ESEKSSHpYGMFQPQK; Y766, SLHSDCDpYVTLPLGD). Rabbit anti-phospho Yki (S168) was generated by Abgent (Suzhou, China) with antigen peptide HHSRARpSSPAC.
For Luciferase reporter assay, the 3xSd2-Luc reporter was described previously [40]. The Dual-Glo™ luciferase assay system (Promega, Madison, WI, USA) was used according to the manufacturer's instructions.
RNA interference in Drosophila S2 cells
dsRNA was designed and synthesized according to standard protocol. To perform knockdown experiment, S2 cells were diluted into 1×106 cells per ml with serum-free medium for 1 h starvation with 15 μg ml−1 indicated dsRNA.
Mapping Ex phosphorylation sites
To identify Ack-induced Ex phosphorylation sites, S2 cells were transfected with Ex-Myc or Ex-Myc and V5-Ack (10 dishes for each group). Cells were collected 48 h after transfection and were lysed in IP buffer. Ex-Myc protein was immunoprecipitated by 50 μg immobilized mouse anti-Myc antibody (Sigma) and subjected to MS analysis at the Protein Center, SIBCB. The candidate sites were identified by the increased phosphorylation abundance in the Ex-Myc and V5-Ack transfected cells versus Ex-Myc transfected cells.
Statistical analysis
All of the data in this study were expressed as the mean±s.d. and were analyzed using the Student’s t-test. The results were considered as statistically significant if P<0.05.
Microscopy and data analysis
Fluorescent microscopy was performed on a Leica LAS SP5 confocal microscope; confocal images were obtained using the Leica AF Lite system (Leica Microsystems, Wetzlar, Germany). Images were processed in Photoshop CS.
References
Prieto-Echague V, Miller WT . Regulation of ack-family nonreceptor tyrosine kinases. J Signal Transduct 2011; 2011: 742372.
van der Horst EH, Degenhardt YY, Strelow et al. Metastatic properties and genomic amplification of the tyrosine kinase gene ACK1. Proc Natl Acad Sci USA 2005; 102: 15901–15906.
Mahajan K, Mahajan NP . ACK1/TNK2 tyrosine kinase: molecular signaling and evolving role in cancers. Oncogene 2015; 34: 4162–4167.
Mahajan NP, Whang YE, Mohler JL, Earp HS . Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res 2005; 65: 10514–10523.
Mahajan NP, Liu Y, Majumder S et al. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc Natl Acad Sci USA 2007; 104: 8438–8443.
Mahajan K, Coppola D, Rawal B et al. Ack1-mediated androgen receptor phosphorylation modulates radiation resistance in castration-resistant prostate cancer. J Biol Chem 2012; 287: 22112–22122.
Hoare S, Hoare K, Reinhard MK, Lee YJ, Oh SP, May WS Jr . Tnk1/Kos1 knockout mice develop spontaneous tumors. Cancer Res 2008; 68: 8723–8732.
Sem KP, Zahedi B, Tan I, Deak M, Lim L, Harden N . ACK family tyrosine kinase activity is a component of Dcdc42 signaling during dorsal closure in Drosophila melanogaster. Mol Cell Biol 2002; 22: 3685–3697.
Abdallah AM, Zhou X, Kim C et al. Activated Cdc42 kinase regulates Dock localization in male germ cells during Drosophila spermatogenesis. Dev Biol 2013; 378: 141–153.
Schoenherr JA, Drennan JM, Martinez JS et al. Drosophila activated cdc42 kinase has an anti-apoptotic function. PLoS Genet 2012; 8: e1002725.
Yu FX, Guan KL . The Hippo pathway: regulators and regulations. Genes Dev 2013; 27: 355–371.
Harvey KF, Zhang X, Thomas DM . The Hippo pathway and human cancer. Nat Rev Cancer 2013; 13: 246–257.
Yin M, Zhang L . Hippo signaling: a hub of growth control, tumor suppression and pluripotency maintenance. J Genet Genomics 2011; 38: 471–481.
Huang J, Wu S, Barrera J, Matthews K, Pan D . The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005; 122: 421–434.
Yin MX, Zhang L . Hippo signaling in epithelial stem cells. Acta Biochim Biophys Sin (Shanghai) 2015; 47: 39–45.
Yu FX, Zhao B, Guan KL . Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015; 163: 811–828.
Harvey KF, Pfleger CM, Hariharan IK . The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 2003; 114: 457–467.
Pantalacci S, Tapon N, Leopold P . The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat Cell Biol 2003; 5: 921–927.
Udan RS, Kango-Singh M, Nolo R, Tao C, Halder G . Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat Cell Biol 2003; 5: 914–920.
Wu S, Huang J, Dong J, Pan D . Hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 2003; 114: 445–456.
Jia J, Zhang W, Wang B, Trinko R, Jiang J . The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis. Genes Dev 2003; 17: 2514–2519.
Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ . The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev 1995; 9: 534–546.
Jin Y, Dong L, Lu Y et al. Dimerization and cytoplasmic localization regulate Hippo kinase signaling activity in organ size control. J Biol Chem 2012; 287: 5784–5796.
Hao Y, Chun A, Cheung K, Rashidi B, Yang X . Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem 2008; 283: 5496–5509.
Zhang J, Smolen GA, Haber DA . Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res 2008; 68: 2789–2794.
Tapon N, Harvey KF, Bell DW et al. salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 2002; 110: 467–478.
Kango-Singh M, Nolo R, Tao C et al. Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila. Development 2002; 129: 5719–5730.
Lai ZC, Wei X, Shimizu T et al. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell 2005; 120: 675–685.
Lei QY, Zhang H, Zhao B et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol 2008; 28: 2426–2436.
Oh H, Irvine KD . In vivo regulation of Yorkie phosphorylation and localization. Development 2008; 135: 1081–1088.
Yu J, Zheng Y, Dong J, Klusza S, Deng WM, Pan D . Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev Cell 2010; 18: 288–299.
Badouel C, Gardano L, Amin N et al. The FERM-domain protein Expanded regulates Hippo pathway activity via direct interactions with the transcriptional activator Yorkie. Dev Cell 2009; 16: 411–420.
Oh H, Reddy BV, Irvine KD . Phosphorylation-independent repression of Yorkie in Fat-Hippo signaling. Dev Biol 2009; 335: 188–197.
Salah Z, Aqeilan RI . WW domain interactions regulate the Hippo tumor suppressor pathway. Cell Death Dis 2011; 2: e172.
Webb C, Upadhyay A, Giuntini F et al. Structural features and ligand binding properties of tandem WW domains from YAP and TAZ, nuclear effectors of the Hippo pathway. Biochemistry 2011; 50: 3300–3309.
Zhang X, Milton CC, Poon CL, Hong W, Harvey KF . Wbp2 cooperates with Yorkie to drive tissue growth downstream of the Salvador-Warts-Hippo pathway. Cell Death Differ 2011; 18: 1346–1355.
Wang W, Huang J, Wang X et al. PTPN14 is required for the density-dependent control of YAP1. Genes Dev 2012; 26: 1959–1971.
Kwon Y, Vinayagam A, Sun X et al. The Hippo signaling pathway interactome. Science 2013; 342: 737–740.
Oh H, Irvine KD . Yorkie: the final destination of Hippo signaling. Trends Cell Biol 2010; 20: 410–417.
Zhang L, Ren F, Zhang Q, Chen Y, Wang B, Jiang J . The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Dev Cell 2008; 14: 377–387.
Duffy JB . GAL4 system in Drosophila: a fly geneticist's Swiss army knife. Genesis 2002; 34: 1–15.
Hamaratoglu F, Willecke M, Kango-Singh M et al. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol 2006; 8: 27–36.
Thompson BJ, Cohen SM . The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell 2006; 126: 767–774.
Nolo R, Morrison CM, Tao C, Zhang X, Halder G . The bantam microRNA is a target of the hippo tumor-suppressor pathway. Curr Biol 2006; 16: 1895–1904.
Bischof J, Maeda RK, Hediger M, Karch F, Basler K . An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci USA 2007; 104: 3312–3317.
Zahedi B, Shen W, Xu X, Chen X, Mahey M, Harden N . Leading edge-secreted Dpp cooperates with ACK-dependent signaling from the amnioserosa to regulate myosin levels during dorsal closure. Dev Dyn 2008; 237: 2936–2946.
Cho E, Feng Y, Rauskolb C, Maitra S, Fehon R, Irvine KD . Delineation of a Fat tumor suppressor pathway. Nat Genet 2006; 38: 1142–1150.
McCartney BM, Kulikauskas RM, LaJeunesse DR, Fehon RG . The neurofibromatosis-2 homologue, Merlin, and the tumor suppressor expanded function together in Drosophila to regulate cell proliferation and differentiation. Development 2000; 127: 1315–1324.
Pellock BJ, Buff E, White K, Hariharan IK . The Drosophila tumor suppressors Expanded and Merlin differentially regulate cell cycle exit, apoptosis, and Wingless signaling. Dev Biol 2007; 304: 102–115.
Silva E, Tsatskis Y, Gardano L, Tapon N, McNeill H . The tumor-suppressor gene fat controls tissue growth upstream of expanded in the hippo signaling pathway. Curr Biol 2006; 16: 2081–2089.
Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D . Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 2013; 154: 1342–1355.
Wu JS, Luo L . A protocol for mosaic analysis with a repressible cell marker (MARCM) in Drosophila. Nat Protoc 2006; 1: 2583–2589.
Lee T, Luo L . Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci 2001; 24: 251–254.
Rodriguez A, Chen P, Oliver H, Abrams JM . Unrestrained caspase-dependent cell death caused by loss of Diap1 function requires the Drosophila Apaf-1 homolog, Dark. EMBO J 2002; 21: 2189–2197.
Igaki T, Yamamoto-Goto Y, Tokushige N, Kanda H, Miura M . Down-regulation of DIAP1 triggers a novel Drosophila cell death pathway mediated by Dark and DRONC. J Biol Chem 2002; 277: 23103–23106.
Maitra S, Kulikauskas RM, Gavilan H, Fehon RG . The tumor suppressors Merlin and Expanded function cooperatively to modulate receptor endocytosis and signaling. Curr Biol 2006; 16: 702–709.
Angus L, Moleirinho S, Herron L et al. Willin/FRMD6 expression activates the Hippo signaling pathway kinases in mammals and antagonizes oncogenic YAP. Oncogene 2011; 31: 238–250.
Prieto-Echague V, Gucwa A, Craddock BP, Brown DA, Miller WT . Cancer-associated mutations activate the nonreceptor tyrosine kinase Ack1. J Biol Chem 2010; 285: 10605–10615.
Phatak SS, Zhang S . A novel multi-modal drug repurposing approach for identification of potent ACK1 inhibitors. Pac Symp Biocomput 2013, 29–40.
Jin M, Wang J, Kleinberg et al. Discovery of potent, selective and orally bioavailable imidazo[1,5-a]pyrazine derived ACK1 inhibitors. Bioorg Med Chem Lett 2013; 23: 979–984.
Jiao X, Kopecky DJ, Liu J et al. Synthesis and optimization of substituted furo[2,3-d]-pyrimidin-4-amines and 7H-pyrrolo[2,3-d]pyrimidin-4-amines as ACK1 inhibitors. Bioorg Med Chem Lett 2012; 22: 6212–6217.
Mahajan K, Challa S, Coppola D et al. Effect of Ack1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity. Prostate 2010; 70: 1274–1285.
DiMauro EF, Newcomb J, Nunes JJ et al. Discovery of 4-amino-5,6-biaryl-furo[2,3-d]pyrimidines as inhibitors of Lck: development of an expedient and divergent synthetic route and preliminary SAR. Bioorg Med Chem Lett 2007; 17: 2305–2309.
Mahajan K, Mahajan NP . ACK1 tyrosine kinase: targeted inhibition to block cancer cell proliferation. Cancer Lett 2013; 338: 185–192.
Shinmura K, Kiyose S, Nagura K et al. TNK2 gene amplification is a novel predictor of a poor prognosis in patients with gastric cancer. J Surg Oncol 2014; 109: 189–197.
Blaumueller CM, Mlodzik M . The Drosophila tumor suppressor expanded regulates growth, apoptosis, and patterning during development. Mech Dev 2000; 92: 251–262.
LaJeunesse DR, McCartney BM, Fehon RG . Structural analysis of Drosophila merlin reveals functional domains important for growth control and subcellular localization. J Cell Biol 1998; 141: 1589–1599.
Huang HL, Wang S, Yin MX et al. Par-1 regulates tissue growth by influencing hippo phosphorylation status and hippo-salvador association. PLoS Biol 2013; 11: e1001620.
Acknowledgements
We would like to thank Richard G. Fehon, Nicholas Harden, Bruce A. Hay, Allen Lauhgon, Hugo Stocker, Nicolas Tapon, Kenneth D. Irvine, the Bloomington Drosophila center, National institute of Genetics, Vienna Drosophila RNAi Centers, and the Developmental Studies Hybridoma Bank for reagents and fly stocks. This research is supported by grants from the National Basic Research Program of China (973 Program 2012CB945001), the ‘Strategic Priority Research Program’ of the Chinese Academy of Sciences (XDA01010406), the National Natural Science Foundation of China (31171394, 31371462), the State Key Program of National Natural Science of China (31530043), the ‘Cross and cooperation in science and technology innovation team’ project of the Chinese Academy of Sciences and the CAS/SAFEA International Partnership Program for Creative Research Teams.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hu, L., Xu, J., Yin, MX. et al. Ack promotes tissue growth via phosphorylation and suppression of the Hippo pathway component Expanded. Cell Discov 2, 15047 (2016). https://doi.org/10.1038/celldisc.2015.47
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/celldisc.2015.47
- Springer Nature Singapore Pte Ltd.