
Abstract
The heavy metal cadmium (Cd) is nephrotoxic. Recent studies show that autophagy plays an essential role in Cd-induced kidney injury. However, the mechanisms of Cd-induced kidney injury accompanied by autophagy are still obscure. In the present study, we first confirmed that Cd induced kidney damage and dysfunction, along with autophagy, both in vivo and in vitro. Then, we observed that cyclooxygenase-2 (COX-2) and the eIF2α–ATF4 pathway of endoplasmic reticulum (ER) stress were induced by Cd in both kidney tissues and cultured cells. Further studies showed that inhibition of COX-2 with celecoxib or RNA interference (RNAi) inhibited the Cd-induced autophagy in kidney cells. In addition, blocking ER stress with 4-phenylbutyrate or RNAi partially counteracted COX-2 overexpression and autophagy induced by Cd, which suggested that ER stress was required for Cd-induced kidney autophagy. Significantly, our results showed that Cd activated ATF4 and induced its translocation to the nucleus. Knockdown of ATF4 inhibited Cd-induced COX-2 overexpression. While COX-2 overexpression is involved in renal dysfunction, there is no prior report on the role of COX-2 in autophagy regulation. The results of the current study suggest a novel molecular mechanism that the ER stress eIF2α–ATF4 pathway-mediated COX-2 overexpression contributes to Cd-induced kidney autophagy and injury. The present study implies that COX-2 may be a potential target for therapy against Cd-induced nephrotoxicity.
Similar content being viewed by others

Main
Cadmium (Cd) is a toxic heavy metal and ubiquitous environmental pollutant. Human exposure to Cd comes from several sources, such as metal industries, battery production, electroplating processes and cigarette smoking.1, 2 Cd exposure has serious detrimental effects on human health, such as bone softening, hepatic injury and renal dysfunction.3, 4 Previous studies showed that Cd-induced nephrotoxicity may occur through several mechanisms, such as oxidative stress,5 DNA damage,6 alteration of transcriptional regulation7 or apoptosis.2, 8 Toxic compounds usually kill cells via one of the three types of cell death: apoptosis, autophagy-dependent cell death and necrosis.9, 10 Recently, Cd has also been reported to induce autophagy in the kidney.7, 10, 11 As an adaptive response to stimuli, autophagy degrades macromolecules and organelles to maintain cell energy homeostasis and survival,12 but the molecular mechanisms of Cd-induced kidney autophagy are little understood.
Endoplasmic reticulum (ER), an organelle in cells, is involved in protein folding and trafficking and maintaining calcium homeostasis.13 When ER function is altered by intracellular or extracellular stimuli, ER stress occurs, with the accumulation of unfolded and misfolded proteins in the ER lumen, which activates an adaptive response commonly named the unfolded protein response (UPR). ER stress is transduced by three ER transmembrane proteins: protein kinase RNA-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1α (IRE1α) and activating transcription factor 6 (ATF6).14 Normally, the ER lumenal domains of the three transducers bind to glucose-regulated protein 78 (GRP78 or BiP). When UPR occurs, GRP78 prevents protein aggregation and helps them fold properly. As one major transducer of ER stress, PERK directly phosphorylates eukaryotic initiation factor 2 α-subunit (eIF2α), decreases most mRNA transcription and promotes transcription factor ATF4 translocation and activation. Accumulating evidence has indicated that ER stress triggers autophagy, which disposes of unfolded proteins in cells.15, 16, 17 However, there is no research exploring the role of ER stress in Cd-induced autophagy.
As the rate-limiting enzyme for prostaglandin E2 (PGE2) synthesis, COX-2 (encoded by the PTGS2 gene) has been implicated in renal dysfunction and inflammation. Many lines of evidence indicate that COX-2 can be induced by Cd in several experimental models.18, 19 Nevertheless, there is no report on the role of COX-2 in autophagy induced by drugs or pollutants, especially Cd.
In the present study, we found Cd triggered autophagy accompanied by ER stress and COX-2 upregulation, both in mice kidney tissues and cultured human kidney cells. We hypothesized that ER stress and/or COX-2 would regulate Cd-induced kidney autophagy and wanted to provide insight into potential molecular mechanisms for intervention in Cd-induced nephrotoxicity. We showed, for the first time, that blocking COX-2 could inhibit Cd-induced autophagy. Furthermore, we found that Cd-induced COX-2 overexpression along with autophagy was inhibited by an ER stress inhibitor or RNAi. Notably, we revealed that Cd-induced kidney autophagy was mediated by COX-2 via eIF2α-ATF4 signaling transcriptional activation.
Results
Cd causes kidney injury both in vivo and in vitro
As shown in Figure 1a, the body weights of the 0.2 and 1 mg/kg Cd groups did not change compared with those of day 1, while those of the 5 mg/kg Cd group showed a significant decline from day 5 to day 7. We also calculated the ratios of liver and kidney to body weight and found that Cd significantly increased the ratio of liver to body in the 1 and 5 mg/kg Cd exposure groups, while there was no alteration in that of kidney to body weight (Figure 1b). Moreover, the mouse metallothionein (MT) genes (mMT1 and mMT2) mRNA was significantly elevated both in livers and kidneys of mice exposed to Cd (Supplementary Figure S1a). Following this discovery, serum renal function parameters were analyzed. As shown in Figure 1c, we observed that serum creatinine (CRE) was raised in the 0.2 and 1 mg/kg Cd groups, but not changed in the 5 mg/kg group; serum blood urea nitrogen (BUN) was not significantly changed in the 0.2 and 1 mg/kg Cd groups, but significantly declined in the 5 mg/kg Cd group (Figure 1d); while the ratio of BUN/CRE was decreased in all three Cd groups, significantly in the 5 mg/kg Cd (Figure 1e). Further, histological changes were observed using hematoxylin and eosin (HE) staining. As shown in Figure 1f, shrinkage of glomeruli and the degeneration of tubules were observed in the 1 and 5 mg/kg Cd groups compared with the control group, which indicated that Cd damaged the kidney tissues, explaining the renal dysfunction demonstrated by the altered blood chemistry. These data indicated that renal function was damaged by Cd exposure in vivo.

Cd causes kidney injury and cytotoxicity. (a) Body weight curve. (b) Ratios of liver and kidney weight to body weight. (c) CRE, (d) BUN and (e) BUN/CRE: renal function parameters; (f) histological changes (HE staining) of kidney after Cd exposure to mice for 7 days (magnification, × 400). Arrows indicate the histological changes of the glomerulus (black) and tubules (white). The scale bar is 50 μm. (g) Cell growth of HEK cells using MTT assay. HEK cells are exposed to the indicated (log scale) Cd concentrations for 12 or 24 h, and the blue and red dotted lines stand for a nonlinear curve fitting as their IC50 values, respectively. (*Significantly different from untreated control and #significantly different from different times at the same concentration. Data represent the mean±S.D.; (a–f), n=5–6; (g), n=3, P<0.05)
In vitro, hMT1B increased in a time-dependent manner in HEK cells exposed to 40 μM Cd (Supplementary Figure S1b). The MTT assay was used to evaluate cell growth after 12 and 24 h exposure to Cd (0–160 μM). As shown in Figure 1g, cell growth decreased in a concentration- and time-dependent manner after Cd exposure compared with control (0.9% saline), and the 50% growth inhibition concentration (IC50) at 24 h was 51.6 μM.
Cd induces kidney cell autophagy both in vivo and in vitro
The major characteristics of autophagy are formation of autophagosomes and processing of LC3-I to LC3-II.20 In mice kidney tissues, LC3-II protein was sharply increased in all Cd exposure groups (Figure 2a). As a selective substrate of autophagy, p62 protein was significantly depleted (Figure 2a). Moreover, immunohistochemical analysis (Figure 2b) showed that LC3-II protein was significantly increased by Cd exposure in kidney tubules, which was quantitatively measured using Image Pro (Supplementary Figure S2). Furthermore, Cd exposure resulted in p-AKT elevation and p-mTOR decrease in HEK cells (Figures 2c and d). Along with these changes, the appearance of LC3-II and degradation of p62 increased in a concentration- and time-dependent manner after Cd treatment (Figures 2c and d). Next, the LC3-II loci were imaged after Cd exposure for 12 h using immunofluorescence analysis. As shown in Figure 2e, specific punctuate, LC3-II was observed as more dots visible in Cd-exposed cells than in control cells. To determine the role of autophagy in Cd-induced cytotoxicity, an autophagy inducer, rapamycin (rapa), and a lysosome inhibitor, chloroquine (CQ), were used. We found CQ, not rapa, rescued cell growth inhibition induced by Cd (Figure 2f). Moreover, rapa could enhance Cd-induced autophagy, as evidenced by the decrease of p-mTOR and increase of p-AKT and LC3-II (Figure 2g).

Cd induces kidney autophagy both in vivo and in vitro. (a) Autophagy-related proteins (LC3-II and p62) levels (treated with Cd for 7 days). (b) Immunohistochemical analysis for LC3 in kidneys of Cd-exposed mice (magnification, × 400). Arrows indicate the increase of LC3 in the tubules. Scale bar is 50 μm. (c and d) Autophagy-related proteins (p-AKT, AKT, p-mTOR, mTOR, LC3 and p62) are analyzed using western blotting (c, as a function of Cd concentrations for 12 h; d, as a function of exposure time with 40 μM Cd). (e) Immunofluorescence analysis for LC3 loci in Cd-exposed (40 μM) HEK cells for 12 h. Scale bar is 20 μm. (f) CQ, not rapa, enhances cell growth inhibited by Cd. (g) Rapa enhances autophagy induced by Cd (40 μM) treatment for 12 h. (*Significantly different from untreated control, #significantly different from Cd treatment. Data represent the mean±S.D.; n=3, P<0.05)
Cd elevates COX-2 expression both in vivo and in vitro
To explore the role of COX-2 in Cd-induced renal dysfunction, COX-2 expression was assessed after Cd exposure. In vivo, as shown in Figure 3a, Cd increased mPTGS2 mRNA and COX-2 protein levels in a dose-dependent manner. Furthermore, immunohistochemical analysis confirmed that COX-2 was significantly induced in kidney tubules by Cd exposure (Figure 3b and Supplementary Figure S2).

Cd upregulates COX-2 expression both in vivo and in vitro. (a) mPTGS2 mRNA and COX-2 protein levels and (b) immunochemical analysis for COX-2 in the kidney tissues of Cd-exposed mice for 7 days (magnification, × 400). Arrows indicate the COX-2 signals in the kidney tubules. Scale bar is 50 μm. (c and d) hPTGS2 mRNA and COX-2 protein levels in HEK cells (c, as a function of exposure time with 40 μM Cd; d, as a function of Cd concentrations for 12 h). (e) PGE2 production and (f) Immunofluorescence analysis for COX-2 are elevated in Cd-exposed HEK cells for 12 h (40 μM). Scale bar is 20 μm. (*significantly different from untreated control. Data represents the mean±S.D.; n=3, P<0.05)
In vitro (in HEK cells), hPTGS2 mRNA and COX-2 protein were significantly increased in a concentration- and time-dependent manner after Cd treatment (Figures 3c and d). PGE2 was also increased in Cd-treated cells (Figure 3e). Furthermore, immunofluorescence analysis revealed that COX-2 signals were stronger in the cytoplasm and perinuclear space of Cd-treated cells than in that of control cells (Figure 3f), which was consistent with other reports that COX-2 expressed in the perinuclear space in granulocytes21 and HaCaT cells.22
Inhibition of COX-2 reverses Cd-induced autophagy
To elucidate whether COX-2 overexpression is required for Cd-induced autophagy, COX-2 was inhibited using gene knockdown or drugs. First, we found that cell growth was partially restored in HEK-shPTGS2 (COX-2 knockdown) cells compared with HEK-shctrl cells upon 10–80 μM Cd exposure for 12 and 24 h (Figure 4a). Additionally, the expression level of hMT1B mRNA was also rescued in HEK-shPTGS2 cells in Supplementary Figure S3a. In Figure 4b, the increase of p-AKT and LC3-II, while the decrease of p-mTOR and p62 induced by Cd, was attenuated in HEK-shPTGS2 cells compared with the control cells, which was verified with the PTGS2 small interference RNA (siRNA) transfection assay (Supplementary Figure S3b). Consistently, PGE2 was also decreased in HEK-shPTGS2 cells upon Cd exposure (Figure 4c). Next, celecoxib, a clinical COX-2 inhibitory drug, was used to treat HEK cells in combination with Cd. As shown in Figure 4d, celecoxib partially restored the cell growth inhibition caused by 40 μM Cd. Celecoxib markedly inhibited COX-2 overexpression and subsequent autophagy in Cd-exposed HEK cells (Figure 4e). Additionally, we observed similar phenomena with other COX-2 inhibitors, CAY10404 and NS398 (Supplementary Figures S3c and d). These data demonstrated that Cd-induced autophagy was regulated by COX-2 in vitro.

Cd-induced autophagy can be reversed by inhibition of COX-2. (a–c) Results of COX-2 knockdown using shPTGS2 cells: (a) alteration of cell growth (*significantly different from untreated control, #significantly different from the same time in different cells, P<0.05); (b) COX-2 and autophagy and (c) production of PGE2. (d and e) Blocking COX-2 with celecoxib rescues 40 μM Cd-induced cell growth inhibition with MTT assay (d) and autophagy with western blot analysis (e) in HEK cells for 12 h (*significantly different from untreated control, #significantly different from Cd treatment. Data represent the mean±S.D.; n=3, P<0.05)
Cd-induced COX-2 overexpression depends on ER stress
We evaluated ER stress response upon Cd exposure both in vivo and in vitro. Cd significantly increased the mRNA level of mGRP78 and mCHOP (two ER stress molecular markers) in a dose-dependent manner, but did not affect the mATF6 or mIRE1α mRNA level in mice kidney (Figure 5a). Being consistent with these results, the GRP78 and p-eIF2α proteins were also induced by Cd in kidney tissues (Figure 5a). Moreover, immunohistochemical analysis confirmed that GRP78 was significantly increased in kidney tubules of Cd-exposed mice compared with control (Figure 5b and Supplementary Figure S2).

Cd induces ER stress both in vivo and in vitro. (a) Levels of mRNA (mGRP78, mCHOP, mATF6 and mIRE1α) and proteins (GRP78, p-eIF2α and CHOP) related to ER stress caused by Cd for 7 days and (b) immunochemical analysis for GRP78 in the kidney tissues of Cd-exposed mice (magnification, × 400). Arrows indicate that the GRP78 signals increase in the glomerulus and tubules. Scale bar is 50 μm. (c and d) ER stress reporter genes show ER stress occurs when HEK cells treated by Cd for 6 h. Tg used as a positive control. (e and f) ER stress related gene mRNA (hGRP78 and hCHOP) and protein (GRP78 and p-eIF2α) levels in Cd-exposed HEK cells (e, concentration series of Cd; f, time series with 40 μM Cd). (g) Confocal images for CHOP in the Cd-exposed HEK cells (40 μM). Scale bar is 20 μm (*significantly different from untreated control. Data represents the mean±S.D.; n=3, P<0.05)
We examined the effect of Cd on ER stress by other means. Thapsigargin (Tg), an ER stress inducer, was used as a positive control. As shown in Figures 5c and d, Tg or Cd decreased the secretion of Gaussia luciferase (Gluc) and secreted alkaline phosphatase (SEAP) in a concentration-dependent manner in HEK-Fluc-Gluc and HEK-Fluc-SEAP cells. In addition, Cd elevated hGRP78 and hCHOP mRNA and the GRP78 and p-eIF2α proteins in HEK cells (Figures 5e and f). Additionally, as depicted in Figure 5g, CHOP (CCAAT-enhancer-binding protein homologous protein) increased in Cd-exposed cells compared with control cells as detected using immunofluorescence analysis.
To elucidate whether Cd-induced ER stress is required for overexpression of COX-2, Tg was used to treat HEK cells combining with Cd. Tg significantly enhanced both the overexpression of COX-2 and autophagy induced by Cd, as indicated by an increased LC3-II (Figure 6a). 4-PBA, an ER stress inhibitor, counteracted the cell growth inhibition, COX-2 overexpression, ER stress and autophagy caused by Cd (Figures 6b and c). Moreover, when HEK cells were transfected with GRP78 siRNA, followed by Cd exposure, the knockdown of GRP78 or CHOP also significantly attenuated the Cd-induced COX-2 overexpression, ER stress and autophagy in HEK cells (Figure 6d and Supplementary Figure S4). Taken together, these results clearly indicated that ER stress is required for overexpression of COX-2 induced by Cd.

COX-2 induction in HEK cells by Cd requires ER stress. (a) 40 μM Cd-induced autophagy is enhanced by Tg (1 μM) in HEK cells. (b and c) 5 μM 4-PBA treatment partially counteracts Cd-induced cell growth inhibition, ER stress and autophagy. (d) GRP78 silencing also decreases Cd-induced ER stress and autophagy (*significantly different from untreated control, #significantly different from Cd treatment. Data represents the mean±S.D.; n=3, P<0.05)
Cd induces COX-2 overexpression via ATF4 transcriptional regulation
To investigate the molecular mechanism of Cd-induced overexpression of COX-2 with ER stress, the transcription factor ATF4 was examined. In Figure 7a, Cd slightly increased mATF4 mRNA, although there was no significant change compared with control, and ATF4 protein was elevated by Cd in kidney tissues. In addition, Cd increased the hATF4 mRNA and ATF4 protein in HEK cells (Figures 7b and c). Immunofluorescence analysis revealed that ATF4 protein was translocated from the cytoplasm to the nucleus (Figure 7d). Furthermore, ATF4 siRNA significantly inhibited the activation of ATF4 and COX-2 protein levels in HEK cells (Figure 7e). Finally, we found that ATF4 was translocated into the nucleus and COX-2 was transcriptionally activated upon Cd treatment, changes that were inhibited by blocking ER stress with 4-PBA, whereas ATF4 translocation and COX-2 expression were enhanced by activation of ER stress with Tg (Figure 7f). Interestingly, we found that, by itself, 4-PBA could increase ATF4 translocation into the nucleus, which might be due to the side effect of 4-PBA on cell signaling related to ATF4. We also observed that Cd-induced COX-2 was counteracted by the ATF4 siRNA (Figure 7g). These results indicated that transcriptional activation of ATF4 was essential for COX-2 overexpression in Cd-treated HEK cells.

COX-2 induction by Cd is mediated by ATF4 transactivation. (a) Cd increases the levels of mATF4 mRNA and ATF4 protein in kidney tissues. (b and c) Cd elevates hATF4 mRNA and ATF4 protein levels in HEK cells. Tg is used as a positive control (b, as a function of Cd concentrations for 12 h; c, as a function of time with 40 μM Cd). (d) In HEK cells, Cd causes ATF4 translocation from the cytoplasm to the nucleus as detected using a confocal microscopy. Scale bar is 20 μm. (e) ATF4 silencing restores Cd-induced PTGS2 mRNA and COX-2 protein overexpression. NC is the negative control of siRNA. (f) 4-PBA inhibits ATF4 translocation and COX-2 upregulation induced by Cd. (g) ATF4 silencing also decreases Cd-induced ATF4 translocation and COX-2 upregulation (*significantly different from untreated control, #significantly different from Cd treatment. Data represents the mean±S.D.; n=3, P<0.05)
Discussion
Our study provided evidence that Cd caused kidney injury and dysfunction with autophagy in mice. Cd also induced cell growth inhibition and autophagy in kidney cells, which supported previous studies.10 Next, we found that Cd-induced autophagy was associated with COX-2 overexpression both in vivo and in vitro. Further studies demonstrated that several COX-2 inhibitors, such as celecoxib, rescued the kidney from Cd-induced injury and autophagy, which was further confirmed by PTGS2 RNAi protection against Cd damage in HEK cells. Additionally, ER stress, which was proved to be required for COX-2 overexpression in kidney tissues and cultured cells, occurred upon Cd treatment. Finally, our study demonstrated that the eIF2α–ATF4 pathway activated COX-2 transcription to regulate Cd-induced autophagy. We believe that this study has found a novel molecular mechanism of Cd-induced renal autophagy and injury, which is dependent on COX-2 overexpression regulated by ER stress.
The toxic heavy metal Cd is known to cause itai-itai disease, characterized by bone softening and kidney failure.23 More evidence shows that Cd exposure causes severe liver and kidney damage both in vivo and in vitro.24, 25, 26 Cd first accumulates in the liver after exposure.27 Most of the Cd is captured by MT proteins to form Cd/MT complexes that are gradually released into the blood and reabsorbed from the glomerular filtrate in the kidney.27 Then, the complexes degrade quickly to release the Cd, leading to renal injury.28 Our study showed that Cd exposure resulted in hepatomegaly, which was consistent with previous studies.29, 30 mMTs mRNA was induced by Cd both in livers and kidneys, which indicated that Cd had been transferred and released into the kidney which might result in renal damage. In line with these data, histological analysis showed glomerular shrinkage and tubular degeneration upon Cd exposure. We further examined serum renal function parameters. CRE is cleared from blood mainly by glomerular filtration as well as proximal tubular secretion within the kidney. A rise of serum CRE is observed only in the significantly injured kidney.31 BUN is another indicator of renal function. Once the kidney is damaged and dysfunctional, urea nitrogen is not filtered totally from the blood, resulting in BUN elevation.32 However, since urea is synthesized by liver, BUN will be lower than the normal level when the liver is severely damaged.33 A low BUN/CRE ratio indicates reduction of BUN reabsorption caused by kidney damage.31 In our study, Cd raised the serum CRE and BUN in the 0.2 and 1 mg/kg groups, indicating kidney filtration failure, but not in the 5 mg/kg group, which might have resulted from severe hepatomegaly and liver damage. The ratio of BUN/CRE decreased in all Cd groups, suggesting that Cd caused severe tubule damage and glomerular filtration failure. In support of our results, exposure of mice to 1.5 mg/kg Cd for 1 week also caused the histological alteration and dysfunction of kidney.34
We further found that Cd induced autophagy both in vivo and in vitro, which is consistent with a recent study showing that 0.3 mg/kg Cd induced autophagy in the kidney.10 Growing evidence indicates that autophagy may play a protective role against Cd-induced nephrotoxicity. An animal study showed that inhibition of autophagy with CQ and 3-methyladenine worsened ischemia–reperfusion-induced mice kidney injury and autophagy.35 Atg5-deficient mice had markedly affected kidney function and were more sensitive to kidney ischemic injury.36 Indeed, autophagy is either a survival or a death safeguard mechanism, which depends on the extent of stress. A review by Scarlatti et al.37 stated that once cells are severely damaged, autophagy can remove most of the organelles and kill the cells by a caspase-independent form of cell death, known as autophagy-dependent cell death. For examples, salivary Pa-4 cells treated with baflomycin A1 or CQ rescued the cell viability inhibition induced by H2O2.38 Caspase-8 inhibition could have the untoward effect of exacerbating cell death by activating the autophagic death pathway induced by Z-Val-Ala-Asp-fluoromethylketone (zVAD-FMK).39 In the present study, we found inhibition of autophagy with CQ would prevent the Cd-induced cell growth inhibition. Moreover, blocking autophagy upstream signaling, such as COX-2 and ER stress, also improved cell growth. We therefore deduced that autophagy induced by Cd might cause kidney cell growth inhibition and result in kidney failure.
The kinase mTOR is a major negative regulator of autophagy,40 which is the key effector in the PI3K–AKT–mTOR pathway.41 Autophagy can be activated via inhibition of the PI3K–AKT–mTOR pathway.42 However, inhibition of mTOR by some chemicals, such as rapa, will promote AKT phosphorylation by blocking the phosphorylation of insulin receptor substrate 1 and p70 ribosoma S6 kinase,41, 43 which are known to be a part of a negative feedback mechanism of PI3K–AKT signal transduction.44 Our results showed that Cd inhibited mTOR but actually increased the phosphorylation of AKT, suggesting that Cd inhibited mTOR which might not depend on AKT. Another possible explanation for Cd-induced increased AKT phosphorylation could be due to the loss of mTOR-mediated feedback inhibition on AKT, similar to rapa. Consistently, we discovered that COX-2 knockdown increased p-mTOR leading to a decrease of autophagy and p-AKT.
A previous study showed that Cd induced ER stress along with autophagy in mice kidney.10 In the present study, we also found that autophagy and ER stress were triggered in Cd-exposed kidney tissues and HEK cells. We confirmed that Cd induced ER stress in HEK cells using two ER stress reporter systems. Then, we demonstrated that an ER stress inhibitor counteracted Cd-induced autophagy and cell growth inhibition in HEK cells, which was verified by the GRP78 siRNA transfection assay. Further, previous studies showed that activation of ER stress induces autophagy.15, 45 These studies provided evidence that ER stress was required for Cd-induced autophagy in the kidney. Moreover, Cd-induced COX-2 overexpression was attenuated by GRP78 or ATF4 silencing, which suggested that COX-2-mediated autophagy induced by Cd required ER stress in the kidney.
We found that Cd increased COX-2 and PGE2, along with autophagy and ER stress in vitro. Inhibition of COX-2 by celecoxib or PTGS2 RNAi counteracted Cd-induced autophagy in the kidney. In support of these results, celecoxib has been proved to attenuate rodent renal injury induced by several other chemical or physical factors, such as cisplatin,46 cyclosporine47 or hypoxia.48 As long ago as 1997, celecoxib was developed49 and then used in the clinic.50 However, some case reports showed that long-time use or high doses of celecoxib raised the risk of renal injury in the elderly and in patients with pre-existing renal dysfunction, liver injury or heart diseases.51 Therefore, use of COX-2 inhibitors requires caution. More specific and safer COX-2 inhibitors or therapy strategy should be developed to guard against kidney injury.
Much work has studied the molecular mechanisms by which ER stress regulates autophagy. It has been reported that ER stress induces autophagy through activation of the RAF–MEK–ERK–MAPK cascade in melanoma.52 Qin et al.53 showed that autophagy triggered by ER stress depends on inhibition of the AKT–TSC–mTOR pathway. Furthermore, ER molecular chaperons and pathways, such as GRP78 protein and the IRE1α–JNK–Bcl-2, ATF6–XBP1–ATG and PERK–eIF2α–ATF4 pathways, are involved in ER stress-mediated autophagy.54, 55, 56 In the present study, we observed that Cd affected gene expression of GRP78, ATF4 and CHOP, but not IRE1α or ATF6, in kidney tissues. Further, p-eIF2α and ATF4 proteins were activated by Cd both in vivo and in vitro. Cd-induced autophagy was counteracted by ER stress blocking. Therefore, it is likely that the eIF2α–ATF4 pathway regulates Cd-induced kidney autophagy and injury.
Specifically, the present study demonstrated that Cd promoted ATF4 translocation from the cytoplasm to the nucleus. Silencing of ATF4 inhibited both its translocation and the COX-2 overexpression induced by Cd. Once translocated into the nucleus, ATF4 binds to the promoter region of PTGS2 gene and promotes its transcription.57 These studies revealed a novel pathway by which Cd triggers autophagy via COX-2 overexpression, which is transcriptionally regulated by ATF4.
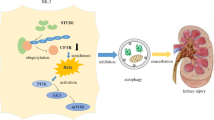
In conclusion, the present study showed that Cd exposure resulted in kidney injury with autophagy. We demonstrated that Cd-induced autophagy was mediated by the eIF2α–ATF4 pathway and its downstream molecule COX-2, as illustrated in Figure 8. COX-2 has been proven to be a potential molecular marker of Cd-induced nephrotoxicity. The present study also suggests that safer COX-2 inhibitors or other therapy strategies should be developed for treatment of nephrotoxicity induced by xenobiotics.

Cd induces kidney autophagy mediated by the eIF2α–ATF4 pathway in ER stress and its downstream molecule COX-2
Materials and Methods
Chemicals and reagents
The reagents used were as follows: cadmium chloride (CdCl2, purity>99%), CQ, rapa, 4-PBA and Tg (Sigma, St. Louis, MO, USA); NS398 and CAY10404 (Cayman, Ann Arbor, MI, USA) and celecoxib (Pfizer, New York, NY, USA).
Animal exposure and sample collection
Adult male ICR mice (30–35 g, 10–12 weeks old) were randomly separated into control and three exposure groups (n=5–6) and housed in a ventilated animal room at 22±2 °C with a 12 h light/dark cycle. The mice were intraperitoneally injected at doses of 0.2, 1 or 5 mg/kg body weight CdCl2 every day for 1 week. The CdCl2 was dissolved in 0.9% physiological saline. In the control group, the mice were injected with 0.9% physiological saline. Twenty-four hours after the last injection, all mice were killed and serum, livers and kidneys were sampled for experimental purposes. The ratios of organ weight to body weight were calculated. Standard hematological renal functions were analyzed using a Cobas 8000 system (Roche, Basel, Switzerland) in Xiamen Zhongshan Hospital. All experiments were approved by the Animal Ethical Committee of Xiamen University and conducted in accordance with the guidelines of the National Institutes of Health.
Histology and immunohistochemistry
After being embedded in paraffin blocks, kidney tissues were sectioned at 5 μm thickness and the sections were placed on microscope slides. HE staining was performed on the slides, which were then observed and photographed with an optical microscope (TS-100, Nikon, Tokyo, Japan). For immunohistochemical analysis, the slides were dewaxed with xylene and rehydrated with graded ethanol. After incubation with 3% hydrogen peroxide to block endogenous peroxidase, the slides were incubated with 10% goat serum for 30 min to block nonspecific proteins. Then, the slides were incubated with specific primary antibodies at 4 °C overnight, followed by horseradish peroxidase-conjugated secondary antibody incubation for 30 min. Slides were then incubated with developing solution (peroxidase substrate containing diaminobenzidine) with hematoxylin counterstaining. Lastly, images were captured. Brown signals, showing immunoreactivity, were analyzed using Image Pro Plus 6.0. (Media Cybernetics,Bethesda, MD, USA).
Cell culture
HEK cells were cultured in HyQ RPMI-1640 medium (Life, New York, NY, USA) with 10% fetal bovine serum. HEK293T cells were used for lentivirus packaging and were maintained in Dulbecco minimum essential medium with 10% fetal bovine serum. All culture media contained 1% (v/v) penicillin–streptomycin (Life). Cells were cultured in a humidified incubator with 5% CO2 at 37 °C.
Cell growth inhibition
To determine cell growth inhibition, 8000 cells were plated in each well of 96-well plates and attached to the substratum for 24 h. The cells were exposed to various concentrations of Cd for 12 and 24 h. Untreated cells served as a control. Cell growth inhibition was determined using a 0.5 mg/ml (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium) bromide (MTT, Sigma) incubation for 4 h. The formazan product was then dissolved in 100 μl lysis buffer. The plates were read against reagent blank at 570 nm using a microplate reader (Multiscan, Thermo, Waltham, MA, USA). Control cells were taken as 100% viability.
Enzyme-linked immunosorbent assay (ELISA)
The production of PGE2 was measured using an ELISA kit from Cayman Chemical (Ann Arbor, MI, USA) according to the manufacturer’s instruction.
Construction of reporter cell lines
The gene expression lentiviral vectors pLv-EF1α-Gluc-IRES-Puro, pLv-EF1α-SEAP-IRES-Puro and plv-EF1α-Firefly luciferase (Fluc)-IRES-Bsd were gifts from Dr. Lei Guo (Food and Drug Administration, USA). Two stable cell lines (HEK-Fluc-Gluc and HEK-Fluc-SEAP) were constructed as described previously.58 Briefly, lentiviral stocks were produced by transfection of HEK293T cells with a mixture of a lentiviral vector and viral packaging plasmids (pMDL-G, pVSV-G and pRSV-REV) in the ratio of 10 : 5 : 2 : 3, using Lipofectamine 2000 (Life). To construct a stable Fluc cell line, plv-EF1α-Fluc-IRES-Bsd plasmid was infected into HEK cells with lentivirus and screened with 10 μg/ml blasticidin. Then, the cells containing Fluc gene were infected with lentivirus carrying Gluc or SEAP. The cells were screened for successful transfection with 0.8 μg/ml puromycin.
Construction of COX-2 knockdown cell lines
Two stable cell lines were generated as described previously.59 Briefly, using the DNA Transfection Reagent X-tremeGENE HP (Roche), the lentiviral vectors described above, and viral packaging plasmids (pCMV-delta 8.9 and VSVG), were co-transfected into HEK293T cells to produce lentiviral stocks. HEK cells were infected with lentivirus carrying pLKO-PTGS2-shRNA recombinant plasmid. Then, the cells were selected with 0.8 μg/ml puromycin. The established PTGS2 knockdown HEK cells were identified and then named HEK-shPTGS2, and the corresponding control cells were called HEK-shctrl.
siRNA transfection
PTGS2, GRP78 and ATF4 siRNA and a negative control (NC) were designed and synthesized by RiboBio Company (Guangzhou, China). HEK cells were seeded at close to 30% confluence without any antibiotics. Twenty-four hours later, specific siRNA or NC (50 nM) was added into cells with Lipofectamine 2000 according to the manufacturer’s instructions.
Luciferase activity assay
Gluc activity was measured by adding 50 μl Gluc assay reagent (Nanolight Technology, Pinetop, AZ, USA) to 20 μl of the Gluc sample and analyzing with a GLOMAX 96 microporous plate light reader (Promega, Madison, WI, USA) to determine the bioluminescence. Fluc activity was similarly analyzed: 20 μl luciferase cell lysis buffer (NEB, Ipswich, MA, USA) was added to cellular extract, and then 100 μl of the Fluc assay reagent (Lulong, Xiamen, China) was added to the cell lysate and read immediately.
SEAP assay
SEAP was detected by using the Phospha-Light EXP assay kit (Life) according to the manufacturer’s manuals. Briefly, an aliquot of 50 μl cell-free medium was mixed with 50 μl of 1 × dilution buffer, heated at 65 °C for 30 min, and then cooled on ice. Fifty microliters of the diluted sample was incubated in a well of a white 96-well assay plate with 50 μl assay buffer for 5 min. Next, 50 μl of reaction buffer was added and incubated for 20 min. The chemiluminescence was determined with the GLOMAX 96 microporous plate light reader.
Quantitative real-time PCR (qRT-PCR)
The total RNA was extracted from the tissues or cells using TRIzol reagent (Life) according to the manufacturer’s protocol. One microgram of total RNA was reverse transcribed into cDNA using TaKaRa reverse transcription reagents (TaKaRa, Otsu, Japan). The expression level of target genes was measured by qRT-PCR using the SYBR Green I kit (TaKaRa) on a Bio-Rad CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). The amplification conditions were: initial denaturation at 95 °C for 30 s, 42 cycles of 95 °C for 5 s and 60 °C for 34 s. hACTB or mGAPDH mRNA was used as the reference for human or mouse gene expression, respectively. The primers’ sequences were listed in Supplementary Table S1.
Western blot analysis
The kidney tissues were homogenized and lysed in RIPA buffer (Beyotime, Nantong, China) with protease inhibitors. The cells were lysed in whole-cell lysate buffer with phosphatase and protease inhibitors (Beyotime). Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Millipore, Boston, MA, USA). The membranes were incubated with primary antibodies including COX-2 (Abcam, Cambridge, UK; dilution 1 : 1000), HistoneH3 (Beyotime; dilution 1 : 1000), β-actin (ProteinTech, Chicago, IL, USA; dilution 1 : 3000), GAPDH (Kangchen, Shanghai, China; dilution 1 : 5000), p-AKT(Ser473), p-AKT(Thr308), p-mTOR(Ser2448), mTOR, GRP78, p-eIF2α, ATF4, CHOP, LC3 and p62 (CST, Danvers, MA, USA; 1 : 1000) overnight at 4 °C, followed by a horseradish peroxidase-conjugated IgG secondary antibody (dilution 1 : 3000) for 1 h at room temperature. Photographs were captured and the relative density of blotting was quantified using Image J software (NIH, Bethesda, MD, USA).
Immunofluorescence assay
HEK cells were grown in six-well plates with or without 40 μM Cd for 12 h, and then fixed in 4% paraformaldehyde (pH 7.2–7.4) for 20 min. The samples were incubated in PBS plus 0.3% Triton X-100 for 5 min on ice. After blocking with 1% BSA for 30 min, the cells were incubated overnight at 4 °C with COX-2, ATF4, CHOP or LC3 antibody (dilution 1 : 200). After washing, the samples were incubated with goat anti-mouse/rabbit IgG secondary antibody (Alexa Fluor 594, Life) for 1 h in the dark. Next, the cells were counterstained with DAPI (Beyotime). Finally, they were imaged using a confocal microscope (Carl Zeiss, Jena, Germany).
Statistics
All results were expressed as the mean±standard deviation (S.D.). The statistical analyses were conducted using an unpaired Student’s t-test with SPSS 18.0 software, and P<0.05 (two tailed) was used as the criterion for significant difference.
Abbreviations
- 4-PBA:
-
4-phenylbutyrate
- AKT:
-
RAC-α serine/threonine-protein kinase
- ATF4:
-
activating transcription factor 4
- ATF6:
-
activating transcription factor 6
- ATG:
-
autophagy-related gene
- BUN:
-
blood urea nitrogen
- Cd:
-
cadmium
- CHOP:
-
CCAAT-enhancer-binding protein homologous protein
- COX-2:
-
cyclooxygenase-2
- CQ:
-
chloroquine
- CRE:
-
creatinine
- eIF2α:
-
eukaryotic initiation factor 2α-subunit
- ELISA:
-
enzyme-linked immunosorbent assay
- ER:
-
endoplasmic reticulum
- ERK:
-
extracellular signal-regulated kinase
- Fluc:
-
firefly luciferase
- Gluc:
-
Gaussia luciferase
- GRP78:
-
glucose-regulated protein 78
- HE:
-
hematoxylin and eosin
- HEK:
-
human embryonic kidney
- IC50:
-
50% growth inhibition concentration
- IRE1α:
-
inositol-requiring enzyme 1α
- JNK:
-
Jun N-terminal kinase
- LC3:
-
microtubule-associated protein 1 light chain 3
- MAPK:
-
mitogen-activated protein kinase
- MEK:
-
mitogen-activated protein/ERK kinase
- mTOR:
-
mammalian target of rapamycin
- MT:
-
metallothionein
- MTT:
-
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium) bromide
- NC:
-
negative control
- PERK:
-
protein kinase RNA-like endoplasmic reticulum kinase
- PGE2:
-
prostaglandin E2
- PTGS2:
-
prostaglandin-endoperoxide synthase 2
- qRT-PCR:
-
quantitative real-time PCR
- rapa:
-
rapamycin
- RNAi:
-
RNA interference
- SEAP:
-
secreted alkaline phosphatase
- siRNA:
-
small interference RNA
- Tg:
-
thapsigargin
- TSC:
-
tuberous sclerosis complex
- UPR:
-
unfolded protein response
- zVAD-FMK:
-
Z-Val-Ala-Asp-fluoromethylketone
References
Jarup L, Akesson A . Current status of cadmium as an environmental health problem. Toxicol Appl Pharm 2009; 238: 201–208.
Thevenod F, Lee WK . Cadmium and cellular signaling cascades: interactions between cell death and survival pathways. Arch Toxicol 2013; 87: 1743–1786.
Wallin M, Sallsten G, Lundh T, Barregard L . Low-level cadmium exposure and effects on kidney function. Occup Environ Med 2014; 71: 848–854.
Johri N, Jacquillet G, Unwin R . Heavy metal poisoning: the effects of cadmium on the kidney. Biometals 2010; 23: 783–792.
Nair AR, Lee WK, Smeets K, Swennen Q, Sanchez A, Thevenod F et al. Glutathione and mitochondria determine acute defense responses and adaptive processes in cadmium-induced oxidative stress and toxicity of the kidney. Arch Toxicol 2015; 89: 2273–2289.
Bork U, Lee WK, Kuchler A, Dittmar T, Thevenod F . Cadmium-induced DNA damage triggers G(2)/M arrest via chk1/2 and cdc2 in p53-deficient kidney proximal tubule cells. Am J Physiol Renal Physiol 2010; 298: F255–F265.
Kim HR, Lee KY, Ahn SG, Lee BH, Jung KT, Yoon JH et al. Transcriptional regulation, stabilization, and subcellular redistribution of multidrug resistance-associated protein 1 (MRP1) by glycogen synthase kinase 3 alpha beta: novel insights on modes of cadmium-induced cell death stimulated by MRP1. Arch Toxicol 2015; 89: 1271–1284.
Gao D, Xu Z, Zhang X, Zhu C, Wang Y, Min W . Cadmium triggers kidney cell apoptosis of purse red common carp (Cyprinus carpio without caspase-8 activation. Dev Comp Immunol 2013; 41: 728–737.
Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ 2007; 14: 1237–1243.
Chargui A, Zekri S, Jacquillet G, Rubera I, Ilie M, Belaid A et al. Cadmium-induced autophagy in rat kidney: an early biomarker of subtoxic exposure. Toxicol Sci 2011; 121: 31–42.
Gobe G, Crane D . Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol Lett 2010; 198: 49–55.
Komatsu M, Ichimura Y . Physiological significance of selective degradation of p62 by autophagy. FEBS Lett 2010; 584: 1374–1378.
Krebs J, Agellon L B, Michalak M . Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: an integrated view of calcium signaling. Biochem Biophys Res Commun 2015; 460: 114–121.
Rutkowski DT, Kaufman RJ . That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci 2007; 32: 469–476.
Yorimitsu T, Nair U, Yang Z, Klionsky DJ . Endoplasmic reticulum stress triggers autophagy. J Biol Chem 2006; 281: 30299–30304.
Liu S, Sarkar C, Dinizo M, Faden AI, Koh EY, Lipinski M M et al. Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell Death Dis 2015; 6: e1582.
Taniguchi M, Yoshida H . Endoplasmic reticulum stress in kidney function and disease. Curr Opin Nephrol Hypertens 2015; 24: 345–350.
Huang YY, Xia MZ, Wang H, Liu XJ, Hu YF, Chen YH et al. Cadmium selectively induces MIP-2 and COX-2 through PTEN-mediated Akt activation in RAW264.7 cells. Toxicol Sci 2014; 138: 310–321.
Morales AI, Vicente-Sanchez C, Jerkic M, Santiago JM, Sanchez-Gonzalez PD, Perez-Barriocanal F et al. Effect of quercetin on metallothionein, nitric oxide synthases and cyclooxygenase-2 expression on experimental chronic cadmium nephrotoxicity in rats. Toxicol Appl Pharmacol 2006; 210: 128–135.
Burada F, Nicoli ER, Ciurea ME, Uscatu DC, Ioana M, Gheonea DI . Autophagy in colorectal cancer: an important switch from physiology to pathology. World J Gastrointest Oncol 2015; 7: 271–284.
Watanabe T, Nagai A, Sheikh AM, Mitaki S, Wakabayashi K, Kim SU et al. A human neural stem cell line provides neuroprotection and improves neurological performance by early intervention of neuroinflammatory system. Brain Res 2016; 1631: 194–203.
Soliman E, Henderson KL, Danell AS, Van Dross R . Arachidonoyl-ethanolamide activates endoplasmic reticulum stress-apoptosis in tumorigenic keratinocytes: role of cyclooxygenase-2 and novel J-series prostamides. Mol Carcinog 2016; 55: 117–130.
Baba H, Tsuneyama K, Kumada T, Aoshima K, Imura J . Histopathological analysis for osteomalacia and tubulopathy in itai-itai disease. J Toxicol Sci 2014; 39: 91–96.
Xu S, Pi H, Chen Y, Zhang N, Guo P, Lu Y et al. Cadmium induced Drp1-dependent mitochondrial fragmentation by disturbing calcium homeostasis in its hepatotoxicity. Cell Death Dis 2013; 4: e540.
Fujiki K, Inamura H, Matsuoka M . PI3K signaling mediates diverse regulation of ATF4 expression for the survival of HK-2 cells exposed to cadmium. Arch Toxicol 2014; 88: 403–414.
Fujiki K, Inamura H, Matsuoka M . Detrimental effects of Notch1 signaling activated by cadmium in renal proximal tubular epithelial cells. Cell Death Dis 2014; 5: e1378.
Dudley RE, Gammal LM, Klaassen CD . Cadmium-induced hepatic and renal injury in chronically exposed rats: likely role of hepatic cadmium-metallothionein in nephrotoxicity. Toxicol Appl Pharmacol 1985; 77: 414–426.
Abouhamed M, Wolff NA, Lee WK, Smith CP, Thevenod F . Knockdown of endosomal/lysosomal divalent metal transporter 1 by RNA interference prevents cadmium-metallothionein-1 cytotoxicity in renal proximal tubule cells. Am J Physiol Renal Physiol 2007; 293: F705–F712.
Rafati A, Hoseini L, Babai A, Noorafshan A, Haghbin H, Karbalay-Doust S . Mitigating effect of resveratrol on the structural changes of mice liver and kidney induced by cadmium: a stereological study. Prev Nutrit Food Sci 2015; 20: 266–275.
Gong P, Chen FX, Wang L, Wang J, Jin S, Ma YM . Protective effects of blueberries (Vaccinium corymbosum L. extract against cadmium-induced hepatotoxicity in mice. Environ Toxicol Pharmacol 2014; 37: 1015–1027.
Stark JL . BUN/creatinine: your keys to kidney function. Nursing 1980; 10: 33–38.
Lee JI, Kim MJ, Park CS, Kim MC . Influence of ascorbic acid on BUN, creatinine, resistive index in canine renal ischemia-reperfusion injury. J Vet Sci 2006; 7: 79–81.
Kari FW, Yoshihara H, Thurman RG . Urea synthesis from ammonia in periportal and pericentral regions of the liver lobule. Effect of oxygen. Eur J Biochem 1987; 163: 1–7.
Guo M, Xu X, Yan X, Wang S, Gao S, Zhu S . In vivo biodistribution and synergistic toxicity of silica nanoparticles and cadmium chloride in mice. J Hazard Mater 2013; 260: 780–788.
Jiang M, Liu K, Luo J, Dong Z . Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol 2010; 176: 1181–1192.
Liu SY, Hartleben B, Kretz O, Wiech T, Igarashi P, Mizushima N et al. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012; 8: 826–837.
Scarlatti F, Granata R, Meijer AJ, Codogno P . Does autophagy have a license to kill mammalian cells? Cell Death Differ 2009; 16: 12–20.
Lin HH, Lin SM, Chung Y, Vonderfecht S, Camden JM, Flodby P et al. Dynamic involvement of ATG5 in cellular stress responses. Cell Death Dis 2014; 5: e1478.
Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004; 304: 1500–1502.
Heras-Sandoval D, Perez-Rojas JM, Hernandez-Damian J, Pedraza-Chaverri J . The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal 2014; 26: 2694–2701.
O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006; 66: 1500–1508.
Zhang C, Jia X, Wang K, Bao J, Li P, Chen M et al. Polyphyllin VII induces an autophagic cell death by activation of the JNK pathway and inhibition of PI3K/AKT/mTOR pathway in HepG2 cells. PloS One 2016; 11: e0147405.
Miyazaki M, McCarthy JJ, Esser KA . Insulin like growth factor-1-induced phosphorylation and altered distribution of tuberous sclerosis complex (TSC)1/TSC2 in C2C12 myotubes. FEBS J 2010; 277: 2180–2191.
Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 2004; 166: 213–223.
Fouillet A, Levet C, Virgone A, Robin M, Dourlen P, Rieusset J et al. ER stress inhibits neuronal death by promoting autophagy. Autophagy 2012; 8: 915–926.
Suddek GM, El-Kenawi AE, Abdel-Aziz A, El-Kashef HA . Celecoxib, a selective cyclooxygenase-2 inhibitor, attenuates renal injury in a rat model of cisplatin-induced nephrotoxicity. Chemotherapy 2011; 57: 321–326.
El-Gowelli HM, Helmy MW, Ali RM, El-Mas MM . Celecoxib offsets the negative renal influences of cyclosporine via modulation of the TGF-beta1/IL-2/COX-2/endothelin ET(B) receptor cascade. Toxicol Appl Pharmacol 2014; 275: 88–95.
Senbel AM, AbdelMoneim L, Omar AG . Celecoxib modulates nitric oxide and reactive oxygen species in kidney ischemia/reperfusion injury and rat aorta model of hypoxia/reoxygenation. Vasc Pharmacol 2014; 62: 24–31.
Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, celecoxib). J Med Chem 1997; 40: 1347–1365.
Whelton A, Maurath CJ, Verburg KM, Geis GS . Renal safety and tolerability of celecoxib, a novel cyclooxygenase-2 inhibitor. Am J Ther 2000; 7: 159–175.
Schoors D . A comparison of the renal and cardiovascular safety of celecoxib and rofecoxib. Clin Ther 2002; 24: 468–469; author reply 469–470.
Meng XX, Yao M, Zhang XD, Xu HX, Dong Q . ER stress-induced autophagy in melanoma. Clin Exp Pharmacol Physiol 2015; 42: 811–816.
Qin L, Wang Z, Tao L, Wang Y . ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010; 6: 239–247.
Kania E, Pajak B, Orzechowski A . Calcium homeostasis and ER stress in control of autophagy in cancer cells. Biomed Res Int 2015; 2015: 352794.
Lee H, Noh JY, Oh Y, Kim Y, Chang J W, Chung CW et al. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum Mol Genet 2012; 21: 101–114.
Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H et al. ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14: 230–239.
Cho HK, Cheong KJ, Kim HY, Cheong J . Endoplasmic reticulum stress induced by hepatitis B virus X protein enhances cyclooxygenase 2 expression via activating transcription factor 4. Biochem J 2011; 435: 431–439.
Chen S, Xuan J, Couch L, Iyer A, Wu Y, Li QZ et al. Sertraline induces endoplasmic reticulum stress in hepatic cells. Toxicology 2014; 322: 78–88.
Liao K, Xia B, Zhuang QY, Hou MJ, Zhang YJ, Luo B et al. Parthenolide inhibits cancer stem-like side population of nasopharyngeal carcinoma cells via suppression of the NF-κB/COX-2 pathway. Theranostics 2015; 5: 302–321.
Acknowledgements
We thank Drs. William Melchior and Lei Guo for English editing. This work was supported by grants from the National Natural Science Foundation of China (NSFC, Nos. 81172705, 81573181, 81402648 and 81472997), NSFC Key Program (No. 81130052), Early-stage Project of National Key Basic Research Program of China (No. 2014CB560710), the Natural Science Foundation of Fujian Province of China (Nos. 2014J01372 and 2015J01344), Project funded by Science Foundation of Xiamen City (No. 3502Z20140045), Project funded by Science Foundation of Fujian Province (No. 2014Y2004), the Education Scientific Research Project of Young Teachers in Fujian Province (No. JA14004), Xiamen Municipal Bureau of Ocean and Fisheries (No. 14PYY051SF04), the Shanhai Fund Project (No. 2013SH007) and principal funding (No. CXB2014012) of Xiamen University.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E Baehrecke
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Luo, B., Lin, Y., Jiang, S. et al. Endoplasmic reticulum stress eIF2α–ATF4 pathway-mediated cyclooxygenase-2 induction regulates cadmium-induced autophagy in kidney. Cell Death Dis 7, e2251 (2016). https://doi.org/10.1038/cddis.2016.78
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2016.78
- Springer Nature Limited
This article is cited by
-
Autophagy: a necessary defense against extreme cadmium intoxication in a multigenerational 2D experiment
Scientific Reports (2020)
-
Drp1 and RB interaction to mediate mitochondria-dependent necroptosis induced by cadmium in hepatocytes
Cell Death & Disease (2019)
-
Sirtuin-1 ameliorates cadmium-induced endoplasmic reticulum stress and pyroptosis through XBP-1s deacetylation in human renal tubular epithelial cells
Archives of Toxicology (2019)
-
Celecoxib alleviates nonalcoholic fatty liver disease by restoring autophagic flux
Scientific Reports (2018)
-
Initial autophagic protection switches to disruption of autophagic flux by lysosomal instability during cadmium stress accrual in renal NRK-52E cells
Archives of Toxicology (2017)