
Abstract
The biological function of E26 transformation-specific (ETS) transcription factor EHF/ESE-3 in human cancers remains largely unknown, particularly gastric cancer. The aim of this study was to explore the role of EHF in tumorigenesis and its potential as a therapeutic target in gastric cancer. By using quantitative RT-PCR (qRT-PCR), immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH) assays, we investigated the expression and copy number of EHF in a cohort of gastric cancers and control subjects. Specific EHF siRNAs was used to determine the biologic impacts and mechanisms of altered EHF expression in vitro and in vivo. Dual-luciferase reporter, chromatin immunoprecipitation (ChIP) and electrophoretic mobility shift assay (EMSA) assays were performed to identify its downstream targets. Our results demonstrated that EHF was significantly upregulated and frequently amplified in gastric cancer tissues as compared with control subjects. Moreover, EHF amplification was positively correlated with its overexpression and significantly associated with poor clinical outcomes of gastric cancer patients. We also found that EHF knockdown notably inhibited gastric cancer cell proliferation, colony formation, migration, invasion and tumorigenic potential in nude mice and induced cell cycle arrest and apoptosis. Importantly, we identified EHF as a new HER2 transcription factor and the modulator of HER3 and HER4 in gastric cancer. Collectively, our findings suggest that EHF is a novel functional oncogene in gastric cancer by regulating the human epidermal growth factor receptor (HER) family of receptor tyrosine kinases and may represent a potential prognostic marker and therapeutic target for this cancer.
Similar content being viewed by others


Main
Gastric cancer is the fourth most common cancer and the second leading cause of cancer-related deaths in the world.1, 2 Given that most of patients are usually diagnosed at advanced stages in developing countries and the outcomes are still poor, the early diagnosis is thus extremely important for good prognosis.3, 4 Like other cancers, multiple genetic and epigenetic alterations have a critical role in the pathogenesis of gastric cancer. Therefore, identification of prognostic and/or therapeutic targets may improve early diagnosis and treatment efficacy for this disease.
The E26 transformation-specific (ETS) transcription factor family is one of the largest transcription factor families, which can be structurally categorized into 11 subfamilies including ETS, ERG, ELG, ELF, ERF, TEL, PEA3, SPI, TCF, PDEF and ESE.5, 6, 7, 8 All ETS factors are characterized by the ETS domain, which is a highly conserved DNA-binding domain comprising ~85 amino acids that form the winged helix-turn-helix DNA-binding motif, and recognizes the core consensus DNA sequence 5′-GGA(A/T)-3′ (ETS-binding site, EBS).9, 10 Some of them have been found to alter expression patterns involving diverse mechanisms, such as gene fusion and chromosomal rearrangements,11, 12 amplification and/or overexpression,13, 14 point mutations.15 Thus, the ETS factors have a key role in various diseases, including cancers.8, 16, 17
EHF/ESE-3 is new member of the ETS transcription factors. Like ESE-1 and ESE-2, it is exclusively expressed in a subset of epithelial cells.18 The previous studies have showed that altered expression of EHF may affect the normal process of epithelial cell differentiation and contribute to cell transformation.18, 19, 20 Moreover, EHF may regulate epithelial growth and differentiation and have an important role in oncogenesis of epithelium-derived tumors.18, 21 EHF has been demonstrated to function as a potential tumor suppressor gene in prostate cancer and esophageal squamous cell carcinoma (ESCC),19, 22, 23 and be frequently downregulated by promoter methylation.22 On the other hand, it is overexpressed in ovarian and mammary cancers24, 25, 26 and may be a predictive marker for poor survival in ovarian cancer.24, 25 However, the roles of EHF in gastric tumorigenesis are not fully understood.
In this study, we found frequent EHF overexpression and genomic amplification in gastric cancers, and demonstrated EHF amplification was one of the major mechanisms for its overexpression. Importantly, we for the first time revealed a close association of EHF amplification with poor patient survival. EHF downregulation significantly reduced in vitro and in vivo oncogenic potential of gastric cancer cells by regulating HER family of receptor tyrosine kinases.
Results
EHF is highly expressed and amplified in gastric cancers
We first examined EHF expression in a cohort of tumor tissues including gastric cancers, gliomas, lung cancers and thyroid cancers and non-cancerous tissues. As shown in Figure 1a and Supplementary Figure 1, compared with matched non-cancerous gastric tissues, EHF was significantly upregulated in gastric cancer tissues at both mRNA and protein levels. Similarly, we found that EHF expression was higher in gastric cancer cell lines than gastric mucosal epithelial cell line GES-1 (Supplementary Figure 2a and b). Moreover, EHF was also significantly upregulated in gliomas (P<0.0001), lung cancers (P=0.005) and thyroid cancers (P=0.001) compared with control subjects. We also investigated EHF expression in a total of 384 stomach adenocarcinomas using TCGA data set from the Cancer Browser database.27 As expected, EHF expression in tumor tissues was significantly higher than that in normal controls (P<0.0001; Figure 1b). In addition, we found that EHF expression was positively correlated with poor survival in gastric cancer patients (data from the Kaplan–Meier Plotter; P=0.003; Figure 1c).

EHF is highly expressed in multiple cancers. (a) qRT-PCR assay was performed to evaluate mRNA expression of EHF in primary gastric cancers (T) and their matched non-cancerous gastric tissues (N; n=30), gliomas (G; n=18) and meningiomas (M; n=19), primary lung cancers (T) and their matched non-cancerous tissues (N; n=18), as well as primary thyroid cancers (T) and their matched non-cancerous tissues (N; n=20), respectively. EHF expression was normalized with 18S rRNA levels. (b) High expression of EHF in gastric cancers (T) compared with normal gastric tissues (N) in TCGA data set. Horizontal lines indicate the median. (c) Significant correlation between EHF expression and the survival of gastric cancer patients in the Kaplan–Meier Plotter database
Given that genomic amplification is one of the major causes of oncogene overexpression in human cancers including gastric cancer,28 we analyzed the copy number of EHF in 131 gastric cancers and 37 control subjects by using qRT-PCR assay. EHF copy number corresponding to each individual case was presented in Figure 2a. Further analysis showed that EHF copy number in gastric cancers was significantly higher than that in control subjects (P<0.0001). We also analyzed EHF copy number in cell lines. The results showed that EHF copy number was higher in BGC823 and SGC7901 cells than GES-1, MGC803 and AGS cells (Supplementary Figure 2c). With a gene copy number of 4 or more defined as gene amplification, EHF amplification was detected in 55 of 131 (41.98%) gastric cancers, but not in control subjects. This finding was supported by fluorescence in situ hybridization (FISH) analysis (Figure 2b).

EHF is frequently amplified in gastric cancers. (a) Real-time quantitative PCR was performed to analyze EHF copy number in a cohort of gastric cancers (T) and control subjects (N). Horizontal lines indicate mean±S.E. (b) Bicolor FISH analysis demonstrates EHF amplification (red signals) in primary gastric cancer tissues by using EHF DNA probe, and reference centromeric probe on chromosome 11 (CEN11) was shown in green. Arrows indicate the cells with EHF amplification. Magnification for each set: × 1000. Scale bars, 200 μm. (c) Increasing extent of specific staining (brown color) was associated with increasing EHF copy number (number inside brackets). Shown are representative images of IHC on gastric cancer histologic slides using anti-EHF antibody. Magnification for each set: × 400. Scale bars, 200 μm. (d) Linear regression analysis was performed to evaluate the relationship between EHF immunohistostaining score and EHF copy number on 16 randomly selected gastric cancer cases (R2=0.71). (e) EHF mRNA expression was analyzed by using qRT-PCR assay in primary gastric cancers (n=30) grouping with EHF amplification and matched normal tissues (N). 18S rRNA was used as a normalized control. Data are presented as mean±S.E. (f) Kaplan–Meier survival curves were used to assess patient survival. The patients with EHF amplification (Am+) had significantly shorter survival times than those without EHF amplification(Am−) (left panel). When the patients with residual cancers were excluded, the patients with EHF amplification still had significantly poor survival compared with those without EHF amplification (right panel). Statistically significant differences were indicated: *P<0.05; ***P<0.001
To test the relationship between of EHF copy number and its expression, we randomly selected 16 gastric cancer cases with different EHF copies and did immunohistochemistry (IHC) for EHF. Increased staining of EHF was seen with increased EHF copies (Figure 2c). Linear regression analysis on the 16 cases revealed a positive correlation between the IHS score and copies of EHF (R2=0.71; Figure 2d). Similarly, our data showed that there was a significantly positive relationship between mRNA expression of EHF and its genomic amplification (P=0.017; Figure 2e). However, EHF expression was still higher in the cases without EHF amplification than matched normal gastric tissues (P<0.001), suggesting the existence of other possible mechanisms leading to its overexpression.
EHF amplification is associated with poor clinical outcomes in gastric cancer
We investigated the association of EHF amplification with clinicopathologic features and clinical outcomes in a cohort of gastric cancers. As shown in Supplementary Table 1, EHF amplification was significantly positively associated with differentiation (P=0.049), lymph node metastasis (P=0.029) and survival status (P=0.001). We further conducted a multiple multivariable logistic regression (Supplementary Table 2). As exppected, after adjustment, EHF amplification was still closely correlated with survival status (OR=3.748, 95% CI=1.525–9.214; P=0.004).
Next, Kaplan–Meier method was used to determine the effect of EHF amplification on patient survival. The data showed that the patients with EHF amplification had significantly shorter median survival times than those without EHF amplification (32.2 months versus 62.0 months; P=0.0002; Figure 2f, left panel). Accumulated evidences have demonstrated that residual tumor after surgery is an independent risk factor for gastric cancer patients.28 Thus, we excluded the cases with residual tumor to evaluate the effect of EHF amplification on patient survival. The results showed that EHF amplification still significantly shortened patient survival times (34.4 months versus 63.8 months, P=0.001; Figure 2f, right panel). Moreover, univariate cox regression analysis indicated that EHF amplification was significantly associated with poor survival with a hazard ratio (HR) of 2.593 (95% CI=1.583–4.249; P=0.0002; Table 1). Multivariate cox regression analysis further demonstrated that EHF amplification might be an independent prognostic factor in gastric cancer (HR=2.426; 95% CI=1.474–3.991; P<0.0001; Table 1).
EHF promotes gastric cancer cell growth
To elucidate the role of EHF in gastric carcinogenesis, we tested the growth-suppressive effect by knocking down EHF expression in gastric cancer cell lines AGS, BGC823 and SGC7901 using siRNA approach. EHF knockdown by two different EHF siRNAs (si-EHF-979 and si-EHF-309) was confirmed by qRT-PCR and western blot assays (Figure 3a). These two specific siRNAs significantly inhibited cell proliferation (Figure 3b) and colony-forming ability in monolayer culture (Figure 3c) compared with control siRNA (si-NC). Conversely, ectopic expression of EHF in GES-1 and MGC803 cells significantly promoted cell proliferation compared with empty vector (Supplementary Figure 3). Altogether, these results suggest the growth-promoting role of EHF in gastric tumorigenesis.

EHF knockdown inhibits cell growth and induces cell cycle arrest and apoptosis in gastric cancer cells. (a) Knockdown of EHF mRNA (left panels) and protein (right panels) by using two different siRNAs (si-EHF-979 and si-EHF-309) in gastric cancer cell lines was evidenced by qRT-PCR and western blot assays, respectively. 18S rRNA was used as a normalized control for qRT-PCR assay. GAPDH was used as loading control in western blot analysis. (b) EHF knockdown significantly inhibited cell proliferation in gastric cancer cells. (c) Left panel shows the representative images of colony formation in cells transfected with si-EHF or si-NC. Quantitative analysis of colony numbers is shown in right panel. (d) The indicated cells were transiently transfected with si-EHF-979 or si-NC. DNA content was measured by flow cytometry to determine cell cycle fractions. (e) Apoptotic cells including early and late apoptotic cells were measured 72 h after transfection by flow cytometry analysis of Annexin V-FITC/PI double-labelled cells. The data were presented as mean±S.E. of values from three independent experiments. Statistically significant differences were indicated: *P<0.05; **P<0.01; ***P<0.001
EHF knockdown induces gastric cancer cell cycle arrest and apoptosis
We next evaluated the effect of EHF knockdown on cell cycle distributions and apoptosis in gastric cancer cells. The results showed that cell cycle was arrested at the S phase in si-EHF-979 transfected cells compared with si-NC transfected cells (Figure 3d). This effect could be reversed by exogenous overexpressing EHF in these three cell lines (Supplementary Figure 4). In addition, si-EHF-979 transfection caused an increase in the numbers of apoptotic cells compared with control cells (Figure 3e).
EHF knockdown inhibits gastric cancer cell growth in nude mice
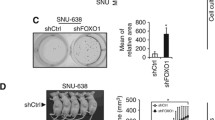
We assessed the effect of EHF knockdown on the growth of xenograft tumors in nude mice. As shown in Figure 4a, tumors induced by si-EHF-979 transfected BGC823 cells showed significantly longer latency and smaller mean tumor volume than tumors induced by control cells. At the end of experiments, tumors were isolated and weighed. The mean weight of si-EHF-979 transfected cell-derived tumors was significantly less compared with control tumors (P=0.002; Figure 4b). Moreover, we analyzed the expression levels of EHF in xenogaft tumors by using qRT-PCR and western blot assays. As shown in Supplementary Figure 5, EHF expression was significantly decreased in si-EHF-979 group compared with si-NC groups at both mRNA and protein levels. These data suggest that transient transfection of si-EHF-979 may knockdown EHF expression and inhibit the growth of xenogaft tumors within 15 days after injection, although there may be off-target effect. To assess the proliferation index in the xenograft tumor, tumor sections were stained with anti-Ki-67 antibody. The percentage of Ki-67-positive cells was significantly decreased in si-EHF-979 transfected cell-derived tumors compared with control tumors (P<0.001; Figure 4c). These observations further support that EHF is a functional oncogene in gastric cancer cells.

EHF knockdown inhibits xenograft tumor growth. (a) Subcutaneous tumor growth curve of si-EHF-979 transfected BGC823 cells in nude mice was compared with si-NC transfected cells. The si-EHF-979 group showed a retarded tumor growth compared to the si-NC group. Data are shown as mean±S.D. (n=6 per group). (b) A representative picture for tumor growth of cells transfected with the indicated siRNA in nude mice (upper panel). Histogram represents mean of tumor weight from the si-EHF-979 and si-NC groups (lower panel). Data are shown as mean±S.D. (n=6 per group). (c) Shown is representative Ki-67 staining of xenograft tumors from the si-EHF-979 and si-NC groups(left panels). Histogram represents mean±S.E. of the percentage of Ki-67-positive cells from five microscopic fields in each group (right panel). Magnification for each set: × 400. Scale bars, 200 μm. Statistically significant differences were indicated: **P<0.01; ***P<0.001
EHF enhances gastric cancer cell migration and invasion
We also attempted to test the effects of EHF on gastric cancer cell migratory and invasive ability. There were a significantly lower number of migrated/invaded cells in si-EHF-979 transfected cells than si-NC transfected cells (Figure 5a). Conversely, ectopic expression of EHF in MGC803 cells significantly enhanced the migration and invasive potential of cancer cells (Supplementary Figure 6). Given an important role of matrix metalloproteinases (MMPs) in cell invasiveness,29 we next tested the effect of EHF on the expression of MMP-2, -7, -9 and -14. EHF knockdown significantly inhibited the expression of these genes in at least one cell lines (Figure 5b), whereas overexpressing EHF in MGC803 cells increased the expression of these genes (Supplementary Figure 7a). These data indicate that metastasis-associated phenotypes may be link to the regulation of MMPs by EHF in gastric cancer.

EHF knockdown inhibits gastric cancer cell migration and invasion. (a) The representative images of migrated/invaded cells (upper panels). Histograms, corresponding to upper panels, show means±S.E. of cell numbers from three independent assays (lower panels). Magnification for each set: × 200. Scale bars, 50 μm. (b) qRT-PCR was performed to test the effect of EHF depletion on the expression of metastasis-related genes MMP2, 7, 9 and 14 in gastric cancer cells. Expression levels of these genes were normalized with 18S rRNA levels. Data were presented as mean±S.E. *P<0.05;**P<0.01; ***P<0.001
We further tested the effect of EHF on the process of epithelial–mesenchymal transition (EMT), which is one of the critical steps during tumor metastasis including gastric cancer.30 As shown in Figure 6a, knocking down EHF expression in AGS, BGC823 and SGC7901 cells increased E-cadherin expression, and decreased Vimentin expression. Conversely, overexpressing EHF in MGC803 cells decreased E-cadherin expression and increased Vimentin expression (Supplementary Figure 7b). These findings were also supported by immunofluorescence (IF) assay in Figure 6b. Collectively, these results suggest that EHF contributes to gastric cancer cell metastasis via promoting EMT process.

EHF knockdown inhibits EMT process in gastric cancer cells. (a) The expression of E-cadherin and Vimentin was determined in the indicated cells by western blot analysis. GAPDH was used as loading control. (b) Immunofluorescence staining was then used to assess the expression of E-cadherin and Vimentin proteins in cells transfected with si-EHF-979 or si-NC. Red color represents target protein fluorescence and blue color represents Hoechst33342 staining for nuclei. Magnification for each set: × 400. Scale bars, 100 μm
EHF modulates the activities of the HER family of receptor tyrosine kinases in gastric cancer cells
The HER family includes four members: EGFR/HER1, HER2, HER3 and HER4.31 Alterations in HER family play a critical role in the progression and survival of many cancers including gastric cancer.32 In addition, we found the core GGAA/T motif (EBS) in the promoters of EGFR, HER2, HER3 and HER4 by analyzing their promoter sequences using MatInspector online software (http://www.genomatix.de/online_help/help_matinspector/matinspector_help). Thus, we attempted to determine whether oncogenic role of EHF in gastric cancer is associated with the activation of HER family. As shown in Figure 7a, EHF expression was significantly positively correlated with the expression of EGFR (R=0.43; P=0.018), HER2 (R=0.51; P=0.004) and HER3 (R=0.39; P=0.034) in gastric cancers. Similarly, we found a positive association of EHF expression with the expression of HER2 (R=0.29; P<0.0001) and HER3 (R=0.36; P<0.0001) by using TCGA dataset from the Cancer Browser database (Supplementary Figure 8). In addition, EHF knockdown dramatically decreased the expression of HERs in the indicated cell lines compared with the controls, particularly HER2 and HER3 (Figure 7b and c). On the other hand, overexpressing EHF in MGC803 cells upregulated the expression of HER2-4 (Supplementary Figure 9). Interestingly, we found that the expression of BMP1, BMP4 and c-Met was decreased by EHF knockdown in at least two cell lines compared with the controls (Supplementary Figure 10). As expected, EBS could also be found in their promoters.

EHF regulates the expression of HER receptors and the activities of their downstream signaling pathways in gastric cancer cells. (a) qRT-PCR assay was used to evaluate mRNA expression of EHF and HER receptors in primary gastric cancers (n=30). Linear regression analysis was performed to assess the correlations between them. 18S rRNA was used as a normalized control. (b) qRT-PCR assay was performed to investigate the effect of EHF knockdown on the expression of HER receptors. Expression levels of these genes were normalized with 18S rRNA levels. Data were presented as mean±S.E. (c) The effect of EHF depletion on the expression of HER2-4 was determined in the indicated cells by western blot analysis. GAPDH was used as loading control. (d) Cells transfected with si-EHF-979 or si-NC were lysed and lysates were subjected to western blot analysis. The antibodies against phospho-Erk (p-Erk), total Erk (t-Erk), phospho-Akt (p-Akt) and total Akt (t-Akt) were used to determine the effect of EHF knockdown on the activities of the MAPK/Erk and PI3K/Akt cascades. GAPDH was used as a loading control
Increasing evidences have demonstrated that overexpression of HER family members leads to the activation of downstream pathways including the MAPK/Erk and PI3K/Akt pathways.33 As expected, EHF knockdown significantly inhibited the activities of both pathways in gastric cancer cells, characterized by reduced phosphorylation of Erk (p-Erk) and Akt (p-Akt) (Figure 7d). This was supported by a very recent study that the levels of p-Erk and p-Akt were inhibited by EHF knockdown in ovarian cancer cells.25
To examine whether EHF was indeed involved in directly regulating promoter activities of HER2-4, we cloned their promoters into a pGL3-Basic luciferase plasmid to construct luciferase reporter plasmids including pGL3-HER2-Luc (−607/+11), pGL3-HER3-Luc (−997/+440) and pGL3-HER4-Luc (−697/+306). The results showed that ectopic expression of EHF was able to significantly increase promoter activity of HER2, but not HER3 and HER4, in BGC823 cells (Figure 8a and Supplementary Figure 11a). Next, to further identify HER2 promoter core region, three different lengths of HER2 promoter region (F1: −607/+11; F2: −175/+11; F3: −607/−175 bp) were inserted into the pGL3-Basic luciferase plasmid, and co-transfected into BGC823 cell with pcDNA3.1(-)A-EHF or empty vector, respectively. The results showed that pGL3-HER2-Luc-F1 and -F2 exhibited strong luciferase activity, but not pGL3-HER2-Luc-F3, compared with pGL3-Luc-Basic (Figure 8a). On the other hand, EHF knockdown significantly decreased the promoter activity of HER2 (P<0.01) (Figure 8b), but not HER3 (Supplementary Figure 11b), in BGC823 cells. In addition, we found that the promoter activity of HER2 in HEK293T cells was increased with the increased amounts of EHF-expressing plasmid (Figure 8c). Taken together, these observations suggest that EHF may be a potential transcription factor of HER2, and the regulation site of EHF is located at −175/+11 of HER2 gene.

EHF is identified as a new HER2 transcription factor and the modulator of HER3 and HER4 in gastric cancer. (a) BGC823 cells were transiently transfected with pGL3-Basic or luciferase reporter constructs containing various lengths of the promoter region of HER2 gene, as indicated (F1: −607/+11; F2: −175/+11; F3: −607/−175) (left panels). Cotransfection with empty vector was used as a control. The ratio of the Luc/Renilla activity is shown as means±S.E. of three independent assays (right panels). (b) The luciferase reporter gene assay was performed to evaluate the effect of EHF knockdown on promoter activity of HER2 in BGC823 cells. The ratio of the Luc/Renilla activity is shown as means±S.E. of three independent assays. (c) HEK293T cells were cotransfected pGL3-HER2-Luc-F1 and various amounts of pcDNA3.1(-)A-EHF or empty vector, respectively. Promoter activities of HER2 were measured by luciferase reporter gene assays. All the ratio of the Luc/Renilla activity is shown as means±S.E. of three independent assays. (d) Putative promoter regions of HER2 (−607/+11), HER3 (−997/+440) and HER4 (−697/+306) were inserted into the pGL3-Basic to construct the luciferase reporter plasmid pGL3-HER2-Luc, pGL3-HER3-Luc and pGL3-HER4-Luc (upper panels). P1-P7 represent the regions analyzed by ChIP assays for HER2, HER3 and HER4, respectively. BGC823 cells were transiently transfected with pcDNA3.1/myc-His(-)A-EHF or empty vector, and were subjected to ChIP-qRT-PCR assays using anti-Myc tag antibody. Flod enrichment was shown as means±S.E. of three independent assays (lower panels). (e) EMSA assay was performed to confirm the interaction between EHF and HER2 promoter. Shown are specific DNA-binding of in vitro translated EHF protein to an oligonucleotide sequence (SH2) containing ETS responsive element (GAGGAA) from the HER2 promoter. Unlabeled mutated probes contain specific mutations in the GGAA ETS core or flanking nucleotides of core sequence, as indicated by MT1 and MT2. Unlabeled wild-type (WT) and mutated (MT1 or MT2) competitor probes were added at 100-fold molar excess. Statistically significant differences were indicated: *P<0.05; **P<0.01; ***P<0.001
Next, we attempted to explore whether the activity of HER2 was regulated by EHF through directly binding to its promoter. Thus, the chromatin immunoprecipitation (ChIP) assay was performed in BGC823 cells transfected with pcDNA3.1/myc-His(-)A-EHF and empty vector using anti-Myc tag antibody, followed by qRT-PCR targeting their promoter regions. As expected, EHF strongly bound to HER2 promoter and weakly bound to HER3 promoter, but not HER4 in BGC823 cells (Figure 8d). Three different fragments within HER2 promoter (P1: −604/−484; P2: −274/−155; P3: −147/-37) were all enriched by 8.14-fold on average in pcDNA3.1/myc-His(-)A-EHF-transfected cells compared with vector-transfected cells (P<0.001; Figure 8d). To be consistent with the dual luciferase findings, the ChIP assays further support HER2 as a target of EHF.
To test whether EHF directly interacts with HER2 promoter, an oligonucleotide sequence (SH2) containing the putative Ets binding site (EBS; GAGGAA) from HER2 promoter was used to assess DNA-binding and transactivation by in vitro-translated full-length EHF protein using EMSA assay. This oligonucleotide sequence has previously been demonstrated to be responsive to another ETS factor ESE1/ESX.34 As shown in Figure 8e, full-length EHF protein exhibited high-affinity, sequence-specific binding to SH2. Moreover, unlabeled specific competitor probes (WT) completely competed with SH2 for EHF binding. As seen with other ETS factors, unlabeled competitor probes (MT1; 100 fold) with mutations in the GGAA ETS core of SH2 failed to compete against SH2 for EHF binding, whereas those (MT2; 100 fold) with mutations in flanking nucleotides of core sequence were relatively effective at competing for EHF binding. To determine the role of HER2 in growth-promoting effect of EHF on gastric cancer cells, we knocked down HER2 expression in GES-1 and MGC803 cells overexpressing EHF. The results showed that proliferation-promoting effect of EHF were significantly attenuated upon HER2 depletion (Figure 9). Altogether, our data suggest that EHF promotes gastric tumorigenesis through transcriptionally regulating HER2 expression via binding to GGAA core sequence within its promoter.

HER2 depletion attenuates proliferation-promoting effect of EHF in gastric cancer cells. Inhibitory effect of HER2 depletion on cell proliferation in GES-1 and MGC803 cells overexpressing EHF were evaluated by MTT assay. The data were presented as mean±S.E. Statistically significant differences were indicated: *P<0.05; **P<0.01; ***P<0.001
Discussion
In this study, we first provided strong evidences supporting the oncogenic activities of EHF in gastric cancer. First, EHF was frequently overexpressed and amplified in gastric cancers compared with matched non-cancerous gastric tissues. Second, EHF amplification (or overexpression) was significantly associated with poor clinical outcomes and may be used as a potential prognostic marker for gastric cancer patients. Third, knocking down EHF expression in gastric cancer cells significantly inhibited cell growth and invasiveness. Fourth, EHF was identified to be a new transcription factor of HER2, and also modulated the expression of HER3 and HER4 in gastric cancer.
It has been well documented that genomic amplification is one of the major causes of oncogene overexpression in human cancers.28, 35 To identify the mechanisms that may contribute to EHF overexpression in gastric cancers, we examined EHF copy number and its mRNA/protein expression in gastric cancers, and demonstrated a significantly positive relationship between them, suggesting that EHF amplification is one of the major mechanisms of EHF overexpression in gastric cancers. In addition, our data showed that EHF amplification dramatically affected patient survival, implicating that it may be used as a potential prognostic marker for gastric cancer patients. However, our data also showed that EHF overexpression did not always coincide with its genomic amplification, suggesting that there are other possible mechanisms may contribute to its overexpression.
Although a previous study has reported frequent EHF amplification in gastric cancers,36 the role and mechanisms of EHF in gastric tumorigenesis remain totally unknown. We thus tested its oncogene function in gastric cancer by a series of in vitro and in vivo studies. As expected, EHF knockdown in gastric cancer cells showed significant growth-inhibitory effect by inhibition of cell proliferation and colony formation in vitro and tumorigenic potential in nude mice in vivo. EHF knockdown induced cell cycle arrest and apoptosis, and inhibited cell migration, invasion and EMT process in gastric cancer cells. We also demonstrated that knocking down EHF expression in gastric cancer cells significantly inhibited expression of MMP-2, -7, -9 and -14 genes, suggesting that the decrease in the metastasis-related phenotypes may be associated with suppression of expression or activities of MMPs. This is supported by previous studies that MMPs genes are key targets of ETS proteins and Ets-mediated induction of these genes contributes to the invasive and angiogenic phenotypes of malignant cells, such as PEA3, ETS1, and ETS2.37, 38 These findings further suggest that EHF may be a potential oncogene in gastric cancer cells.
As a member of the ETS family, downstream targets/pathways of EHF in gastric cancer remain to be identified. A previous study has reported that there is a conserved ETS-responsive element (GAGGAA) within a DNase I hypersensitive site in the proximal HER2 promoter and demonstrated that it can be recognized by an ETS-immunoreactive factor in HER2-expressing breast cancer cells.39 However, although more than 10 different ETS factors have been found to be co-expressed with HER2 in cancer cells, only a few ETS family members such as Elf-1, PEA3 and ESE1 have been studied as potential HER2 transactivators.34, 40, 41, 42 The core GGAA/T motif (ETS binding site, EBS) was found in the HER2 promoter by using MatInspector online software. To be consistent with this, we found that there were positive correlations between the expression of EHF and HER receptors in a cohort of gastric cancers including EGFR, HER2 and HER3, particularly HER2, in a cohort of gastric cancers, as supported by the information from TGCA database. Down-regulating EHF expression in gastric cancer cells significantly reduced mRNA expression of HER2-4. However, the luciferase reporter gene assays demonstrated that ectopic expression of EHF only enhanced promoter activity of HER2, but not HER3 and HER4, in gastric cancer cells. These data suggest that HER2 may be a potential downstream target of EHF, whereas HER2 and HER4 may be indirect targets of EHF, as supported by the ChIP assay. Accordingly, EHF knockdown remarkably inhibited the activity of their downstream signaling pathways such as the MAPK/Erk and PI3K/Akt pathways. Collectively, EHF may be a new transcription factor for HER2 in gastric cancer by binding to a functional EBS within its promoter, and promotes gastric tumorigenesis by activating HER family of receptor tyrosine kinase.
EHF has been demonstrated to be a potential tumor-suppressor in prostate cancer and ESCC,19, 22, 23 whereas our data suggest that EHF may be a functional oncogene in gastric cancer. We thus speculate that EHF derived from different types of cells or tissues has strikingly distinct functional activities in tumorigenesis. It is the fact that the DNA binding domains of ETS factors are very similar, thus their specific roles in tumorigenesis are largely dependent on other factors including interaction with other nuclear factors such as transcription factors, co-activators or co-repressor.43, 44 Moreover, the activities of ETS proteins are also regulated by post-translational modifications such as phosphorylation, acetylation, sumoylation, ubiquitinylation and glycosylation.45
In summary, we found frequent overexpression and amplification of EHF in gastric cancers and revealed a strong association of EHF overexpression/amplification with poor patient outcomes. Moreover, our data support an oncogenic role of EHF as a HER2 transcription factor and the modulator of HER3 and HER4 in gastric tumorigenesis. A better understanding of the physiologic and pathologic function of EHF will significantly improve our knowledge of the pathogenesis of gastric cancer, and targeting this frequently overexpressed/amplified oncogene may elucidate the effective treatment of this cancer in the future.
Materials and Methods
Clinical samples
A cohort of primary cancer tissues and matched non-cancerous tissues or benign tumors were obtained from patients who underwent surgery at the First Affiliated Hospital of Xi’an Jiaotong University, including gliomas, gastric, lung and thyroid cancers. Moreover, a total of 131 paraffin-embedded gastric cancer tissues were randomly obtained from the First Affiliated Hospital of Xi’an Jiaotong University between January 1999 and December 2005, with a follow-up period of 10 years after the surgery. Normal controls from 37 patients with chronic gastritis who underwent endoscopic biopsy were also obtained from the same hospital. Informed consent was obtained from each patient before the surgery. All patients did not receive chemotherapy and radiotherapy before the surgery, and all sections were histologically examined by a senior pathologist at Department of Pathology of the Hospital based on World Health Organization criteria. Clinicopathological data were collected from the patients’ files or by interview with the patients or their relatives and were summarized in Supplementary Table 1. The study protocol was approved by the Human Ethics Committee of the First Affiliated Hospital of Xi’an Jiaotong University.
Cell culture and short interfering RNA (siRNA) cell transfection
Human gastric cancer cell lines AGS, BGC823, MGC803 and SGC7901, human immortalized gastric mucosal epithelial cell line GES-1 and embryonic kidney cell line 293T were used in this study. These cells were maintained in RPMI medium 1640 or Dulbecco's modified Eagle medium (Gibco, Grand Island, NY) with 10% fetal bovine serum (FBS; Hyclone, Logan, UT). Oligonucleotides of target-specific and control siRNAs were obtained from GenePharma (Shanghai, P.R. China) and the sequences were presented in Supplementary Table 3. Cells were transfected at 70% confluence using Lipofectamine 3000 (Invitrogen, Grand Island, NY), with a final siRNA concentration of 50 nM. Specific oligonucleotides with maximal knockdown efficiency were selected among three different sequences until use. All silencing experiments were performed in three replicates.
EHF expression plasmid construction
To construct EHF expression plasmid, total RNA from GES-1 cell line was isolated by TRIzol Reagent following manufacturer’s instruction (Invitrogen, Grand Island, NY). The cDNA was reverse transcribed by using PrimeScript™ II 1st Strand cDNA Synthesis Kit (Takara Inc., Dalian, P.R. China). The full-length open reading frame (ORF) of human ESE with or without stop codon TGA was amplified and then cloned into pcDNA3.1(−)A mammalian expression vector with a Myc-His tag (Invitrogen, Grand Island, NY), which was designated as pcDNA3.1/myc-His(−)A-EHF or pcDNA3.1(−)A-EHF.
RNA extraction and quantitative RT-PCR (qRT-PCR)
Total RNA from tissues and cell lines were extracted using Trizol reagent (Takara Inc., Dalian, P.R. China) following the manufacturer’s protocol. The cDNA was synthesized with 500 ng total RNA by using PrimeScript RT reagent Kit (Takara Inc., Dalian, P.R. China). Quantitative RT-PCR (qRT-PCR) was carried out on a CFX96 Thermal Cycler DiceTM real-time PCR system (Bio-Rad Laboratories, Inc., CA) using SYBR Premix Ex TaqTM (Takara Inc., Dalian, P.R. China). The mRNA expression of the indicated genes was normalized to 18S rRNA cDNA. Each sample was run in triplicate. The primer sequences were presented in Supplementary Table 4.
Tissues and DNA preparation
Paraffin-embedded serial sections were cut at intervals of 5 μm. One of sections was stained by hematoxylin and eosin (H&E) and was marked as a tumor representative tissue by a senior pathologist for gastric cancer. Tumor tissues were isolated by manual microdissection under an inverted microscope using the marked H&E section as target tissue identification. DNA was extracted from isolated tissues as previously described.28 The fresh gastric tissues were collected and immediately frozen in liquid nitrogen and stored at −80 °C prior to DNA extraction. DNA was then isolated using standard phenol/chloroform protocol.
Copy number analysis
Real-time quantitative PCR was performed to analyze EHF copy number in gastric cancers and control subjects as described previously.28, 46 The TaqMan probe for EHF was 5′-6FAM-AAC CTG CCT TTCTGC TTT TCA TCA GAC CC-TAMRA-3′, and the primers were 5′-CCT ATC TTTGCT GTG ACT TAG ATC ATT AG-3′ (forward) and 5′-CGG ATG AAT TCCCAT AAG TGA GT-3′ (reverse). The TaqMan probe and primers for β-actin were described previously.28 Each sample was run in triplicate, and β-actin was run in parallel to normalize the input DNA. Standard curves were established using serial dilutions of normal leukocyte DNA. EHF amplification was defined by a copy number ≥4.
Fluorescence in situ hybridization
The FISH analysis was performed on formalin-fixed, paraffin-embedded gastric cancer tissues and matched non-cancerous tissues using the EHF DNA probe/CEN11 probe mixture (Exon Biotechnology Inc, Guangzhou, P.R. China). Briefly, the paraffin-embedded tissue slides were deparaffinized through xylene, and rehydrated in an ethanol series (100, 85 and 70%), and treated with protenase K solution (200 μg/ml) and pepsin (0.005% in 0.01 M HCl solution) at 37 °C, respectively. The slides were then dehydrated in an ethanol series (70, 85 and 100%), and the probe mixture was added to the slides and immediately covered by coverslips and sealed the edges with rubber cement. The slides were subsequently denatured at 85 °C for 5 minutes and incubated at 37 °C overnight. After hybridization, the slides were washed in 2 × SSC, 2 × SSC/0.1% NP-40 buffer at 37 °C for 5 min each, and were counterstained with DAPI antifade solution. FISH signals in 20–30 cells for each specimen were counted, and the criteria for gene amplification were defined when FISH signals were detected by tested probes compared with control probes ≥1.5. Fluorescence images were captured with Olympus IX71 microscope (Olympus, Tokyo, Japan), which enables simultaneous detection of both FITC and Texas Red fluorescence. The color mergence was performed using ImageJ image software (ImageJ version 1.44p, NIH, MD).
Immunohistochemistry
Immunohistochemical analysis was performed as previously described.28, 46 Briefly, after dewaxing in xylene and rehydrating in a gradient concentration of ethanol, the paraffin-embedded tissue slides were incubated in 0.3% hydrogen peroxide in distilled water to block endogenous peroxidase activity, and treated with an antigen retrieval method by heating, and were then incubated with rabbit anti-EHF antibody (Abcam, Inc; 1 : 100) or mouse anti-Ki67 antibody (BD Biosciences, Inc; 1 : 200) overnight at 4 °C. Subsequently, the slides were incubated with biotinylated goat anti-rabbit IgG or goat anti-mouse IgG (ZSGB-bio, Beijing, P.R. China). Immunodetection was performed with the Streptavidin-Peroxidase system (ZSGB-bio, Beijing, P.R. China) according the manufacture’s protocol. After washing, diaminobenzidine and hematoxylin were respectively added to detect immunoreactive proteins. EHF protein expression was scored in double-blinding way (ie, without knowing the EHF copy number of the case), and 0, 1, 2, 3 reprents negative, weak positive, positive and strong positive, respectively.
Western blot analysis
Cells were lysed in prechilled RIPA buffer containing protease inhibitors. The protein lysates were separated on SDS–PAGE and then transferred to PVDF membranes (Roche Diagnostics, Mannheim, Germany). The membranes were blocked for 2 h in 5% bovine serum albumin (BSA) in 1 × TBS-T (0.5% Tween-20) and incubated with the indicated primary antibodies, including anti-EHF (Abcam, Inc), anti-total-Erk1/2 (Abcam, Inc), anti-phospho-Erk1/2 (Epitomics, Inc), anti-phospho-AktSer473 (Bioworld Technology, co, Ltd), anti-total-Akt (Bioworld Technology, co, Ltd), anti-HER2 (Sino Biological, Inc), anti-HER3 (Sino Biological, Inc), anti-HER4 (Sino Biological, Inc), anti-E-cadherin (Epitomics, Inc), anti-Vimentin (Epitomics, Inc) and anti-GAPDH (Abgent, Inc). The membranes were then incubated with species-specific HRP-conjugated secondary antibodies from ZSGB-BIO, and immunoblotting signals were visualized using the Western Bright ECL detection system (Advansta, CA).
Cell proliferation, cell cycle and apoptosis assays
Cell proliferation, colony formation, cell cycle and apoptosis assays were similarly performed as previously described.46
In vivo tumorigenicity
Four- to five-week-old male athymic nude mice were purchased from SLAC laboratory Animal Co., Ltd. (Shanghai, PR. China) and housed in a specific pathogen-free (SPF) environment. The mice were randomly divided into two groups (six mice per group). Tumor xenografts were established by subcutaneously injecting 4 × 106 BGC823 cells transfected with the indicated siRNAs into the right armpit region of nude mice. From day 3 post-injection, tumor size was measured every 2 days. Tumor volumes were calculated by the formula (length × width2 × 0.5). The mice were sacrificed after 15 days. Tumors were harvested and weighted. Tumors obtained from representative animals were embedded in paraffin, sectioned at 4 μm, and stained with H&E. Ki-67 staining was used to evaluate cell proliferation. All experimental procedures were approved by the Animal Ethics Committee of Xi’an Jiaotong University.
Cell migration and invasion assays
Cell migration and invasion assays were similarly performed as previously described.46
Immunofluorescence staining
The process of IF staining was similarly performed as described previously.46
Dual-luciferase reporter assay
To construct luciferase reporter plasmids, the promoter regions of HER2-4 genes were amplified from genomic DNA of BGC823 cells. The amplification products were digested with restriction enzymes and inserted into pre-digested pGL3-Basic luciferase vector (Promega Corp., Madison, WI, USA) to produce the luciferase reporter plasmids pGL3-HER2-Luc, pGL3-HER3-Luc and pGL3-HER4-Luc. All of the constructs were verified by Sanger sequencing. The primers for plasmid constructs were presented in Supplementary Table 5.
To test the promoter activity of HER2-4 genes modulated by EHF, BGC823 or 293T cells were transfected with pcDNA3.1(-)A-EHF or empty vector in six-well plates and were cotransfected with pGL3-HER2-Luc, pGL3-HER3-Luc or pGL3-HER4-Luc, and pRL-TK plasmids (Promega Corp., Madison, WI, USA) using Lipofectamine 3000 (Invitrogen, Grand Island, NY, USA). The pRL-TK plasmid, containing Renilla luciferase, was used to normalize transfection efficiency. Cells were collected 36 h post-transfection, and luciferase activities were analyzed on EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA, USA) using the dual-luciferase reporter assay system (Promega Corp., Madison, WI, USA) according to the manufacturer’s instructions. Data were expressed as relative luciferase activity (Firefly luciferase activity/Renilla luciferase activity). Each experiment was performed in triplicate.
Chromatin immunoprecipitation assay
The ChIP assay was used to evaluate transcription factor EHF binding to its target DNA by using the Pierce Magnetic ChIP Kit (Pierce Biotechnology, Rockford, IL, USA). In brief, BGC823 cells were transfected with pcDNA3.1/myc-His(-)A-EHF and empty vector. After 2 days, the indicated cells (1 × 107cells) were cross-linked with formaldehyde (final concentration 1% vol/vol) and the cross-linking reaction was stopped by the addition of glycine. The harvested cells were then lysed and digested by using membrane extraction buffer and MNase digestion buffer, and the chromatin was sonicated by using sonics VCX-130PB (Sonics & Materials, Inc., Newtown, CT, USA). Next, 10% of the chromatin from each lysate was saved as an input control. The remaining chromatin was immunoprecipitated by using mouse monoclonal anti-Myc tag, clone 4A6 antibody (Millipore, Temecula, CA, USA). The same amount of non-specific IgG was used as control. Immunoprecipitated protein DNA complex was then captured with ChIP Grade Protein A/G Magnetic Beads. After reversal of the cross-link, digestion of proteins with proteinase K and DNA recovery, the DNA fragments were used as templates for qRT-PCR analysis using the primers presented in Supplementary Table 6, and the data were normalized by respective 5% input. Each experiment was performed in triplicate.
Electrophoretic mobility shift assays
EHF protein was obtained when 1 μg of pcDNA3.1(-)A-EHF or empty vector as DNA template was used according to the protocol of the TNT T7/SP6 Coupled Reticulocyte Lysate System (Promega Corp., Madison, WI, USA). EMSA was performed by using LightShift Chemiluminescent EMSA Kit (Pierce Biotechnology, Rockford, IL, USA) following the manufacturer’s protocol. The binging reaction mixtures contained 2 μl of the translation products, 100 fmol of biotin-labeled oligonucleotide probes (Sangon, Shanghai, China), 2.5% glycerol, 5 mM MgCl2, 50 ng/μl Poly (dI-dC) and 1% NP-40 were incubated in binding buffer at room temperature for 20 min. Unlabeled wild type or mutant oligonucleotides (10 pmol) were incubated with the translation products at room temperature for 15 min prior to the addition of biotin-labeled probes. The mixtures containing loading buffer were separated on a 6% non-denaturing polyacrylamide gel in 0.5 × TBE buffer at 100 V, and oligonucleotides were electrophoretic transferred to a nylon membrane (Roche Diagnostics, Mannheim, Germany). After cross-linking with HL-2000 HybriLinker Hybridization Oven (UVP, Upland, CA, USA), the membrane was detected using a LightShift Chemiluminescent EMSA Kit. The sequences of the double-stranded oligonucleotides used to detect the DNA-binding activity of EHF were presented in Supplementary Table 7.
Statistical analysis
Statistical analysis was performed using the SPSS statistical package (11.5, Chicago, IL, USA). EHF expression and amplification in cancer tissues and control subjects were compared by the paired-samples t-test, Mann–Whitney U test or Wilcoxon Signal-Rank Test. The correlation between EHF amplification and clinicopathological characteristics of gastric cancer patients was analyzed by Fisher’s exact test or Pearson’s χ2-test. Multivariate models that adjusted for the most important covariates were analyzed by logistic regression test. Survival curves were constructed according to the Kaplan–Meier method and statistical analysis was performed using the log-rank test. Univariate survival analysis was performed to investigate the effect of EHF amplification on the survival of gastric cancer patients. Multivariate cox regression analysis was used to examine the effect of EHF amplification on survival of independently of gender, age, differentiation and tumor stage. P<0.05 were considered significant.
Abbreviations
- ChIP:
-
chromatin immunoprecipitation
- qRT-PCR:
-
quantitative RT-PCR
- EBS:
-
ETS-binding site
- EMSA:
-
electrophoretic mobility shift assay
- EMT:
-
epithelial–mesenchymal transition
- ESCC:
-
esophageal squamous cell carcinoma
- ETS:
-
E26 transformation-specific
- FISH:
-
fluorescence in situ hybridization
- H&E:
-
hematoxylin and eosin
- HER:
-
human epidermal growth factor receptor
- IF:
-
immunofluorescence
- IHC:
-
immunohistochemistry
- MMPs:
-
matrix metalloproteinases
- PI3K:
-
phosphatidylinositol-3-kinase
References
Siegel R, Ma J, Zou Z, Jemal A . Cancer statistics, 2014. CA Cancer J Clin 2014; 64: 9–29.
Ajani JA, Bentrem DJ, Besh S, D'Amico TA, Das P, Denlinger C et al. Gastric cancer, version 2.2013: featured updates to the NCCN Guidelines. J Natl Compr Canc Netw 2013; 11: 531–546.
Takahashi T, Saikawa Y, Kitagawa Y . Gastric cancer: current status of diagnosis and treatment. Cancers 2013; 5: 48–63.
Pasechnikov V, Chukov S, Fedorov E, Kikuste I, Leja M . Gastric cancer: prevention, screening and early diagnosis. World J Gastroenterol 2014; 20: 13842–13862.
Cooper CD, Newman JA, Gileadi O . Recent advances in the structural molecular biology of Ets transcription factors: interactions, interfaces and inhibition. Biochem Soc Trans 2014; 42: 130–138.
Hollenhorst PC, McIntosh LP, Graves BJ . Genomic and biochemical insights into the specificity of ETS transcription factors. Annu Rev Biochem 2011; 80: 437–471.
Gutierrez-Hartmann A, Duval DL, Bradford AP . ETS transcription factors in endocrine systems. Trends Endocrinol Metab 2007; 18: 150–158.
Kar A, Gutierrez-Hartmann A . Molecular mechanisms of ETS transcription factor-mediated tumorigenesis. Crit Rev Biochem Mol Biol 2013; 48: 522–543.
Sharrocks AD . The ETS-domain transcription factor family. Nat Rev Mol Cell Biol 2001; 2: 827–837.
Sementchenko VI, Watson DK . Ets target genes: past, present and future. Oncogene 2000; 19: 6533–6548.
Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005; 310: 644–648.
Kumar-Sinha C, Tomlins SA, Chinnaiyan AM . Recurrent gene fusions in prostate cancer. Nat Rev Cancer 2008; 8: 497–511.
Baldus CD, Liyanarachchi S, Mrozek K, Auer H, Tanner SM, Guimond M et al. Acute myeloid leukemia with complex karyotypes and abnormal chromosome 21: Amplification discloses overexpression of APP, ETS2, and ERG genes. Proc Natl Acad Sci USA 2004; 101: 3915–3920.
Ghadersohi A, Sood AK . Prostate epithelium-derived Ets transcription factor mRNA is overexpressed in human breast tumors and is a candidate breast tumor marker and a breast tumor antigen. Clin Cancer Res 2001; 7: 2731–2738.
Mueller BU, Pabst T, Osato M, Asou N, Johansen LM, Minden MD et al. Heterozygous PU.1 mutations are associated with acute myeloid leukemia. Blood 2003; 101: 2074.
Seth A, Watson DK . ETS transcription factors and their emerging roles in human cancer. Eur J Cancer 2005; 41: 2462–2478.
Oikawa T, Yamada T . Molecular biology of the Ets family of transcription factors. Gene 2003; 303: 11–34.
Kas K, Finger E, Grall F, Gu X, Akbarali Y, Boltax J et al. ESE-3, a novel member of an epithelium-specific ets transcription factor subfamily, demonstrates different target gene specificity from ESE-1. J Biol Chem 2000; 275: 2986–2998.
Albino D, Longoni N, Curti L, Mello-Grand M, Pinton S, Civenni G et al. ESE3/EHF controls epithelial cell differentiation and its loss leads to prostate tumors with mesenchymal and stem-like features. Cancer Res 2012; 72: 2889–2900.
Stephens DN, Klein RH, Salmans ML, Gordon W, Ho H, Andersen B . The Ets transcription factor EHF as a regulator of cornea epithelial cell identity. J Biol Chem 2013; 288: 34304–34324.
Tugores A, Le J, Sorokina I, Snijders AJ, Duyao M, Reddy PS et al. The epithelium-specific ETS protein EHF/ESE-3 is a context-dependent transcriptional repressor downstream of MAPK signaling cascades. J Biol Chem 2001; 276: 20397–20406.
Cangemi R, Mensah A, Albertini V, Jain A, Mello-Grand M, Chiorino G et al. Reduced expression and tumor suppressor function of the ETS transcription factor ESE-3 in prostate cancer. Oncogene 2008; 27: 2877–2885.
Wang L, Xing J, Cheng R, Shao Y, Li P, Zhu S et al. Abnormal localization and tumor suppressor function of epithelial tissue-specific transcription factor ESE3 in esophageal squamous cell carcinoma. PLoS One 2015; 10: e0126319.
Brenne K, Nymoen DA, Hetland TE, Trope' CG, Davidson B . Expression of the Ets transcription factor EHF in serous ovarian carcinoma effusions is a marker of poor survival. Hum Pathol 2012; 43: 496–505.
Cheng Z, Guo J, Chen L, Luo N, Yang W, Qu X . Knockdown of EHF inhibited the proliferation, invasion and tumorigenesis of ovarian cancer cells. Mol Carcinog 2015; 55: 1048–1059.
Sprater F, Hovden AO, Appel S . Expression of ESE-3 isoforms in immunogenic and tolerogenic human monocyte-derived dendritic cells. PLoS One 2012; 7: e49577.
Goldman M, Craft B, Swatloski T, Ellrott K, Cline M, Diekhans M et al. The UCSC Cancer Genomics Browser: update 2013. Nucleic Acids Res 2013; 41: D949–D954.
Shi J, Yao D, Liu W, Wang N, Lv H, Zhang G et al. Highly frequent PIK3CA amplification is associated with poor prognosis in gastric cancer. BMC Cancer 2012; 12: 50.
Egeblad M, Werb Z . New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2002; 2: 161–174.
De Craene B, Berx G . Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 2013; 13: 97–110.
Roskoski R Jr. . The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res 2014; 79: 34–74.
Begnami MD, Fukuda E, Fregnani JH, Nonogaki S, Montagnini AL, da Costa WL Jr et al. Prognostic implications of altered human epidermal growth factor receptors (HERs) in gastric carcinomas: HER2 and HER3 are predictors of poor outcome. J Clin Oncol 2011; 29: 3030–3036.
Lemmon MA, Schlessinger J . Cell signaling by receptor tyrosine kinases. Cell 2010; 141: 1117–1134.
Chang CH, Scott GK, Kuo WL, Xiong X, Suzdaltseva Y, Park JW et al. ESX: a structurally unique Ets overexpressed early during human breast tumorigenesis. Oncogene 1997; 14: 1617–1622.
Albertson DG . Gene amplification in cancer. Trends Genet 2006; 22: 447–455.
Zhang D, Wang Z, Luo Y, Xu Y, Liu Y, Yang W et al. Analysis of DNA copy number aberrations by multiple ligation-dependent probe amplification on 50 intestinal type gastric cancers. J Surg Oncol 2011; 103: 124–132.
de Launoit Y, Baert JL, Chotteau-Lelievre A, Monte D, Coutte L, Mauen S et al. The Ets transcription factors of the PEA3 group: transcriptional regulators in metastasis. Biochim Biophys Acta 2006; 1766: 79–87.
Watabe T, Yoshida K, Shindoh M, Kaya M, Fujikawa K, Sato H et al. The Ets-1 and Ets-2 transcription factors activate the promoters for invasion-associated urokinase and collagenase genes in response to epidermal growth factor. Int J Cancer 1998; 77: 128–137.
Scott GK, Daniel JC, Xiong X, Maki RA, Kabat D, Benz CC . Binding of an ETS-related protein within the DNase I hypersensitive site of the HER2/neu promoter in human breast cancer cells. J Biol Chem 1994; 269: 19848–19858.
Scott GK, Chang CH, Erny KM, Xu F, Fredericks WJ, Rauscher FJ 3rd et al. Ets regulation of the erbB2 promoter. Oncogene 2000; 19: 6490–6502.
Benz CC, O'Hagan RC, Richter B, Scott GK, Chang CH, Xiong X et al. HER2/Neu and the Ets transcription activator PEA3 are coordinately upregulated in human breast cancer. Oncogene 1997; 15: 1513–1525.
Eckel KL, Tentler JJ, Cappetta GJ, Diamond SE, Gutierrez-Hartmann A . The epithelial-specific ETS transcription factor ESX/ESE-1/Elf-3 modulates breast cancer-associated gene expression. DNA Cell Biol 2003; 22: 79–94.
Li R, Pei H, Watson DK . Regulation of Ets function by protein - protein interactions. Oncogene 2000; 19: 6514–6523.
Verger A, Duterque-Coquillaud M . When Ets transcription factors meet their partners. Bioessays 2002; 24: 362–370.
Tootle TL, Rebay I . Post-translational modifications influence transcription factor activity: a view from the ETS superfamily. Bioessays 2005; 27: 285–298.
Shi J, Liu W, Sui F, Lu R, He Q, Yang Q et al. Frequent amplification of AIB1, a critical oncogene modulating major signaling pathways, is associated with poor survival in gastric cancer. Oncotarget 2015; 6: 14344–14359.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81272933, 81372217, 81402340 and 81472622) and Science and Technology Project of Shaanxi Province (No. 2012K13-01-01).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by R Johnstone
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Shi, J., Qu, Y., Li, X. et al. Increased expression of EHF via gene amplification contributes to the activation of HER family signaling and associates with poor survival in gastric cancer. Cell Death Dis 7, e2442 (2016). https://doi.org/10.1038/cddis.2016.346
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2016.346
- Springer Nature Limited
This article is cited by
-
SLC39A10 promotes malignant phenotypes of gastric cancer cells by activating the CK2-mediated MAPK/ERK and PI3K/AKT pathways
Experimental & Molecular Medicine (2023)
-
Helichrysetin inhibits gastric cancer growth by targeting c-Myc/PDHK1 axis-mediated energy metabolism reprogramming
Acta Pharmacologica Sinica (2022)
-
ESE3-positive PSCs drive pancreatic cancer fibrosis, chemoresistance and poor prognosis via tumour–stromal IL-1β/NF–κB/ESE3 signalling axis
British Journal of Cancer (2022)
-
NCOA3 is a critical oncogene in thyroid cancer via the modulation of major signaling pathways
Endocrine (2022)
-
Synergistic activation of mutant TERT promoter by Sp1 and GABPA in BRAFV600E-driven human cancers
npj Precision Oncology (2021)