
Abstract
Immune surveillance through Foxp3+ regulatory T cells plays a crucial role in bone homeostasis. Scurfy, the mouse model of autoimmune IPEX syndrome, bears a loss-of-function mutation in Foxp3 that leads to multi-organ inflammation. Herein, we report that scurfy mice exhibit severe bone loss mediated by accelerated osteoclastogenesis. Mechanistically, Foxp3 deficiency results in the upregulation of NF-κB in T helper cells through the loss of repressive Foxp3/NEMO interaction, thereby unleashing NF-κB-mediated over-production of pro-osteoclastogenic cytokines. Flow cytometry analysis shows marked increase in lin-Sca-1+c-kit+ hematopoietic stem cells (LSK HSCs) and granulocyte/macrophage progenitors (GMPs) in bone marrow of scurfy mice with corresponding exacerbated osteoclastogenic potential, implying that osteoclast progenitors are affected at a very primitive stage in this disorder. Scurfy LSK HSCs exhibit greater sensitivity to M-CSF and contain abundant PU.1+ Sf LSK HSCs compared with WT. Accordingly, genetic or pharmacological inhibition of M-CSF or mTOR signaling, but not IL-17 signaling, attenuates osteoclastogenesis and osteopenia in scurfy. Thus, our study suggests that Foxp3 deficiency leads to osteopenia owing to dysregulated NF-κB activity and subsequent cytokine-mediated hyper-proliferation of myeloid precursors, and positions the NF-κB pathway as a potential target for therapeutic intervention for this disorder.
Similar content being viewed by others


Main
Skeletal homeostasis is primarily controlled by bone-resorbing osteoclasts and bone-forming osteoblasts.1 The former cells originate from hematopoietic lineage and require M-CSF and RANKL to differentiate into multi-nucleated bone-resorbing cells.2, 3 The differentiation factors are derived from several sources, chiefly mesenchymal cells and activated T cells, suggesting that bone homeostasis is regulated by various systems, including the immune network. Regulatory T cells (Tregs) are crucial for maintaining immune self-tolerance.4 The development and survival of these cells depend on the expression of the transcription factor Foxp3.5 The functional significance of this transcription factor in Tregs was recognized in humans with immune dysregulation polyendocrinopathy and enteropathy X-linked (IPEX) syndrome.6 These patients, similar to scurfy (Sf) mice, lack Tregs owing to mutations in the Foxp3 gene, which leads to the development of fatal autoimmune hyper-proliferative disease.7 In contrast, forced expression of Foxp3 alleviates autoimmune responses, such as inflammatory bowel disease.7, 8
T-cell immune regulation of skeletal homeostasis has been widely studied owing to inflammatory bone disparities.9, 10 The initial direct evidence for such regulation was the identification of T-cell-derived RANKL, the primary osteoclast differentiation factor.11, 12 Subsequent studies identified IL-17, product of Th17 cells, as the principal potent cytokine that induces the osteoclastogenic factors RANKL, IL-1 and tumor necrosis factor α.13 On the other hand, strong evidence suggests that Th-cell-derived IFNγ and IL-4 are potent inhibitors of osteoclastogenesis.14 Likewise, potentiating Treg cells by forced expression of Foxp3 increased bone mass in mice by impairing osteoclast differentiation.15 However, additional cellular and molecular details have been offered to describe the mechanisms underlying T-cell regulation by Foxp3 and suppression of osteoclastogenesis. In this regard, it was reported that transcriptional (NFAT and NF-κB),16 cell–cell interaction (via CTLA-4)17 and cytokine-dependent18 mechanisms contribute to this response.
To further elucidate the cellular and molecular mechanism underlying T-cell regulation of osteoclast differentiation, we examined the skeletal development of the Sf mouse in which Treg differentiation and function are dysregulated owing to Foxp3 mutation. Our findings show that the skeleton of Sf mice deteriorates because of elevated osteoclastogenesis. This phenomenon appears to be the result of increased frequency of a myeloid sub-population, namely GMPs, that gives rise to osteoclasts. The high frequency of these progenitors was maintained by higher levels of M-CSF as depletion of this factor reversed the osteoporotic bone phenotype. We further provide evidence that these cellular changes are likely subsequent to hyper-activation of NF-κB signaling in CD4+ T cells and GMPs, suggesting that Foxp3 is an endogenous regulator of NF-κB-mediated inflammatory and osteoclastogenic factors.
Results
Sf mice exhibit bone loss
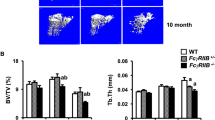
The effect of autoimmune response in many organs of Sf mice has been extensively studied.19 Given the pronounced inflammation-induced skeletal manifestations, we decided to investigate how lack of immune surveillance by Tregs in Sf mice affects bone. Bone morphometric and histological analyses on 4-week-old Sf as well as wild type (WT) littermates show significant reduction of bone volume in both trabecular (Figure 1a) and cortical (Figure 1b, arrows) bones of Sf mice. Quantitative analysis further indicated that all bone parameter changes (Figures 1c–f) denote bone loss in Sf mice. TRAP staining of histological sections of long (Figures 1g and h) and metatarsal bones (Figures 1i and j) revealed increased marrow cellularity (Figure 1i; asterisk) and elevated number of TRAP-positive cells per bone surface area in Sf mice (Figures 1h and j), suggesting that the bone loss phenotype of these mice is due to increased numbers of osteoclasts and their progenitors. Indeed, we established that Sf-derived whole bone marrow (WBM) cultures, plated at similar cell density to WT in the presence of RANKL and M-CSF, gave rise to increased number of osteoclasts compared with WT cultures (Figures 1k and l), which was also confirmed by semi-quantitative analysis of osteoclast marker expression (cathepsin K, MMP9 and β3 integrin) (Supplementary Figure S1A).

Sf mice exhibit severe bone loss and increased number of osteoclasts in bone. (a) Representative 3-D reconstructed images of the trabecular bone region in the proximity to the growth plate were captured by μCT analysis. (b) Representative images of the proximal and midshaft cortex of the long bones are shown. (c–f) Quantification of bone parameters: (c) μCT analysis depicts ratio of cancellous bone volume/tissue volume (BV/TV) in the same areas shown in panel a, (d) trabecular numbers (Tb N), (e) trabecular separation (Tb Sp), (f) and trabecular thickness (Tb Th). (g–j) Histological examination of osteopenia in Sf mice. (g) Representative H&E- and TRAP-stained histological section near the growth plate. (h) Quantifications of osteoclast number per surface area were plotted for the trabecular bone region. (i) Histological section of ankle bones. (j) Quantifications of osteoclast number per surface area were plotted for the sections near the ankle region. (k) WBM cultures from WT and Sf mice were subjected to vehicle or RANKL stimulation for 4 days before TRAP staining to visualize osteoclast formation. (l) Bar graphs were plotted by counting numbers of multi-nulceated (MNC) TRAP+ osteoclasts (OCs) from each of the 4 wells of each RANKL-stimulated groups. All bar graphs were depicted by the average of the numbers, ±S.D. Statistics: n=4 per group; *P<0.05 and **P<0.01 by Student T-test. Scale bars: 200 μm in (a), (i) and (k); 1 mm in (b); 100 μm in (g)
NF-κB activation and over-production of proinflammatory cytokines in Sf-derived T cells
The transcription factor NF-κB is considered the master regulator of inflammatory responses as it modulates the expression of a wide range of inflammatory cytokines, besides being also essential for osteoclastogenesis.20 We tested protein and mRNA expression of NF-κB genes, and found that the expression of RelA/p65 and IKKγ/NEMO, the central modulators of NF-κB signal transduction pathway, was predominately upregulated 2.1- and 5.6-fold for protein and 1.8- and 1.6-fold for mRNA, respectively (Figure 2a and Supplementary Figures S1B and S1C). Using intracellular flow assay, we found that the proportion of NEMO-expressing cells was distinctively increased in Sf bone marrow (Figure 2b, solid line). Further examination of NEMO-expressing cells by multicolor flow cytometry revealed that a subset of myeloid cells (CD11b+c-kit+) bore high NEMO expression (Supplementary Figure S2A). In addition, analysis of NEMO-expressing cells by CD11b and Gr1 resulted in the enrichment of CD11b+Gr1- population by more than 2.5-fold (Supplementary Figure S2B). These findings implied the importance of NEMO upregulation in the myeloid proliferative disease in Sf mice. Using co-immunoprecipitations from primary CD4+ cells (WT and Sf) and from transiently transfected 293T cells, we show that NEMO and Foxp3 physically interact (Figures 2c and d). Further, using NF-κB reporter assay, Foxp3 protein was able to suppress the reporter gene luciferase by 30% (Supplementary Figure S3A). In addition, increased NEMO expression was also found in the lymphoid tissues, including lymph nodes and thymus, but not in the peritoneal cavity, which was mostly comprised of macrophages (Supplementary Figures S3B–S3F). We next tested mRNA expression of pro-inflammatory/osteoclast cytokines in magnetically isolated T helper (TH) cells. As shown in Figure 2e, RANKL, M-CSF, IL-17 and tumor necrosis factor α were all found to be upregulated in Sf-derived CD4+ TH cells, which were also more activated than the WT cells as evidenced by increased expression of the transcription factor NFAT. Intracellular flow assay further confirmed that both bone marrow (Figure 2f) and spleen-derived (Figure 2g), PMA/ionomycin-stimulated CD4+ TH cells produced significantly higher levels of RANKL, M-CSF and GM-CSF, with spleen (but not bone marrow)-derived CD4+ TH cells also producing higher IL-17. Similarly, when spleen-derived CD4+ TH cells were stimulated with anti-CD3/CD28 antibody (Ab) beads, which is more physiologically relevant, we observed 48 × more expression of M-CSF in Sf-derived cells compared with WT controls (Figure 2h). Accordingly, multiplex ELISA showed elevated serum levels of a wide range of chemokines, inflammatory and growth factors in Sf mice compared with WT controls (Supplementary Figure S4). Taken together, Foxp3 deficiency resulted in the loss of Foxp3–NEMO interaction and led to the overexpression of NEMO and subsequent activation of NF-κB in TH cells. These pro-inflammatory cytokine-secreting T cells were hyperactivated, thereby were suspected as culprit inducers of higher osteoclastogenesis in Sf mice.

Foxp3 deficiency in Sf mice resulted in the activation of NF-κB pathway and over-production of pro-osteoclast cytokines in CD4+ T lymphocytes. (a) Western blot of NEMO and Rel A protein expression in splenic CD4+ T cells using WT and Sf-derived CD4+ T cells. Images shown were blots probed with anti-Rel A and anti-NEMO Ab separately. β-actin was used as internal loading control. (b) Intracellular flow analysis was performed on red-cell-depleted WBMs. Cells were fixed, permeabilized and stained with anti-NEMO Ab followed by FITC-conjugated secondary Ab and analyzed on flow cytometer. Histogram of the middle fluorescence intensity (MFI) of FITC was shown. (c) Co-immunoprecipitation of Foxp3 and NEMO protein in primary splenic CD4+T cells. CD4+T-cell proteins derived from WT and Sf pulled down by anti-NEMO Ab were blotted with either anti-NEMO or anti-Foxp3 Ab. Total lysate from Foxp3+Tregs used as positive control. (d) Co-immunoprecipitation of Foxp3 and NEMO protein in transiently transfected 293 T cells. 293 T cells were transfected with both Foxp3 and NEMO (tagged with c-myc) plasmids. Cells transfected with either Foxp3 or NEMO plasmid alone were used as controls. Immunoprecipitation (IP) and immunoblots (IB) were conducted as indicated. In addition, two clones of anti-Foxp3 Ab were used for IP, by which K-13 generated stronger immunoprecipitation than H190. (e) qPCR analysis (fold change) of the indicated factors in MACS purified CD4+ T cells from WT and Sf mice. (f) Cytokine production following PMA stimulation was assessed by intracellular flow analysis on MACS-purified, BM-derived CD4+ T cells using appropriate antibodies. (g) Intracellular flow analysis on MACS-purified, PMA-stimulated spleen-derived CD4+ T cells. (h) qPCR analysis (fold change) of M-CSF expression in MACS-purified, anti-CD3/CD28 stimulated CD4+ T cells from WT and Sf mice. Bar graphs were depicted by the average of the numbers, ±S.D. Statistics for all bar graphs: n=3 per group; *P<0.05 and **P<0.01 by Student T-test
Enhanced myelopoiesis/myeloproliferative disease in the Sf bone marrow leads to increased osteoclastogenic potential
Next, we interrogated whether the increased osteoclast burden and osteopenia in Sf mice is due to skewed myelopoeisis secondary to hypercytokinemia. Because osteoclasts differentiate from the myeloid lineage, we hypothesized that dysregulation of lineage commitment or differentiation of myeloid progenitor cells significantly contribute to increased osteoclastogenic potential of Sf bone marrow. To test this, we performed flow cytometric analysis of the myeloid compartment in the bone marrow, the spleen and blood using monocytic marker CD11b and granulocytic marker gr1. As shown in Figure 3a, the percentage of monocytic CD11b+gr1-/lo cells was significantly increased in bone marrow, whereas percentage of granulocytic CD11b+gr1Hi cells was decreased. We further examined the proliferative activity of Sf-derived cultures using BrdU flow assay following 3 days of culture. Sf cultures exhibited nearly twice of BrdU incorporation (Figures 3b and c) and proliferation of Sf cells was much faster than WT cells in the absence (Supplementary Figure S5A) or presence of RANK ligand (Supplementary Figure S5B). Taken together, increased osteoclast formation in Sf mice is likely because of hyperproliferative osteoclast precursor cells in Sf mice.

Bone marrow CD11b-, not CD11b+, cells from Sf are highly proliferative and osteoclastogenic. (a) Flow analysis was performed on bone marrow myeloid populations from 12 Sf and 11 WT controls stained with anti-CD11b and anti-Gr1 antibodies. Statistics was generated by Student T-test. (b) BrdU incorporation in Sf WBM cultures was carried out as described under methods. (c) Bar graph derived from BrdU flow assays shown in panel b. n=4 per group; *P<0.05 by Student T-test. (d) MACS was performed first with biotin-conjugated lineage antibodies and anti-biotin microbeads followed by anti-CD11b microbeads to obtain lin-CD11b-/lo and lin-CD11bHi fractions. Osteoclatogenic potential of each MACS fraction was subsequently tested by ex vivo osteoclastogenesis assay. WBM cultures were used as control. Scale bars: 200 μm in (d)
Through separating CD11bhi population from CD11blo after lineage depletion (i.e., T and B cells, erythroid), we found that CD11blo population exhibited far greater osteoclastogenesis potential than CD11bhi population (Figure 3d). In addition, CD11blo population isolated from Sf bone marrow possessed hyperactive potency for osteoclastogenesis. In fact, almost an equal level of osteoclast formation could be achieved by plating 10 000 cells of CD11blo cells to 80 000 WBM cells (Figure 3d; compare right with left panels, respectively). We have also tested potential contribution of myeloid-derived suppressor cells (MDSCs) to the Sf osteopenic bone phenotype, as reported in murine breast cancer model, wherein tumor-associated MDSCs, marked as CD11b+Gr-1+ cells,21 and MDSCs derived from murine inflammatory arthritis model22 can be induced into osteoclasts. We found that Sf bone marrow contained slightly less frequency of Gr-1hiLy6G+ MDSCs and much higher frequency of Gr-1dimLy6G- MDSCs (Supplementary Figure S6A). However, both cell populations exhibited poor osteoclastogenic potential (Supplementary Figures S6B and C) as compared with WBM cultures (Supplementary Figure S6D) and cell fractions devoid of the two MDSC populations (Supplementary Figure S6E). Taken together, although all immature myeloid cells were hyperproliferative in Sf compared with WT mice, only CD11blo/- cells exhibited high osteoclastogenic potential.
Alteration of hematopoietic progenitor populations in Sf mice
To test how the number of myeloid progenitor cells is altered in Sf mice, we generated a comprehensive profile of myeloid/osteoclast progenitors from the most primitive (i.e., long-term or LT-HSCs) to the more committed (i.e., GMPs) cells.23 Frequency of the early lineage precursors lineage-negative, Sca-1-postive and c-kit-positive (LSK-HSCs) cells was significantly elevated in Sf compared with WT mice (Figure 4a). Among the more committed myeloid precursors, frequency of GMPs derived from Sf bone marrow was twice as much as from WT counterparts, whereas the frequency of other progenitors was not significantly changed (Figure 4b). Further characterization of LSKs by the SLAM marker (CD150) revealed a significant increase for both short-term HSC (ST-HSC) and mutipotent progenitor frequency, with greater change for the latter (Figure 4c). Taken together, these results suggested that under the inflammatory condition of Sf bone marrow, myelopoiesis was skewed toward granulocyte/macrophage lineage. The primitive hematopoietic progenitor cells at the stage as early as the ST-HSCs respond to the inflammatory cues by moving from quiescence to active cell division, resulting in increasing numbers of myeloid/osteoclast precursors. This phenomenon appeared to be lineage-specific, as GMP frequency observed in Sf bone marrow did not seem to be at the expense of megakaryocyte/erythroid progenitors.

Alteration of hematopoietic progenitor pool in Sf bone marrow. (a, b) Flow cytometry of HSC progenitors was performed as described in Material and Methods. Percentage of LSK hematopoietic progenitor cells were first gated and analyzed, followed by further sub-gating of the lin-c-kit+ cell population (LK) into megakaryocyte/erythroid progenitors/CMP/GMPs. (c) LT-HSC, ST-HSCs and mutipotent progenitors were sub-gated from LSKs for analysis. Bar graphs were depicted by the average of the numbers, ±S.D. Statistics for all bar graphs: n=6 for WT group and n=7 for Sf group; *P<0.05 and **P<0.01 by Student T-test
Sf LSK HSCs are potent osteoclast founder cells
Although the number of osteoclast progenitors is significantly higher in Sf, the osteoclastogenic potential of each progenitor cell may be also exacerbated. Hence, we examined whether myeloid progenitors derived from Sf mice possess higher 'osteoclast founder' potential. Therefore, we FACS-sorted lin-c-kit+CD11blogr-1lo cells (MPs) and LSK cells from WT and Sf mice (Supplementary Figure S7) and combined equal number of each cell population them with total bone marrow cells from WT at the ratio of 1 : 10 (Figure 5) or 1 : 5 (not shown) to make the culture with 10 000 and 5000 cells per well density, without or with RANKL. Total bone marrow cultures served as controls. As shown in Figure 5, WT-derived LSKs supplemented with WT WBMs resulted in a mild increase of osteoclastogenic potential, whereas Sf-derived LSKs supplemented with WT WBMs exhibited higher osteoclastogenic potential comparable with Sf WBMs. These data indicate that Sf LSK HSCs were indeed highly potent 'founders' for osteoclastogenesis. Together, these findings indicate that the ontology of increased osteoclastogenic potential came at an early and primitive stage of differentiation and commitment of Sf BM HSCs.

Sf LSK HSCs are potent osteoclast founders. One thousand FACS-sorted LSK and MPs (lin-c-kit+CD11blo described in Supplementary Figure S4) from WT and Sf bone marrows and mixed with 9000 total bone marrow cells from WT and treated with RANKL. WBM cultures were used as control. Bar graph was depicted by the average of OC numbers, ±S.D. Statistics; n=3 per group; *P<0.05 and **P<0.01 by Student T-test. Scale bar: 200 μm
Inhibition of M-CSF significantly rescued bone erosion in Sf mice
At the mechanistic level, this observation correlates with a recent report suggesting that HSCs can be a direct target for cytokine signaling.24 We reasoned that Sf-derived HSCs might be hypersensitive to M-CSF, one of the essential cytokines for osteoclast development. LSK HSCs were FACS-sorted, plated for osteoclastogenesis assay at 2000 cells per well and stimulated with serial concentrations of M-CSF. As shown in Supplementary Figure S8A, Sf -derived LSKs responded to M-CSF in a much greater manner than the WT controls. Furthermore, detection of PU.1 (which is the target of M-CSF signaling and crucial for osteoclastogenesis) expression in LSKs via intracellular flow revealed that its protein level is significantly increased in Sf LSKs (Supplementary Figure S8B), confirming the activation of M-CSF signaling cascade in these progenitor cells.
To investigate the contribution of M-CSF hyper-production in osteo-pathogenesis of Sf mice, we administrated M-CSF-neutralizing Ab or carrier solution into Sf mice and WT male siblings for 2 weeks. As expected, WT mice exhibited osteopetrosis upon anti-MCSF Ab treatment assessed by μCT analysis (Figure 6a). Interestingly, anti-M-CSF Ab-treated Sf mice also developed increased BV/TV value comparable with WT mice. Ex vivo osteoclastogenesis assay further demonstrated the rescue of osteopenia/hyper-osteoclastogenesis in Sf via M-CSF neutralization (Figure 6b). In fact, osteoclastogenesis derived from anti-MCSF-treated Sf mice was reduced to levels comparable with that derived from WT mice (Figure 6b; right panel). Interestingly, as anti-M-CSF Ab treatment significantly reduced the frequency of LSKs in Sf bone marrow, the frequencies of GMPs and mutipotent progenitors were moderately decreased (Figure 6c and Supplementary Figure S9). In addition, ex vivo osteoclastogenesis of Sf WBMs post M-CSF Ab treatment was significantly blocked.

Neutralization of M-CSF significantly rescued osteopenia in Sf mice. (a) Sf and WT mice were injected with M-CSF-neutralizing Ab or PBS as described in Material and Methods. Cross-sections of femur bone extending from the growth plate were generated by 3-D reconstructed images through μCT analysis. BV/TV ratios were depicted as bar graph. (b) Ex vivo osteoclastogenesis assay post in vivo administration of M-CSF-neutralizing Ab. (c) Flow cytometric analysis of WBM hematopoietic progenitors were performed on WBMs derived from M-CSF Ab-administrated Sf mice with the rest of experimental control mice. Statistics for all bar graphs: n=3 for WT group and n=4 for Sf group; *P<0.05 and **P<0.01 by Student T-test. Scale bars: 1 mm in (a); 200 μm in (b)
M-CSF haploinsufficiency significantly rescued the osteopenic phenotype of Sf mice
To further address the contribution of M-CSF signaling in the osteopenic phenotype of Sf, we crossed Sf hemizygous female (Sf/x) with Op heterozygous (Op/+) male to generate Op/+::Sf mice and asked how osteoclast progenitors derived from Sf would respond to M-CSF happloinsufficiency. As shown in Figure 7a, μCT analysis revealed that introduction of one op allele in Sf mice (i.e., Op/+, Sf/y) was able to bring the trabecular BV/TV value equivalent to the WT control. In addition, WBMs derived from Op/+::Sf mice only gave rise to half osteoclast formation in number as compared with cells derived from Sf alone (Figure 7b). Interestingly, although the frequency of LSK HSCs did not change significantly (Figure 7c), frequency of CMPs and GMPs were reduced in Op/+::Sf (Figure 7d), suggesting that commitment or fate determination of HSCs in Sf bone marrow was altered by haploinsufficiency of M-CSF, resulting in attenuation of hyper-osteoclastogenic activity.

Rescue of the osteopenic phenotype in Sf via genetic ablation of M-CSF. (a) Representative μCT scanned 3-D images of the femur trabecular area of WT, Sf and Op/+:: Sf mice. (b) Ex vivo osteoclastogenesis assay were performed using WBMs from WT, Sf, Op/+ and Op/+::Sf mice. (c) Frequency of LSK HSCs in Op/+ Sf mice using flow cytometry. (d) Profile of megakaryocyte/erythroid progenitors/CMP/GMPs of WBMs derived from WT, Sf, Op/+ and Op/+ Sf mice, *P<0.05 and **P<0.01 by Student T-test. Scale bars: 200 μm in (a and b)
To probe the therapeutic value of targeting M-CSF, we tested whether blocking mTOR signaling (which is downstream M-CSF) would counteract the osteopenic disease in Sf mice. Indeed, following 2 weeks of intraperitoneal rapamycin injections, bone parameter of rapamycin-treated Sf mice was comparable with that of vehicle-treated WT controls (Figures 8a and b). Consistently, in vivo rapamycin treatment reduced osteoclastogenesis activity of Sf-derived bone marrow cells by almost twofold (Figure 8c). Interestingly, the frequency of GMP in Sf BM was significantly reduced to the level as in vehicle-treated WT controls (Figure 8d). Taken together, inhibition of mTOR pathway appeared to an effective approach on ameliorating Sf’s osteopenic phenotype.

In vivo administration of rapamycin ameliorated osteopenia in Sf mice through reduction of osteoclastogenic activity in the bone marrow. (a) Representative images from μCT analysis of rapamycin versus vehicle-treated Sf mice. (b) Trabecular BV/TV values of vehicle- and rapamycin-treated mice. (c) Ex vivo osteoclastogenesis assay. (d) Hematopoietic progenitor cell profiling by flow cytometry depicting megakaryocyte/erythroid progenitors, CMP and GMP cells from the various experimental groups described in panel a. Statistics for all bar graphs: n=4 for per group; *P<0.05 and **P<0.01 by Student T-test. Scale bars: Scale bars: 200 μm in (a and c)
Discussion
In this study, we report severe osteopenia in Sf mice subsequent to the action of Foxp3-deficient lymphocytes. Mechanistically, we provide evidence that Foxp3 deficiency leads to enhanced activity of NF-κB, exemplified by elevated expression of RelA/p65 and NEMO, the scaffold and regulator of NF-κB signaling. As a result, numerous osteotropic and osteoclastogenic factors are produced, which mobilize hematopoietic progenitors pools, culminating with hyper-myeloproliferation of osteoclast progenitors, exacerbated osteoclastogenesis and osteopenia.
In recent years, several reports have suggested that Treg cells play an inhibitory role in osteoclast formation.25, 26 These studies focused on the interaction between Treg cells and osteoclast precursors, either through direct cell–cell contact or paracrine mechanisms. In this report, we provide evidence that lack of immune surveillance, due to Treg cell defeciency, leads to hyperactivation of effector immune cells. Specifically, we identified exuberant expression of RelA/p65 and NEMO as markers of hyperactivated NF-κB in pro-inflammatory cytokine-producing T effector (Foxp3-deficient) cells. Subsequently, production of pro-inflammatory cytokines is enhanced and in turn stimulates repopulation and/or renewal of hematopoietic progenitors. The cytokines produced by the T effector cells directly target hematopoietic stem cell pools and accelerate proliferation, mobilization and differentiation of myeloid progenitors into osteoclasts. This is further evident by the activation of NF-κB in OC progenitors, which is a crucial osteoclastogenic factor. Indeed, inhibition of NF-κB signaling by administration of NBD peptide halted osteoclastogenesis by Sf-derived progenitors (data not shown).
More importantly, through Ab neutralization and genetic ablation, we identified the critical role of M-CSF signaling rather than IL-17 in this process. We demonstrate that the commitment of HSCs is driven toward the macrophage/preosteoclast lineage and ultimately results in increased osteoclast formation and osteopenia. Our findings depict important changes to characteristics of osteoclast progenitors in the absence of immune surveillance. Specifically, LSK HSCs derived from Sf bone marrow were highly potent precursors and founders of osteoclasts. This finding can be illustrated as more osteoclast precursors are present in Sf LSKs and/or osteoclast precursors in the LSK pool are more sensitive to osteoclastogenic stimuli. This is further supported by our observations that more PU.1 LSKs and more GMPs were present in Sf bone marrow. Interestingly, the pharmacologic and genetic approaches we conducted here to inhibit the M-CSF/PI3K/mTOR axis markedly reduced the frequency of GMPs in Sf bone marrow. These signals, as our findings suggest, target the very primitive stage of hematopietic progenitor cells.
The apparent myeloproliferation and abundance of osteoclast progenitors is consistent with higher expression of M-CSF in Sf mice. This factor is obligatory for proliferation and survival of osteoclast precursors, and genetic ablation of the corresponding csf gene or its receptor c-fms obliterates osteoclastogenesis and leads to osteopetrosis.27 Our findings are supported by a recent report that demonstrated that under inflammatory conditions, high expression of M-CSF induces the myeloid master regulator PU.1 leading to changes in HSCs cell fate and skewing myeloid differentiation.24 Hence, modulating this pathway in Sf mice using pharmacologic and genetic approaches relieved the osteoclastogenic burden and significantly attenuated osteopenia in Sf mice. At the cellular level, these M-CSF inhibitory approaches clearly attenuated the increased frequency of the LSK and GMP cells in Sf mice leading to decreased osteoclastogenesis.
M-CSF activates mTOR/PI3K/Akt pathway, and it has been shown that rapamycin blocks M-CSF and RANKL-induced osteoclastogenesis.28 Our study offers novel insights pointing to inhibition of frequency of osteoclast progenitor pool by rapamycin in Sf mice leading to amelioration of osteopenia. Our findings suggest that mTOR signaling plays a key role in mediating bone loss in Sf mice and highlight the significance of M-CSF/mTOR/PI3K axis in autoimmune-mediated osteolytic diseases. This concept is further supported by recent evidence demonstrating significance of mTOR signaling in joint destruction via robust osteoclast activity,29 thereby positioning M-CSF signaling through mTOR as a significant target to combat immune-mediated bone destruction.
Materials and Methods
Mice
C57/B6 WT, Sf and Op mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA). All mice were housed in CSRB NT or BJCIH animal facilities under the guidelines of the Animal Studies Committee at Washington University School of Medicine.
Structural and quantitative analysis of bone deformities
Long bones were dissected from animals and fixed in 10% neutral buffered formalin for 2 days followed by three washes in PBS. Then muscle and connective tissues were carefully removed from bone specimen before they were proceeded to experiments or archived in 70% EtOH.
For μCT scan, long bone specimens were rehydrated with PBS and μCT scan was performed. Long bone specimens were then retrieved from cylindrical vials, rehydrated with PBS and placed in decalcifying solution for 7 days before histological analysis. Histomorphometry was performed by TRAP staining on paraffin-embedded sections to visualize osteoclasts lining on the bone surface followed by counterstained with H&E.
Assessment of cell-specific cytokine production
In vitro cell stimulation and intracellular flow analysis for MACS-purified lymphocytes: bone marrow- or spleen-derived CD4+ T-cells and CD8+ T-cells were purified by MACS according to the manufacturer’s protocols. For stimulation of cytokine production, 500 000 purified cells were placed in modified complete RPMI media containing 5 μg/ml brefeldin A, 50 ng/ml PMA, 0.5 μg/ml ionomycin and cultured at 37 °C for 4 h. To assess cytokine production, stimulated cells were harvested into ice-old PBS, washed and fixed according to eBiosciences (San Diego, CA, USA) intracellular flow cytometry protocol before stained with anti-cytokine FACS antibodies and analyzed on flow cytometer.
RNA isolation and qRT-PCR
Single mononucleated, red blood cell lysed cell suspension derived from bone marrow, spleen, thymus or lymph nodes or MACS-purified cells were lysed in Trizol reagent (Invitrogen, Carlsbad, CA, USA) for isolation of total RNA. cDNA synthesis was performed from 1 ug of total RNA with a mixture of random hexamers and poly-T oligos using Roche (Indianapolis, IN, USA) cDNA synthesis kit. To assess the abundance of mRNA transcripts for cytokine or transcription factors that mediate cytokine production, qPCR analysis was performed with SYBR Green master mix (Bio-Rad, Hercules, CA, USA) with primers corresponding to the gene of interest. GAPDH, tubulin or Cyclophilin A gene expression was monitored as endogenous control. All real-time PCR reactions were run on BioRad’s CFX96 Real-Time System.
Multiplex cytokine/chemokine profiling for serum
Mouse peripheral blood was collected in Microtainer serum separator tube (BD Bioscience, Franklin Lakes, NJ, USA) before centrifugation to obtain serum. All serum samples were archived in −80 °C freezer before being subjected to cytokine/chemokine measurement.
Multiplex cytokine/chemokine assay was performed according to the manufacturer’s protocol (Millipore, St Charles, MO, USA). Briefly, 25 ul serum samples were loaded onto the 32-plex Mouse Cytokine/Chemokine Magnetic Bead Panel 96-well plate (MCYTOMAG-70 K-PX32). Following the incubation with 25 ul of magnetic beads at 4 °C overnight, biotin-conjugated antibodies and PE-conjugated streptavidin were subsequently added before the plate was processed with an automated washer and analyzed on a Bioplex system (Bio-Rad)
Ex vivo osteoclast proliferation and differentiation assay
BrdU flow assay
After being cultured overnight on petridish with 1/50 diluted CMG, stromal cells were removed from WBM culture by re-plating suspension cells on tissue culture-treated plates to initiate macrophage culture. BrdU was added to macrophage culture with or without the presence of RANKL (50 ng/ml) for 4 days. For BrdU flow assay, cells were detached by Accutase and fixed with Foxp3 fix/perm buffer after washing with PBS. Fixed cells were permeabilized, digested with DNAase I, labeled with Alexa647-conjugated anti-BrdU Ab, washed and resuspended in the presence of 7-AAD before analyzing on a flow cytometer.
Assessment and quantification of osteoclast formation
After removal of adherent stromal cells, WBM cells were plated on 96-well microplates at a series of seeding densities from 200 000, 100 000, 50 000, 25 000, 12 500 and 6250 per well (or as otherwise mentioned). After stimulation with RANKL for 3–4 days, fixation and TRAP staining was performed to visualize multinucleated osteoclasts using the Leukocyte Acid Phosphatase kit from Sigma (St. Louis, MO, USA). Cultures without the presence of RANKL were served as the negative control.
FACS-based profiling for hematopoietic progenitor cells, osteoclast progenitors and myeloid lineage cells
For assessment of hematopoietic progenitor cells in the bone marrow, WBMs were suspended in FACS buffer at the maximum concentration of 10 million cells per 100 ul and stained with biotin-conjugated lineage Ab cocktail (anti-B220, anti-CD3e, anti-Gr1, anti-Ter119 and anti-CD41) and HSC Ab cocktail (anti-Sca1 PerCP Cy5.5, anti-c-Kit APC eFluor 780, anti-CD34 FITC, anti-FLK2 APC, anti-CD48 PE Cy7, anti-CD150 PE and CD16/32 eFluor450) before red cell lysis. All FACS antibodies were purchase from either eBioscience, BioLegend (San Diego, CA, USA) or BD Bioscience (San Diego, CA, USA). Following incubation on ice for 45 min, Ab-labeled cells were washed twice with cold FACS buffer and either fixed with 2% PFA or immediately subjected to flow cytometric analysis.
MACS and FACS sorting of bone marrow subpopulations for ex vivo osteoclastogenesis assay
To obtain lineage-negative CD11b+ and CD11b- cells, WBM cells were red cell-lysed, first labeled with biotin-conjugated lineage antibodies and then incubated with anti-biotin magnetic microbeads to deplete lineage cells. Anti-CD11b microbeads was subsequently used to separate CD11b+ from CD11b- cells. These MACS-purified cells were subjected to osteoclastogenesis assay as routinely done for WBMs or bone marrow macrophages, except they were seeded at a much lower series of density to obtain the optimal number of multinucleated osteoclast formation. MDSC isolation kit was used to obtain bone marrow cells that immuno-phenotypically resembled MDSCs.
For isolation of LSK HSCs, CMPs and GMPs, WBM cells were labeled with FACS antibodies as described above for progenitor analysis and sorted by either Sony Synergy (Sony Biotechnology, San Jose, CA, USA) or MoFlo sorter (Beckman Coulter, Indianapolis, IN, USA). These progenitor cells were sorted into ice-old culture alpha MEM supplemented with 20% FBS to maximize their survival rate. After sorting, cell counts were determined after resuspending cell pellets in a small volume of culture media before plating on 96-well microplates for osteoclastogenesis assay.
Osteoclast founder cell assay
FACS-sorted HSCs, CMPs and GMPs were mixed with WBM cells at 1 : 9 ratio before subjecting to osteoclastogenesis assay. To test their sensitivity to M-CSF, recombinant M-CSF (Peprotech, Rocky Hill, NJ, USA) instead of CMG was used in the culture, and standard ex vivo osteoclastogenesis protocol was followed.
Neutralizing Ab anti-M-CSF
Seven-day-old mice were i.p. injected with M-CSF neutralizing Ab (Bio X Cell, West Lebanon, NH, USA) twice a week for 3 weeks before killing for end-point analysis. Control mice were injected with equal volume of sterile PBS in parallel.
Pathway inhibitors
Rapamycin working solution was prepared by diluting 10 mg/ml stock solution with 5% Tween80/5% PEG-400 by fivefold. Seven-day-old mice were then i.p. injected with 20 ug rapamycin through a 25 ul Hamilton syringe (Hamilton Co., Reno, NV, USA). Mice were re-administered five more times for the course of 3 weeks before end-point analysis. Control mice were injected with the vehicle solution.
Western blot analysis and co-immunoprecipitation
For sample preparation, primary tissues (e.g., bone marrow, spleen, lymph nodes, thymus, peritoneal cavity) were processed into single mononuclear cell suspension after red blood cells were removed. To obtain protein lysates for western analysis, CLB buffer was used. For co-immunoprecipitation studies, RIPA buffer was used instead. Lamieli sample buffer (5 × ) was added to each protein lysate with β-mercaptoethanol before boiling for 5 min for protein denaturation and reduction. Samples were then resolved on 8% SDS-PAGE gels, blotted on PVDF membranes using Bio-Rad’s Trans-Blot Turbo system before probes with antibodies.
Luciferase NF-κB reporter assay
293T cells were co-transfected with Luciferase reporter and Renillia luciferase plasmids with pcDNA plasmids carrying NEMO and Foxp3 cDNA. Renilla luciferase activity was used as internal loading control. Luficerase activity was assessed by Promega’s Stop-and-Go system. For each sample, Luciferase activity was normalized by Renilla luciferase activity.
Abbreviations
- Sf:
-
scurfy
- WT:
-
wild type
- Op:
-
osteopetrotic
- WBM:
-
whole bone marrow
- RANKL:
-
RANK ligand
- LSK:
-
lineage negative Sca-1 positive c-Kit positive
- MPs:
-
myeloid precursors
- LT-HSCs:
-
long-term hematopoietic stem cells
- ST-HSCs:
-
short-term hematopoietic stem cells
- CMPs:
-
common myeloid progenitors
- GMPs:
-
granulocyte/macrophage progenitors
References
Harada S-i, Rodan GA . Control of osteoblast function and regulation of bone mass. Nature 2003; 423: 349–355.
Teitelbaum SL . Osteoclasts: what do they do and how do they do it? Am J Pathol 2007; 170: 427–435.
Xing L, Schwarz EM, Boyce BF . Osteoclast precursors, RANKL/RANK, and immunology. Immunol Rev 2005; 208: 19–29.
Sakaguchi S, Yamaguchi T, Nomura T, Ono M . Regulatory T cells and immune tolerance. Cell 2008; 133: 775–787.
Zheng Y, Rudensky AY . Foxp3 in control of the regulatory T cell lineage. Nat Immunol 2007; 8: 457–462.
Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 2001; 27: 68–73.
Hori S, Nomura T, Sakaguchi S . Control of regulatory T cell development by the transcription factor Foxp3. Science 2003; 299: 1057–1061.
Sharma R, Sharma PR, Kim YC, Leitinger N, Lee JK, Fu SM et al. IL-2-controlled expression of multiple T cell trafficking genes and Th2 cytokines in the regulatory T cell-deficient scurfy mice: implication to multiorgan inflammation and control of skin and lung inflammation. J Immunol 2011; 186: 1268–1278.
Walsh MC, Kim N, Kadono Y, Rho J, Lee SY, Lorenzo J et al. Osteoimmunology: interplay between the immune system and bone metabolism. Annu Rev Immunol 2006; 24: 33–63.
Caetano-Lopes J, Canhao H, Fonseca JE . Osteoimmunology—the hidden immune regulation of bone. Autoimmun Rev 2009; 8: 250–255.
Kong Y, Fiege U, Sarosi I, Bolon B, Tafuri A, Morony S et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegrin ligand. Nature 1999; 402: 304–309.
Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 2000; 408: 600–605.
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 2006; 203: 2673–2682.
Abu-Amer Y . IL-4 abrogates osteoclastogenesis through STAT6-dependent inhibition of NF-kappaB. J Clin Invest 2001; 107: 1375–1385.
Zaiss MM, Sarter K, Hess A, Engelke K, Böhm C, Nimmerjahn F et al. Increased bone density and resistance to ovariectomy-induced bone loss in FoxP3-transgenic mice based on impaired osteoclast differentiation. Arthritis Rheum 2010; 62: 2328–2338.
Bettelli E, Dastrange M, Oukka M . Foxp3 interacts with nuclear factor of activated T cells and NF-κB to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci USA 2005; 102: 5138–5143.
Zaiss MM, Axmann R, Zwerina J, Polzer K, Guckel E, Skapenko A et al. Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum 2007; 56: 4104–4112.
Kim YG, Lee CK, Nah SS, Mun SH, Yoo B, Moon HB . Human CD4+CD25+ regulatory T cells inhibit the differentiation of osteoclasts from peripheral blood mononuclear cells. Biochem Biophys Res Commun 2007; 357: 1046–1052.
Ramsdell F, Ziegler SF . FOXP3 and scurfy: how it all began. Nat Rev Immunol 2014; 14: 343–349.
Abu-Amer Y . NF-κB signaling and bone resorption. Osteoporos Int 2013; 24: 2377–2386.
Sawant A, Deshane J, Jules J, Lee CM, Harris BA, Feng X et al. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res 2013; 73: 672–682.
Charles JF, Hsu LY, Niemi EC, Weiss A, Aliprantis AO, Nakamura MC . Inflammatory arthritis increases mouse osteoclast precursors with myeloid suppressor function. J Clin Invest 2012; 122: 4592–4605.
King KY, Goodell MA . Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nature Rev Immunol 2011; 11: 685–692.
Mossadegh-Keller N, Sarrazin S, Kandalla PK, Espinosa L, Stanley ER, Nutt SL et al. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature 2013; 497: 239–243.
Mario M Zaiss, Axmann Roland, Zwerina Jochen, Polzer Karin, Gückel Eva, Skapenko Alla et al. Treg cells suppress osteoclast formation: A new link between the immune system and bone. Arthritis Rheum 2007; 56: 4104–4112.
Schett G, David JP . The multiple faces of autoimmune-mediated bone loss. Nat Rev Endocrinol 2010; 6: 698–706.
Yoshida H, Hayashi S-I, Kunisada T, Ogawa M, Nishikawa S, Okamura H et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990; 345: 442–443.
Glantschnig H, Fisher JE, Wesolowski G, Rodan GA, Reszka AA . M-CSF, TNF[alpha] and RANK ligand promote osteoclast survival by signaling through mTOR//S6 kinase. Cell Death Differ 2003; 10: 1165–1177.
Cejka D, Hayer S, Niederreiter B, Sieghart W, Fuereder T, Zwerina J et al. Mammalian target of rapamycin signaling is crucial for joint destruction in experimental arthritis and is activated in osteoclasts from patients with rheumatoid arthritis. Arthritis Rheum 2010; 62: 2294–2302.
Acknowledgements
This work was supported by grants from the Shriners Hospital for Children (grant 85600) and the National Institute of Health (grant # AR054326, AR049192) to YA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by Y Shi
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Chen, TP., Swarnkar, G., Mbalaviele, G. et al. Myeloid lineage skewing due to exacerbated NF-κB signaling facilitates osteopenia in Scurfy mice. Cell Death Dis 6, e1723 (2015). https://doi.org/10.1038/cddis.2015.87
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2015.87
- Springer Nature Limited
This article is cited by
-
Inflammatory Responses Reprogram TREGS Through Impairment of Neuropilin-1
Scientific Reports (2019)
-
The interplay between inflammation and metabolism in rheumatoid arthritis
Cell Death & Disease (2015)