
Abstract
Metalloprotease-processed CD95L (cl-CD95L) is a soluble cytokine that implements a PI3K/Ca2+ signaling pathway in triple-negative breast cancer (TNBC) cells. Accordingly, high levels of cl-CD95L in TNBC women correlate with poor prognosis, and administration of this ligand in an orthotopic xenograft mouse model accelerates the metastatic dissemination of TNBC cells. The molecular mechanism underlying CD95-mediated cell migration remains unknown. Here, we present genetic and pharmacologic evidence that the anti-apoptotic molecules BclxL and Bcl-2 and the pro-apoptotic factors BAD and BID cooperate to promote migration of TNBC cells stimulated with cl-CD95L. BclxL was distributed in both endoplasmic reticulum (ER) and mitochondrion membranes. The mitochondrion-localized isoform promoted cell migration by interacting with voltage-dependent anion channel 1 to orchestrate Ca2+ transfer from the ER to mitochondria in a BH3-dependent manner. Mitochondrial Ca2+ uniporter contributed to this flux, which favored ATP production and cell migration. In conclusion, this study reveals a novel molecular mechanism controlled by BclxL to promote cancer cell migration and supports the use of BH3 mimetics as therapeutic options not only to kill tumor cells but also to prevent metastatic dissemination in TNBCs.
Similar content being viewed by others
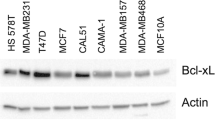
Main
Apoptosis occurs through extrinsic and intrinsic signaling pathways. Whereas death receptors trigger the extrinsic apoptotic signal, mitochondria and their regulation by the Bcl-2 family implement the intrinsic pathway. Members of the Bcl-2 family (which share at least one of the four domains of Bcl-2 homology) are key regulators of the balance between cell life and death. They control mitochondrial membrane permeabilization allowing the liberation into the cytoplasm of apoptogenic factors including cytochrome c, which is responsible for the cascade of caspase activation. The functional activity of pro-survival members of the family (Bcl-2, BclxL, Bcl-w, Mcl-1, and A1) is to sequester the pro-apoptotic members BAX and BAK, which are the executioner molecules of mitochondrial membrane permeabilization. BH3-only proteins such as BAD release BAX/BAK from their sequestration by Bcl-2/BclxL, and thereby implement the cell death program.1 Bcl-2 family proteins also have cellular functions beyond regulation of apoptosis. For instance, Bcl-2 and BclxL proteins regulate Ca2+ release from the endoplasmic reticulum (ER) by interacting with IP3R (for review see Monaco et al.2). Moreover, independent of its anti-apoptotic function, BclxL can exert a pro-metastatic role in breast and pancreatic cancer cells through a molecular mechanism that remains to be defined.3, 4, 5
CD95 (APO1/Fas) is a death receptor that has a pivotal role in immune homeostasis6, 7, 8 and the elimination of infected and transformed cells in mice and humans (for review see Strasser et al.9). Its cognate ligand, CD95L (FasL), belongs to the tumor necrosis factor family. Full-length transmembrane CD95L can be cleaved by metalloproteases,10 releasing a soluble ligand whose pathophysiological functions have been investigated only recently. A growing body of evidence indicates that this soluble ligand aggravates inflammation in chronic inflammatory disorders11, 12 and promotes oncogenesis13, 14, 15, 16 by inducing non-apoptotic signaling pathways such as NF-κB11 and PI3K.12
Breast cancer represents a heterogeneous pathology in terms of molecular profiles and clinical outcomes. Triple-negative breast cancers (TNBCs) are defined by a lack of expression of estrogen, progesterone, and HER2 receptors. Because of the absence of identified targets, treatment guidelines for patients with TNBC include only conventional chemotherapy, and thus prognosis remains significantly worse for these women than for other breast tumor patients benefiting from tailored therapies.17, 18 Accordingly, the development of new and targeted therapeutic strategies is required to improve the treatment efficiency of TNBC patients. We recently observed that among TNBC patients, high amounts of serum CD95L are correlated with poor prognosis and premature metastasis.15 Of interest, soluble metalloprotease-cleaved CD95L (cl-CD95L) induces non-apoptotic signaling pathways that promote metastatic dissemination of TNBCs.15 CD95L can also promote migration of colorectal cancer cells13 and glioblastoma cells.14 Nonetheless, the molecular mechanisms by which this receptor promotes oncogenesis remain to be identified.
Evasion from the apoptotic cell death program is a hallmark of cancer cells and is also an important mechanism to explain therapeutic failure.19 To achieve this, cancer cells exhibit deregulation of many apoptotic factors. Of interest, the molecular mechanisms by which these molecules prevent cell death have been extensively studied, regardless of their non-apoptotic functions that may also help to accelerate carcinogenesis. Herein, we demonstrate that the anti-apoptotic factors Bcl-2 and BclxL collaborate to induce CD95-mediated mitochondrial Ca2+ upload, increasing ATP production and thereby promoting cell migration in TNBC.
Results
BclxL and Bcl-2 are instrumental in the CD95-induced cell migration
Because BclxL and Bcl-2 are instrumental in controlling CD95-mediated apoptotic signaling, we wondered whether these molecules also modulated CD95-mediated non-apoptotic signaling pathways. To address this question, we examined the effect of ABT-737 in TNBC cells exposed to cl-CD95L. The BH3 mimetic ABT-737 exhibits high-affinity binding to the hydrophobic BH3-binding groove of BclxL and Bcl-2, whereas it has a low affinity for Mcl-1.20 Two TNBC cell lines were selected according to their resistance (BT549) or sensitivity (MDA-MB-231) to the ABT-737-mediated cell death pathway (Supplementary Figure S1A). Using the Boyden chamber assay, we observed that a non-toxic dose of ABT-737 abolished the migration of these TNBC cells stimulated with cl-CD95L (Figure 1a). Of interest, although ABT-737 treatment did not alter CD95-mediated Akt phosphorylation at serine 473 or threonine 308, two hallmarks of PI3K activation (Figure 1b), it abrogated the Ca2+ response (Figure 1c). To further investigate the role of BclxL and Bcl-2 in CD95-mediated cell migration, we next silenced Bcl-2 and BclxL in TNBC cells using shRNAs. To rule out misinterpretations due to off-target effects, we evaluated the molecular and biological effects of three different shRNAs targeting distinct sequences of the Bcl-2 and BclxL genes. Downregulation of Bclx in MDA-MB-231 (Figure 1d) and BT549 (Supplementary Figure S1B) cells did not affect Bcl-2 expression but inhibited cell migration when these TNBC cells were exposed to cl-CD95L (Figure 1e and Supplementary Figure S1C). Similarly, a reduction in Bcl-2 expression did not alter the BclxL expression level (Figure 1f and Supplementary Figure S1D) but inhibited CD95-induced cell motility (Figure 1g and Supplementary Figure S1E). In agreement with the BH3 mimetic data, downregulation of BclxL and Bcl-2 in TNBC cells did not inhibit the CD95-mediated PI3K signaling pathway and even enhanced it (Supplementary Figure S1F), suggesting that BclxL and Bcl-2 exert a BH3-independent negative regulatory function in the CD95-mediated PI3K response. Because downregulation of Bcl-2 and BclxL expression abrogated the CD95-mediated Ca2+ response (Figure 1h), these findings suggest that Bcl-2 and BclxL promote cell migration by modulating this Ca2+ response.


CD95 induces a BH3-dependent calcium signal that promotes TNBC cell motility. (a) MDA-MB-231 and BT549 cells were pre-incubated for 1 h in the presence of 0.1 and 10 μM ABT-737, respectively, and then treated or untreated with cl-CD95L (100 ng/ml) for 24 h. Cell migration was evaluated by the Boyden chamber assay. Migrating cells were fixed and stained with Giemsa. For each experiment, five images of random fields were acquired. To quantify cell migration, migrating cells were lysed and absorbance was measured at a wavelength of 560 nm. Values represent the mean±standard deviation of three independently performed experiments (***P<0.001; two-way Mann-Whitney). Bars=50 μm. (b) MDA-MB-231 and BT549 cells were pre-incubated for 1 h with or without non-toxic doses of ABT-737 (0.1 and 10 μM, respectively) and then treated or untreated with cl-CD95L (100 ng/ml) for the indicated amount of time. Cells were lysed and immunoblotting was performed as indicated. Akt was used as a loading control. (c) MDA-MB-231 and BT549 cells were pre-incubated for 1 h in the presence or absence of ABT-737 (0.1 μM). Cells were loaded with the Ca2+ probe FuraPE3 and then stimulated with cl-CD95L (100 ng/ml). The cytosolic calcium concentration was monitored via the ratio F/F0 (relative Ca2+[CYT]). Data represent the mean±s.e. of the mean of F/F0 (n>50 cells, at least three independent experiments). (d) MDA-MB-231 cells were infected with lentiviruses encoding shRNAs targeting different regions of Bclx mRNA. After puromycin selection, cells were lysed and the amounts of BclxL and Bcl-2 were evaluated by immunoblotting. β-Actin served as a loading control. (e) Migration of cells depicted in (d) was assessed using the Boyden chamber assay in the presence or absence of cl-CD95L (100 ng/ml) for 24 h. Migrating cells were fixed, stained (Giemsa), and lysed. Absorbance of migrating cells was measured at a wavelength of 560 nm. Values represent the mean±standard deviation of three independently performed experiments (**P<0.01, *P<0.05; two-way Mann-Whitney). (f) MDA-MB-231 cells were transduced with lentiviruses encoding shRNAs targeting different regions of Bcl-2. After puromycin selection, cells were lysed and the expression levels of Bcl-2 and BclxL were evaluated by immunoblotting. β-Actin served as a loading control. (g) Migration of cells depicted in (f) was evaluated by the Boyden chamber assay in the presence or absence of cl-CD95L (100 ng/ml) for 24 h. Migrating cells were fixed, stained (Giemsa), and lysed. Absorbance of migrating cells was measured at a wavelength of 560 nm. Values represent the mean±s.e. of the mean of three independently performed experiments (**P<0.01; two-way Mann-Whitney). (h) The calcium response was assessed in MDA-MB-231 cells infected with lentiviruses encoding various shRNAs targeting BclxL (left panel) or Bcl-2 (right panel). Cells were loaded with the Ca2+ probe Fluo2 and then stimulated with cl-CD95L (100 ng/ml). The cytosolic calcium concentration was monitored via the ratio F/F0 (relative Ca2+[CYT]). Data represent the mean±s.e. of the mean of F/F0 measured in GFP-positive cells (n>50 cells, at least three independent experiments)
BclxL localization in mitochondrial membranes triggers CD95-mediated cell migration
Bcl-2 and BclxL prevent the release of cytochrome c in response to many apoptotic stimuli.21, 22, 23 However, Bcl-2 and BclxL also associate with other cytoplasmic membranes, notably those of the ER,24 raising the possibility that they exert different functions according to their cellular distribution. It is noteworthy that loss of BclxL or its reconstitution in Bclx-knockout (KO) MEFs did not affect the expression level of CD95 (Figure 2a). BclxL was targeted to the outer mitochondrial membrane by replacing its membrane anchor with the mitochondrial insertion sequence of the listerial protein ActA (BclxL-MT) and to the ER membrane with the ER-specific sequence of cytochrome b5 (CytB5; BclxL-ER).25 To validate the cellular localization of these constructs, we fused each membrane sequence to GFP (Supplementary Figure S2A) and analyzed the distribution of GFP compared with that of ER and mitochondrion trackers using confocal microscopy (Supplementary Figure S2B). GFP-ActA co-localized with the mitochondrion tracker but not with the ER tracker, whereas the opposite was observed with the GFP-fused CytB5 sequence (Supplementary Figure S2B), confirming that these sequences selectively distributed BclxL to the expected organelles.


BclxL-MT is instrumental in cell migration. (a) Bclx-KO MEFs and their counterparts reconstituted with BclxL-ER, BclxL-MT, or BclxL-WT were lysed, and the expression levels of the indicated proteins were assessed by immunoblotting. Numbers indicate different clones. (b) The indicated MEFs were loaded with the Ca2+ probe FuraPE3 and then stimulated with cl-CD95L (100 ng/ml). The cytosolic calcium concentration was monitored via the ratio F/F0 (relative Ca2+[CYT]). Data represent the mean±s.e.of the mean of F/F0 in GFP-positive cells (n>50 cells, at least three independent experiments). (c) Migration of the indicated MEFs treated with or without cl-CD95L (100 ng/ml) was evaluated using the Boyden chamber assay. After 24 h, migrating cells were fixed, stained with Giemsa, and lysed. Absorbance was measured at a wavelength of 560 nm. Values represent the mean±s.e. of the mean of three independently performed experiments (**P<0.01, *P<0.05; two-way Mann-Whitney). (d) Parental MEFs were pre-incubated for 1 h in the presence or absence of a non-toxic dose of BAPTA-AM (5 μM) and then stimulated with or without cl-CD95L (100 ng/ml) for 24 h. Cell migration was analyzed using the Boyden chamber assay. Migrating cells were stained with Giemsa. Data are representative of three independently performed experiments. (e) MEFBclxL-MT were loaded with the Ca2+ probe FuraPE3 and pre-incubated for 1 h in the presence or absence of a non-toxic dose of ABT-199 (1 μM). Cells were then stimulated with cl-CD95L (100 ng/ml) and the cytosolic calcium concentration was monitored via the ratio F/F0 (relative Ca2+[CYT]). Data represent the mean±standard error of F/F0 (n>50 cells, at least three independent experiments). (f) MEFBclxL-WT and MEFBclxL-MT were pre-treated for 1 h with or without (control) a non-toxic dose of ABT-199 (1 μM) and then stimulated with cl-CD95L (100 ng/ml) for 24 h. Cell migration was assessed by the Boyden chamber assay. Migrating cells were fixed with methanol and stained with Giemsa. Absorbance of migrating cells was measured at a wavelength of 560 nm. Values represent the mean±standard deviation of three independently performed experiments (**P<0.01; two-way Mann-Whitney)
The trivial cytosolic calcium response observed in MEFBclxL−/− stimulated with cl-CD95L was increased in cells reconstituted with BclxL-WT (Figure 2b). BclxL-ER evoked a CD95-mediated cytosolic Ca2+ response similar to that in MEFs reconstituted with BclxL-WT (Figure 2b). By contrast, BclxL-MT failed to restore the cytosolic Ca2+ response in these cells (Figure 2b). Importantly, although MEFBclxL−/− did not migrate in the presence of cl-CD95L (Figure 2c), reconstitution of these cells with BclxL-WT restored CD95-mediated cell motility (Figure 2c). Unexpectedly, although BclxL-ER induced a cytosolic Ca2+ response similar to that provoked by BclxL-WT, it failed to trigger cell migration in the presence of cl-CD95L (Figure 2c). By contrast, BclxL-MT did not restore the CD95-mediated cytosolic Ca2+ response, but induced cell migration in a manner similar to BclxL-WT-expressing MEFs (Figure 2c). These latter results encouraged us to re-evaluate the role of Ca2+ in CD95-mediated migration of MEFs. The Ca2+ chelator BAPTA-AM completely inhibited motility of MEFs stimulated with cl-CD95L (Figure 2d), confirming that Ca2+ was instrumental in this cellular process.
ABT-199 is a BH3 mimetic that selectively targets Bcl-2.26 ABT-199 treatment abrogated the residual intracellular Ca2+ response observed in BclxL-MT-expressing MEFs stimulated with cl-CD95L (Figure 2e) and prevented migration of both BclxL-WT- and BclxL-MT-expressing MEFs (Figure 2f), indicating that Bcl-2 and mitochondrial BclxL cooperate to induce CD95-mediated cell motility. Because BclxL localization to mitochondria is mandatory to trigger CD95-mediated cell migration and mitochondria have a calcium-buffering ability,27 we wondered whether BclxL orchestrated CD95-mediated cell migration by promoting mitochondrial Ca2+ uptake. The Ca2+ concentration in the mitochondrial matrix was assessed using confocal microscopy and the mitochondrion-restricted Ca2+ probe Rhod2 (Figure 3a). Whereas the Ca2+ concentration in mitochondria of MEFBclxL−/− did not change in the presence of cl-CD95L, rapid and transient Ca2+ accumulation was observed in mitochondria of BclxL-WT-reconstituted cells (Figure 3b). Although BclxL-ER restored the CD95-mediated cytosolic Ca2+ response (Figure 2b), it failed to trigger mitochondrial Ca2+ uptake (Figure 3c). Importantly, reconstitution of MEFBclxL−/− with BclxL-MT restored CD95-mediated mitochondrial Ca2+ loading (Figure 3d), which was abolished by ABT-199 (Figure 3e). These findings clearly indicated that mitochondrial distribution of BclxL is mandatory for CD95-mediated cell migration and Bcl-2 and BclxL cooperate to elicit mitochondrial Ca2+ uptake in cells exposed to cl-CD95L.

BclxL-MT orchestrates CD95-mediated calcium influx in the mitochondrial matrix. Bclx-deficient MEFs (a) reconstituted with BclxL-WT (b), BclxL-ER (c), or BclxL-MT (d) were loaded with the mitochondrial Ca2+ probe (Rhod2, red) and cell-permeable Mitotracker (green) to label mitochondria. Left panels: confocal images of cells stimulated with cl-CD95L (100 ng/ml) for the indicated amount of time. Right panels: a region of interest was drawn in the Mitotracker-positive region (mitochondria) and fluorescence of the Ca2+ probe Rhod2 was monitored in this region. Graphs depict the overlay of the mitochondrial calcium response of 10 representative cells exposed to cl-CD95L. (e) The same experimental protocol was used as in (d), except that cells were pre-incubated for 1 h with a non-toxic dose of ABT-199 (1 μM). Images show t0, t0+35 s, t0+85 s, and t0+290 s of cells stimulated with cl-CD95L (100 ng/ml)
BID and BAD participate in CD95-mediated mitochondrial Ca2+ influx
BAD is a BH3-only protein that forms a heterodimer with BclxL to displace its interaction with BAX and induces the mitochondrion-dependent apoptotic response.28 BAD also contributes to glucose-driven mitochondrial respiration by modulating glucokinase activity.29 Because BclxL and Bcl-2 prevented Akt activation in cells exposed to cl-CD95L (Supplementary Figure S1F) and Akt phosphorylates BAD to sequester it in the cytosol and block its redistribution to mitochondria,30, 31 we next examined whether BAD contributed to CD95-driven cell migration. Knockout of BAD (Supplementary Figure S3A) enhanced CD95-mediated Akt activation (Supplementary Figure S3B), suggesting that BclxL, Bcl-2, and BAD exert a negative regulatory activity on Akt phosphorylation. More importantly, BAD-KO cells stimulated with cl-CD95L did not exhibit mitochondrial Ca2+ uptake (Supplementary Figure S3C). In cells stimulated with cl-CD95L, BclxL interacted with BAD (Supplementary Figure S3D) and this association was inhibited using the BH3 mimetic ABT-737 (Supplementary Figure S3D). In addition, BAD-KO MEFs failed to migrate in the presence of cl-CD95L (Supplementary Figure S3E), showing that CD95 recruits part of the classical apoptotic machinery in the presence of cl-CD95L to trigger a non-apoptotic signaling pathway that promotes cell migration. BID is another BH3-only protein involved in CD95-mediated cell death.32, 33 Indeed, CD95 activation leads to BID truncation (tBID) through a caspase-8-driven process and tBID is pivotal to induce mitochondrial outer membrane permeabilization and cell death.32 In agreement with the role of BID in the CD95-mediated apoptotic signal, BID-KO MEF cells did not die in the presence of a cytotoxic CD95L designated Ig-CD95L34 (Supplementary Figure S3F). Cells lacking BID triggered a cytosolic Ca2+ response similar to that observed in parental MEF cells when exposed to cl-CD95L (Supplementary Figure S3G) indicating that BID is not involved in Ca2+ release from the ER to the cytosol in cells exposed to cl-CD95L. By contrast, the mitochondrial Ca2+ uptake observed in parental MEF cells was abrogated in BID-KO MEF (Supplementary Figure S3H), which also failed to migrate in presence of cl-CD95L (Supplementary Figure S3I). Finally, although stimulation of MEF or BT549 cells with Ig-CD95L induced BID cleavage (Supplementary Figures S3J and S3K), BID was not processed in cells exposed to cl-CD95L (Supplementary Figures S3J and S3K), indicating that unlike the apoptotic signal, BID cleavage is not a pre-requisite to trigger CD95-mediated cell migration.
BAX and BAK are two pro-apoptotic Bcl-2 members orchestrating mitochondrial outer membrane permeabilization and thereby implementing the apoptotic signal. Although these molecules contributed to the CD95-mediated apoptotic signal (Supplementary Figure S3F), the loss of BAX and BAK did not impair the CD95-mediated cytosolic response (Supplementary Figure S3L) and even seemed to enhance this signal (Supplementary Figure S3L). Also, the CD95-mediated mitochondrial Ca2+ upload in MEF cells lacking BAX and BAK remained unaffected (Supplementary Figure S3M). Finally, BAX/BAK double KO MEF cells as well as parental MEF cells migrated when exposed to cl-CD95L (Supplementary Figure S3I) indicating that BAX and BAK were not involved in the CD95-mediated non-apoptotic signaling pathway. Overall, these findings highlighted that unlike BAX and BAK, BAD and BID contributed to the CD95-mediated non-apoptotic signaling pathway by promoting the Ca2+ flux into mitochondria.
Downregulation of BclxL in TNBC cells inhibits CD95-mediated mitochondrial Ca2+ uptake
To address whether similarly to MEFs, BclxL orchestrated CD95-mediated migration in TNBC cells, we silenced endogenous BclxL in a TNBC cell line (BT549ShBclx) using shRNAs targeting its 3′ untranslated region and reconstituted cells with BclxL-WT, BclxL-MT, or BclxL-ER constructs devoid of the 3′ untranslated region (Figure 4a). Reconstitution of BT549ShBclx with BclxL-WT or BclxL-ER restored the CD95-mediated cytosolic Ca2+ response (Figure 4b), while BclxL-MT failed to implement this signal (Figure 4b). Silencing of BclxL expression in BT549 cells inhibited CD95-mediated cell migration in comparison with parental TNBC cells (Figure 4c). On the other hand, BT549ShBclx reconstituted with BclxL-WT or BclxL-MT migrated when stimulated with cl-CD95L, whereas BclxL-ER failed to restore migration of BT549ShBclx (Figure 4c). In agreement with the MEF data, BclxL-WT and BclxL-MT led to mitochondrial Ca2+ loading in TNBC cells exposed to cl-CD95L (Supplementary Figure S4A), whereas BclxL-ER did not (Supplementary Figure S4A), confirming that BclxL residing in the outer membrane of mitochondria was mandatory to trigger Ca2+ loading of the organelle. Importantly, ABT-199 treatment abrogated the CD95-mediated residual cytosolic Ca2+ response (Figure 4d), ion accumulation in the mitochondrial matrix (Supplementary Figure S4B), and migration (Figure 4e) in BclxL-MT-expressing BT549ShBclx exposed to cl-CD95L. These results clearly demonstrated that Bcl-2 and BclxL cooperate to induce mitochondrial Ca2+ loading and migration of TNBC cells exposed to cl-CD95L.

Localization of BclxL in the outer mitochondrial membrane is pivotal to induce CD95-mediated cell migration. (a) BT549 cells infected with a shRNA targeting the Bclx 3′ untranslated region (ShBclx#54) were reconstituted with BclxL-WT, BclxL-ER, or BclxL-MT devoid of the 3′ untranslated region. Cells were lysed and the expression levels of the indicated proteins were evaluated by immunoblotting. (b) Cells in (a) were loaded with the Ca2+ probe Fluo2 and stimulated with cl-CD95L (100 ng/ml). [Ca2+]i was monitored via the ratio F/F0 (relative Ca2+[CYT]). Data represent the mean±s.e. of the mean of F/F0 in GFP-positive cells (n>50 cells, at least three independent experiments). (c) Migration of cells depicted in (a) was analyzed by the Boyden chamber assay for 24 h in the presence (100 ng/ml) or absence of cl-CD95L. Migrating cells were stained with Giemsa and lysed. Absorbance was measured at a wavelength of 560 nm. Values represent the mean±standard deviation of three independently performed experiments (*P<0.05; two-way Mann-Whitney). (d) Bclx-deficient BT549 cells (BT549ShBclx) reconstituted with BclxL-MT were loaded with the Ca2+ probe Fluo2 and pre-incubated for 1 h in the presence or absence of a non-toxic dose of ABT-199 (1 μM). Cells were then stimulated with 100 ng/ml cl-CD95L (black arrow), and [Ca2+]i was monitored via the ratio F/F0 (relative Ca2+[CYT]). Data represent the mean±standard error of the mean of F/F0 in GFP-positive cells (n>50 cells, at least three independent experiments). (e) BT549ShBclx-BclxL-MT were pre-incubated for 1 h with or without ABT-199 (1 μM) and then treated or untreated with cl-CD95L (100 ng/ml) for 24 h. Cell migration was analyzed using the Boyden chamber assay. Migrating cells were fixed with methanol, stained with Giemsa, and lysed. Absorbance was measured at a wavelength of 560 nm. Values represent the mean±standard deviation of three independently performed experiments (**P<0.01; two-way Mann-Whitney)
BclxL interacts with voltage-dependent anion channel (VDAC) 1 and modulates mitochondrial Ca2+ uniporter (MCU) to promote cell migration
Recent data showed that BclxL interacts with isoforms 1 and 3 of VDAC at the outer mitochondrial membrane to activate mitochondrial Ca2+ uptake and modulate cell death.35, 36 BclxL immunoprecipitation revealed that VDAC3 was not present in the immune complex formed in cells stimulated with cl-CD95L (data not shown). On the other hand, although no association between BclxL and VDAC1 was detected in BclxL-ER-expressing MEFs (Figure 5a), BclxL interacted with VDAC1 in BclxL-MT-expressing MEFs stimulated with cl-CD95L (Figure 5a). Using co-immunoprecipitation (Figure 5b) and proximity ligation assay (Supplementary Figure S5A), we observed that a residual level of BclxL (wild type and MT) interacted with VDAC1 in unstimulated BT549ShBclx reconstituted with BclxL-WT or BclxL-MT cells (Supplementary Figure S5A), whereas no such interactions were detected in BT549ShBclx reconstituted with an empty vector or with BclxL-ER (Supplementary Figure S5A). Furthermore, the number of VDAC1/BclxL interactions significantly increased in BclxL-WT (Supplementary Figure S5A)- and BclxL-MT-expressing BT549ShBclx cells exposed to cl-CD95L (Figure 5b and Supplementary Figure S5A), whereas we did not observe VDAC1/BclxL interaction in empty vector or BclxL-ER-expressing BT549ShBclx (Figure 5b and Supplementary Figure S5A). Also, cell-permeant N-terminal (amino acids 10–26 of VDAC1) and L14-15 (amino acids 208–218 of VDAC1) peptides, which alter binding of BclxL to VDAC1/3,35 completely inhibited CD95-mediated mitochondrial Ca2+ influx (Figure 5c and Supplementary Figure S5B) and cell migration (Figure 5d and Supplementary Figure S5C) in MEFs and TNBC cells. Ca2+ can accumulate in the mitochondrial matrix through activation of MCU residing in the inner mitochondrial membrane.37, 38 Pre-treatment of BT549ShBclx reconstituted with BclxL-WT or BclxL-MT with a selective MCU inhibitor, Ru360,38 not only prevented CD95-mediated mitochondrial Ca2+ influx (Figure 6a) but also abrogated cell migration (Figure 6b). Inhibition of MCU activity also inhibited CD95-mediated mitochondrial Ca2+ loading (Supplementary Figure S6A) and cell migration (Supplementary Figure S6B) in MEF cells. Because it was recently reported that short-term Ca2+ loading in mitochondria can drive ATP production,39 we asked whether mitochondrial Ca2+ accumulation observed in cancer cells stimulated with cl-CD95L was instrumental in producing the energy required for cell migration. BclxL-KO MEFs stimulated with cl-CD95L failed to increase ATP production (Supplementary Figure S7A), while reconstitution of these cells with BclxL-WT or BclxL-MT restored the increased ATP production (Supplementary Figure S7A). By contrast, redistribution of BclxL to the ER membrane failed to generate ATP in the presence of cl-CD95L (Supplementary Figure S7A). Of interest, inhibition of the interaction between BclxL and VDAC1 using the L14-15 peptide (Supplementary Figure S7B) or of MCU activity using Ru360 (Supplementary Figure S7B) abrogated CD95-induced ATP production. This shows that the BclxL/VDAC1/MCU complex in cells stimulated with cl-CD95L orchestrates ATP production necessary for cell migration (Figure 7).

Calcium entry into mitochondria is driven by a BclxL/VDAC1 interaction, which promotes cell motility. (a) Bclx-KO MEFs reconstituted with BclxL-MT (MEFBclxL-MT) or BclxL-ER (MEFBclxL-ER) were stimulated with cl-CD95L (100 ng/ml) for the indicated amount of time. Cells were lysed and BclxL was immunoprecipitated. The immunoprecipitated complex was resolved by SDS-PAGE and immunoblotted with anti-BclxL and anti-VDAC1 antibodies. MEFBclxL−/− were used as a negative control. (b) The experiment in (a) was performed with BT549ShBclx reconstituted with BclxL-MT or BclxL-ER. (c) BclxL-deficient MEFs reconstituted with BclxL-WT were loaded with the mitochondrial Ca2+ probe (Rhod2) and cell-permeable Mitotracker Green. Representative graphs (10 cells) depicting the relative mitochondrial calcium concentration in control cells (left panel) and L14-15- (middle panel) and N-Ter (right panel)-treated cells (1 μM, 1 h) are shown. (d) BclxL-KO MEFs reconstituted with BclxL-WT (MEFBclxL-WT) or BclxL-MT (MEFBclxL-MT) were pre-incubated with the indicated cell-penetrating peptides (1 μM) for 1 h and then stimulated with or without cl-CD95L (100 ng/ml). Cell migration was evaluated by the Boyden chamber assay. Migrating cells were fixed and stained with Giemsa. For each experiment, five images of random fields were acquired. To quantify cell migration, migrating cells were lysed and absorbance was measured at a wavelength of 560 nm. Values represent the mean±standard deviation of three independently performed experiments (**P<0.01, *P<0.05; two-way Mann-Whitney)

MCU orchestrates Ca2+ entry into mitochondria of cells exposed to cl-CD95L. (a) BT549ShBclx cells reconstituted with BclxL-WT (BT549-BclxL-WT, upper panels) or BclxL-MT (BT549-BclxL-MT, lower panels) were loaded with a mitochondrial Ca2+ probe (Rhod2, red) and cell-permeable Mitotracker (green) to label mitochondria. Cells were pre-incubated for 1 h with a non-toxic amount of Ru360 (10 μM) and stimulated with cl-CD95L (100 ng/ml). Graphs depict the relative mitochondrial calcium concentrations in the indicated cells pre-treated with vehicle (left panel) or Ru360 (10 μM, right panel). (b) BT549ShBclx reconstituted with BclxL-WT and BclxL-MT cells were pre-incubated with Ru360 (10 μM) for 1 h and then stimulated with or without cl-CD95L (100 ng/ml). Cell migration was evaluated by the Boyden chamber assay. Migrating cells were fixed and stained with Giemsa. For each experiment, five images of random fields were acquired. To quantify cell migration, migrating cells were lysed and absorbance was measured at a wavelength of 560 nm. Values represent the mean±standard deviation of three independently performed experiments (**P<0.01, *P<0.05; two-way Mann-Whitney)

CD95-mediated cell migration occurs through a Bcl-2/BclxL/VDAC1/MCU-driven mitochondrial calcium loading. Death-inducing signaling complex (DISC) and motility-inducing signaling complex (MISC) are formed through protein–protein interaction. For the CD95-mediated apoptotic signaling pathway, type II cells (e.g., hepatocytes) rely on the release of apoptotic factors (i.e., cytochrome c, smac/Diablo, Omi/Hrt2A) by the mitochondria to implement the apoptotic cue while Type I (e.g., T lymphocytes) does not. The CD95-mediated calcium response is initiated by PLCγ1 activation leading to release of the ER-stored Ca2+ in the cytosol (1), followed by a store-operated calcium entry (SOCE) through activation of the Ca2+ channel Orai1 (2). This study shows the pivotal role played by the calcium transfer from ER to mitochondria (3) in TNBC cell migration induced by cl-CD95L
Discussion
This study revealed that the anti-apoptotic members Bcl-2 and BclxL contribute to the migration of TNBC cells exposed to cl-CD95L, recapitulating TNBC women with the poorest prognosis.15 We provide novel insights into an original molecular mechanism by which BclxL and Bcl-2 cooperate to promote cell migration (Figure 7). Of note, the activity of pyruvate dehydrogenase, an enzyme linking glycolysis to the tricarboxylic acid cycle and ATP production, is positively regulated by Ca2+ in the mitochondrial matrix.40 In this regard, whereas pyruvate dehydrogenase phosphorylation by pyruvate kinase suppresses its activity, its dephosphorylation by Ca2+-dependent pyruvate phosphatase enhances its activity.41 Therefore, we envision that CD95-mediated mitochondrial Ca2+ loading enhances pyruvate phosphatase activity, which in turn activates pyruvate dehydrogenase to promote mitochondrial respiration and ATP production.
Our findings demonstrate that although BclxL expression at the ER membrane restores a cytosolic Ca2+ signal, it cannot evoke Ca2+ passage from ER stores to the mitochondrial matrix. By contrast, its mitochondrion-restricted isoform promotes this mitochondrial Ca2+ uptake but fails to restore the ‘wild-type’ cytosolic Ca2+ signal, indicating that mitochondrion- and ER-localized BclxL contribute to different Ca2+ cues and that both distributions are mandatory to evoke a complete Ca2+ response in malignant cells exposed to cl-CD95L. Interestingly, the Bcl-2 selective BH3 mimetic ABT-199 abrogated both cytosolic and mitochondrial Ca2+ signals. Because the CD95-mediated Ca2+ response is initiated by IP3R-dependent depletion of ER Ca2+ stores followed by extracellular Ca2+ influx via activation of the plasma membrane channel ORAI1,12, 15, 42 these findings suggest that Bcl-2 can control IP3R activity in a BH3-dependent manner (see Figure7). However, this contradicts various studies showing that Bcl-2 and BclxL mainly modulate the release of Ca2+ from ER through an interaction of their BH4 domain with IP3R.43 Nonetheless, it is noteworthy that Bcl-2 and BclxL participate in hubs that modulate various cellular outcomes such as cell survival, proliferation, and metabolism.44 Most, if not all, studies evaluating Bcl-2- and BclxL-dependent regulation of IP3R focused on its effect in the implementation of the apoptotic signaling pathway.43 Therefore, we postulate that similar to death receptors that can implement opposite signaling pathways according to the cellular context (normal versus transformed cells13, 14, 45, 46) or the ligand with which they interact (soluble versus transmembrane11, 12, 15), IP3Rs are involved in different molecular complexes, which might be regulated in a different manner by Bcl-2 family members.
Our data also provide insight into how loss of BclxL in the ER membrane impairs the CD95-mediated cytosolic Ca2+ response, showing that even if Bcl-2 participates in Ca2+ release from ER stores, its presence is not sufficient to evoke a ‘wild-type’ cytosolic Ca2+ response. To explain why there is no redundancy among the Bcl-2 family in the cl-CD95L-mediated Ca2+ signal, we envision that Bcl-2 and BclxL coordinate the efficient release of Ca2+ from the ER by acting on different partners in a large IP3R(s) complex or by modulating the activity of different IP3R isoforms necessary for transmitting a complete Ca2+ response. Our study also reveals the complex interplay between these two Bcl-2 members in the regulation of Ca2+ flux from the ER to the mitochondrial matrix.
Of note, high-calcium microdomains are observed in the lamella of migrating cells and are involved in guiding cells.47 Accordingly, Ca2+ buffering by mitochondria and its control by Bcl-2 and BclxL may be essential to steer cells across a cl-CD95L gradient in TNBC tissues.
IP3R can be phosphorylated by Akt, resulting in reduced Ca2+ efflux from the ER and less apoptosis.48 Of note, whereas the BH3 mimetic ABT-737 did not affect CD95-mediated Akt phosphorylation (Figure 1b), silencing of BclxL or Bcl-2 enhanced Akt activation (Supplementary Figure S1F), indicating that Bcl-2/BclxL-driven PI3K/Akt regulation occurs through a BH3-independent mechanism, which remains to be identified. However, our data led us to hypothesize that by reducing the intensity of PI3K/Akt signaling induced by cl-CD95L in TNBC cells, BclxL and Bcl-2 may enhance IP3R activity and thereby promote Ca2+ flux from the ER to mitochondria to produce more ATP, fueling cell migration.
A phase II clinical trial demonstrated that a decoy CD95 receptor, APG101, can impede the CD95/CD95L interaction in humans suffering from glioblastoma and allow partial response in these patients.49 Although APG101 may represent a short-term therapeutic approach for TNBC patients, its inability to discriminate between the anti-tumor/infectious (i.e., apoptotic signaling) and the pro-inflammatory actions of CD95 may engender, if used in a chronic manner, unexpected adverse events such as infection and tumor relapse.
Overall, this study highlights that the classical ‘apoptotic machinery’ is instrumental in cell migration. Therefore, tumor cells are not only selected according to their ability to resist apoptotic signaling pathways but also their capacity to use this ‘apoptotic machinery’ in order to become more aggressive and thus metastatic. Navitoclax, also designated ABT-263, is an orally bioavailable derivative of ABT-73750 that unfortunately showed side effects such as thrombocytopenia in phase II clinical trials.51 Our results indicate that a combination of low doses of navitoclax with classical chemotherapy applied in TNBC women is an attractive therapeutic option not only to reduce the risk of metastatic dissemination in patients with a high serum concentration of CD95L but also to decrease the side effects observed when this drug is used at cytotoxic doses.52
Materials and Methods
Cell lines and shRNAmir lentiviral transduction
BclxL-KO MEF cells were described earlier53 and BAD-KO MEF cells were from Dr P Juin (Nantes, France). BAX/BAK double KO MEF cells were kindly provided by Dr D Arnoult (Villejuif, France). TNBC cell lines, MDA-MB-231 and BT549, and MEF cell lines, Bcl-2 KO and BID KO, were obtained from ATCC (LGC Standards, Molsheim, France). All cells were cultured in DMEM supplemented with 8% v/v heat-inactivated FCS and 2 mM L-glutamine at 37 °C in a 5% CO2 incubator. Silencing experiments were performed using lentiviral vector encoding shRNA targeting Bclx (RHS4430-101108659; RHS4430-99157776; RHS4430-99890586; RHS4430-99883769; RHS4430-99881577), Bcl-2 (RHS4430-98901868; RHS4430-98851633; RHS4430-101159334) or a scrambled shRNAmir (negative control) (OpenBiosystems, Waltham, MA, USA). Then, cells stably expressing shRNA were selected for 7 days using 0.5 μg/ml of puromycin. To reconstitute Bclx-silenced BT549 cells with our BclxL constructs, shRNA-BclxL-expressing cells were transfected by electroporation with pcDNA3 vector encoding BclxL-WT, BclxL-Acta (MT) or BclxL-CytB5 (ER) or empty vector. Twenty-four hours after transfection, cells were placed in a medium supplemented with 0.6 mg/ml of neomycin. Neomycin-resistant colonies were isolated using cloning cylinders (Bellco Glass, Vineland, NJ, USA).
Antibodies, plasmids, and reagents
ABT-737 and ABT-199 were obtained from Selleckchem (Houston, TX, USA). Anti-β-actin was purchased from Sigma (L’Isle-d’Abeau-Chesnes, France). Anti-BclxL, anti-Bcl-2, anti-Akt, anti-phosphoS473 Akt, anti-phosphoThr308 Akt, and anti-BAD were from Cell Signaling Technology (Boston, MA, USA). Anti-BID mAb was from Millipore (Molsheim, France). Anti-BclxL (clone E18) and anti-VDAC1 (ab14734) were purchased from Abcam (Cambridge, UK). Anti-CD95 (C20), anti-VDAC1, and Ru360 were purchased from Santa Cruz (Santa Cruz, CA, USA). Duolink kits (DUO92102) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
N-terminal peptide (LGKSARDVFTKGYGFG) and L14-15 peptide (LAWTAGNSNTR) (VDAC1 sequences) were synthesized by SynPeptide (Shanghai, China). These peptides were coupled with the cell-permeant sequence of antennapedia homeodomain (RQIKIWFQNRRMKWKK) to their C-terminal domain. IgCD95L and metalloprotease-cleaved CD95L were generated in the laboratory.12 FuraPE3 and Fluo2-AM were from Euromedex (Souffelweyersheim, France); mitotracker green and Rhod2-AM were from Life Technologies SAS (Saint Aubin, France); and BAPTA-AM from Sigma.
GFP sequence lacking stop codon was amplified by PCR from pEGFPN1 sequence using primer pair: sense CGGGATCCATGGTGAGCAAGGGCGAGGAGCTG and antisense CCGCTCGAGCTTGTACAGCTCGTCCATGCCG. The amplicons was digested by Xho1 and BamH1 and inserted into Xho1 and BamH1-cleaved pcDNA3.1(+). Next, pcDNA3.1(+)-GFP was linearized using XhoI/XbaI and sequences corresponding to amino acid residues 107–142 of human CytB5 (ITTIDSSSSWWTNWVIPAISAVAVALMYRLYMAED) or residues 613–639 of Acta (‘Actin Assembly inducing protein’) (LILAMLAIGVFSLGAFIKIIQLRKNN) synthesized by Sigma-Aldrich were inserted in frame with the GFP sequence.
BclxL (amino acid residues 1–209) lacking transmembrane region weas amplified by PCR using pCMV-BclxL (Origene, Rockville, MD, USA) as a template with the following primer pair: (BCLx sense) 5′ CCCAAGCTTATGTCTCAGAGCAACCGGGAGC 3′, (BCLx antisense) 5′ CCCTCGAGGCGTTCCTGGCCCTTTCGGC 3′. Amplicon was digested by HindIII/XhoI and inserted into HindIII/XhoI-digested pCDNA3.1(+)-GFP-Acta or pCDNA3.1(+)-GFP-CytB5 vectors to generate pcDNA3.1(+)-BclxL-Acta (BclxL-MT), pcDNA3.1(+)-BclxL-CytB5 (BclxL-ER) constructs. All constructs were validated by sequencing (GATC Biotech, Mulhouse, France).
Immunoblot analysis
Cells were lysed for 30 min at 4 °C in lysis buffer (25 mM HEPES (pH 7.4), 1% Triton X-100, 150 mM NaCl, and 2 mM EGTA supplemented with a mix of protease and phosphatase inhibitors (Sigma)). Protein concentration was determined by the bicinchoninic acid method (Pierce, Rockford, IL, USA). Proteins were resolved by SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked for 30 min with TBST (50 mM Tris, 160 mM NaCl, 0.05% Tween 20, pH 7.8) containing 5% w/v dried skimmed milk or 5% BSA and then incubated overnight with primary antibody at 4 °C. The membrane was washed (TBST) and secondary HRP-labeled antibody (SouthernBiotech, Birmingham, AL, USA) was added for 1 h. The proteins were visualized with the enhanced chemiluminescence substrate kit (Pierce).
Immunoprecipitation
MEFBclxL-MT and MEFBclxL-ER cells or BT-549shBclx-BclxL-MT and BT-549shBclx-BclxL-ER (107 cells) were stimulated with cl-CD95L for the indicated times and then lysed. Lysate was incubated for 1 h at 4 °C with 1 μg of anti-BclxL mAb (clone E18) and BclxL was immunoprecipitated using proteins A/G-conjugated magnetic beads (Ademtech, Pessac, France). After extensive washing, the immune complex was resolved by SDS-PAGE and immunoblotting were performed.
Measure of cell death
Cell death was assessed using viability MTT assay as described previously.54
Calcium monitoring
Single-cell cytosolic calcium imaging was performed ratiometrically, using FuraPE3-AM calcium dye, as described previously.16 Coverslips were mounted in a recording chamber positioned on the stage of an inverted epifluorescence microscope (IX70, Olympus, Tokyo, Japan) equipped with an × 40 UApo/340-1.15 W objective. Cells were loaded with 5 μM FuraPE3-AM at room temperature in Hank’s Balanced Salt Solution (HBSS) for 30 min. FuraPE3-AM exhibits limited compartmentalization in intracellular stores and is leakage-resistant.55 The cells were rinsed with HBSS and incubated in the absence of the Ca2+ probe for 15 min to complete de-esterification of the dye. Fluorescence micrograph images were captured at 510 nm and at 12-bit resolution by a fast-scan camera (CoolSNAP fx Monochrome, Photometrics, Tucson, AZ, US). FuraPE3-AM was excited at 340 and 380 nm alternately and the ratios of the resulting images were produced at constant 10-s intervals. Regions of interest were drawn on certain recorded cells to restrict data collection to specific regions. Imaging was controlled by Universal Imaging software, including Metafluor and Metamorph. Data were processed using OriginPro 7.5 software (Origin Lab, Northampton, MA, USA).
For experiments on GFP-expressing cell lines, Fluo2-AM was used, because GFP fluorescence disturbs Ca2+ measurement with FuraPE3. ShRNA-transduced cells (GFP expressing cells) were located by their emission of fluorescence at 530 nm for a light excitation at 485 nm. Ca2+ changes were evaluated by exciting Fluo2-AM-loaded cells at 535nm. The values of the emitted fluorescence (605 nm) for each cell (F) were normalized to the starting fluorescence (F0) and reported as F/F0 (relative Ca2+[CYT]). Only GFP-positive cells were considered. Cells were loaded with Fluo2-AM (1 μM) for 30 min and then incubated for 15 min in HBSS to complete de-esterification of the dye.
Single-cell mitochondrial calcium imaging was performed on Rhod2, and mitotracker-loaded cells. Cells were loaded with 3 μM Rhod2-AM and mitotracker green at room temperature in HBSS for 120 min. Mitotracker green and Rhod2 were alternately excited using the 488- and 568-nm lines, respectively, of a krypton-argon laser. Emitted fluorescence was acquired using a Zeiss LSM 510 meta confocal microscope (Zeiss, Göttingen, Germany) equipped with an ApoPLAN × 63 objective. The emitted fluorescence was filtered using a dual bandpass filter set and collected and analyzed using Zeiss software. In some experiments, cells were placed in a Ca2+-free medium (HBSS in which CaCl2 was omitted and 100 μM EGTA). This medium was added to the cells just before recording to avoid leak of the intracellular calcium stores. Data from ~30 cells were acquired per field. One field was acquired from each coverslip and the data pooled from six independent coverslips on three different days. Fluorescence intensity changes were normalized to the initial fluorescence value F0 and expressed as F/F0. Data were summarized as mean +/− S.E.M.
Proximity ligation assay
BclxL-deficient BT549 cells reconstituted with BclxL-WT, BclxL-MT, and BclxL-ER were let adhere to coverslip for 24 h. Then, cells were stimulated with or without cl-CD95L (100 ng/ml) for 15 min. After extensive washes with PBS, cells were fixed and permeabilized with ethanol (70%) for 20 min at −20 °C. Cells were blocked with PBS-5% FCS for 20 min at RT, then incubated for 20 min at 4 °C with a mix of primary antibody consisting of anti-VDAC1 (ab14734) and anti-BclxL (clone E18) diluted to 1:500 in PBS-5% FCS. Following steps were realized according to the manufacturer recommendations.
Motility assays
After membrane hydration of Boyden chambers (Millipore) containing 8 μm pore membranes, 105 cells were added to the top chamber. The bottom chamber was filled with low serum (1%)-containing medium in the presence or absence of cl-CD95L (100 ng/ml). After 24 h, cells were fixed with methanol and stained with Giemsa. Stained cells were then removed from the top side of the membrane using a cotton-tipped swab and five representative pictures were taken of cells that have migrated to the membrane reverse side. For each experiment, invading cells were lysed and absorbance at 560 nm was measured.
Intracellular ATP measurement
Mitochondrial ATP levels were measured by transfecting MEF cells with the mitochondrion-targeted FRET-based probe (Mito-ATEAM), as previously described.56 Briefly, cells seeded on coverslips were washed in a modified Ringer's medium (in mM: 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, and 10 glucose at pH 7.3) and images were collected using 440 nm excitation and alternate 475/535 nm emission on an Axiovert S100 TV microscope through a × 40, 1.3 NA oil-immersion objective (Carl Zeiss, Oberkochen, Germany) equipped with a 16-bit CCD camera, Xenon lamp and filter-based wavelength switcher (Visitron Systems, Puchheim, Germany). Relative changes in mitochondrial ATP are reported as background-subtracted FRET.
Statistical analysis
Two-tailed Mann–Whitney non-parametrical tests were used for comparison of means.
Abbreviations
- Bad:
-
BCL2-associated agonist of cell death
- Bak:
-
BCL2-antagonist/killer
- BAPTA-AM AM:
-
2 bis-(2-aminophenoxy)ethane-N; N; N'; N'-tetraacetic acid acetoxymethyl ester
- Bax:
-
BCL2-associated X protein
- Bid:
-
BH3 interacting domain death agonist
- BH3:
-
Bcl2-homology domain
- GFP:
-
Green fluorescent protein
- IP3R:
-
Inositol 1,4,5-trisphosphate receptor
- MCU:
-
Mitochondrial calcium uniporter
- MEF:
-
Mouse embryonic fibroblast
- NF-κB:
-
Nuclear dactor of kappa light polypeptide gene enhancer in B-cells
- PI3K:
-
Phosphatidyl-inositol-4,5-bisphosphate 3-kinase
- TNBC:
-
Triple-negative breast cancer
- VDAC:
-
Voltage-dependent anion channel
References
Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 2001; 8: 705–711.
Monaco G, Vervliet T, Akl H, Bultynck G . The selective BH4-domain biology of Bcl-2-family members: IP3Rs and beyond. Cell Mol Life Sci 2013; 70: 1171–1183.
Du YC, Lewis BC, Hanahan D, Varmus H . Assessing tumor progression factors by somatic gene transfer into a mouse model: Bcl-xL promotes islet tumor cell invasion. PLoS Biol 2007; 5: e276.
Hager JH, Ulanet DB, Hennighausen L, Hanahan D . Genetic ablation of Bcl-x attenuates invasiveness without affecting apoptosis or tumor growth in a mouse model of pancreatic neuroendocrine cancer. PLoS One 2009; 4: e4455.
Martin SS, Ridgeway AG, Pinkas J, Lu Y, Reginato MJ, Koh EY et al. A cytoskeleton-based functional genetic screen identifies Bcl-xL as an enhancer of metastasis, but not primary tumor growth. Oncogene 2004; 23: 4641–4645.
Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 1995; 81: 935–946.
Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science 1995; 268: 1347–1349.
Drappa J, Vaishnaw AK, Sullivan KE, Chu JL, Elkon KB . Fas gene mutations in the Canale-Smith syndrome, an inherited lymphoproliferative disorder associated with autoimmunity. N Engl J Med 1996; 335: 1643–1649.
Strasser A, Jost PJ, Nagata S . The many roles of FAS receptor signaling in the immune system. Immunity 2009; 30: 180–192.
Tauzin S, Debure L, Moreau JF, Legembre P . CD95-mediated cell signaling in cancer: mutations and post-translational modulations. Cell Mol Life Sci 2012; 69: 1261–1277.
O' Reilly LA, Tai L, Lee L, Kruse EA, Grabow S, Fairlie WD et al. Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature 2009; 461: 659–663.
Tauzin S, Chaigne-Delalande B, Selva E, Khadra N, Daburon S, Contin-Bordes C et al. The naturally processed CD95L elicits a c-yes/calcium/PI3K-driven cell migration pathway. PLoS Biol 2011; 9: e1001090.
Hoogwater FJ, Nijkamp MW, Smakman N, Steller EJ, Emmink BL, Westendorp BF et al. Oncogenic K-Ras turns death receptors into metastasis-promoting receptors in human and mouse colorectal cancer cells. Gastroenterology 2010; 138: 2357–2367.
Kleber S, Sancho-Martinez I, Wiestler B, Beisel A, Gieffers C, Hill O et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell 2008; 13: 235–248.
Malleter M, Tauzin S, Bessede A, Castellano R, Goubard A, Godey F et al. CD95L cell surface cleavage triggers a prometastatic signaling pathway in triple-negative breast cancer. Cancer Res 2013; 73: 6711–6721.
Peter ME, Hadji A, Murmann AE, Brockway S, Putzbach W, Pattanayak A et al. The role of CD95 and CD95 ligand in cancer. Cell Death Differ 2015; 22: 549–559.
Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 2007; 13 (15 Pt 1): 4429–4434.
Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res 2007; 13: 2329–2334.
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435: 677–681.
Kharbanda S, Pandey P, Schofield L, Israels S, Roncinske R, Yoshida K et al. Role for Bcl-xL as an inhibitor of cytosolic cytochrome C accumulation in DNA damage-induced apoptosis. Proc Natl Acad Sci USA 1997; 94: 6939–6942.
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD . The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 1997; 275: 1132–1136.
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 1997; 275: 1129–1132.
Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC . Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res 1993; 53: 4701–4714.
Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW . Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J 1996; 15: 4130–4141.
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013; 19: 202–208.
Gilabert JA, Parekh AB . Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca(2+) current I(CRAC). EMBO J 2000; 19: 6401–6407.
Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ . Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995; 80: 285–291.
Danial NN, Walensky LD, Zhang CY, Choi CS, Fisher JK, Molina AJ et al. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat Med 2008; 14: 144–153.
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997; 91: 231–241.
del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G . Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 1997; 278: 687–689.
Li H, Zhu H, Xu CJ, Yuan J . Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94: 491–501.
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X . Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94: 481–490.
Daburon S, Devaud C, Costet P, Morello A, Garrigue-Antar L, Maillasson M et al. Functional characterization of a chimeric soluble Fas ligand polymer with in vivo anti-tumor activity. PLoS One 2013; 8: e54000.
Huang H, Hu X, Eno CO, Zhao G, Li C, White C . An interaction between Bcl-xL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J Biol Chem 2013; 288: 19870–19881.
Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol 2002; 159: 613–624.
De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R . A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011; 476: 336–340.
Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011; 476: 341–345.
Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA et al. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep 2015; 12: 15–22.
Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010; 142: 270–283.
Patel MS, Korotchkina LG . Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans 2006; 34 (Pt 2): 217–222.
Khadra N, Bresson-Bepoldin L, Penna A, Chaigne-Delalande B, Segui B, Levade T et al. CD95 triggers Orai1-mediated localized Ca2+ entry, regulates recruitment of protein kinase C (PKC) beta2, and prevents death-inducing signaling complex formation. Proc Natl Acad Sci USA 2011; 108: 19072–19077.
Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ 2012; 19: 295–309.
Choe CU, Ehrlich BE . The inositol 1,4,5-trisphosphate receptor (IP3R) and its regulators: sometimes good and sometimes bad teamwork. Sci STKE 2006; 2006: re15.
Barnhart BC, Legembre P, Pietras E, Bubici C, Franzoso G, Peter ME . CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. Embo J 2004; 23: 3175–3185.
Chen L, Park SM, Tumanov AV, Hau A, Sawada K, Feig C et al. CD95 promotes tumour growth. Nature 2010; 465: 492–496.
Wei C, Wang X, Chen M, Ouyang K, Song LS, Cheng H . Calcium flickers steer cell migration. Nature 2009; 457: 901–905.
Pinton P, Giorgi C, Pandolfi PP . The role of PML in the control of apoptotic cell fate: a new key player at ER-mitochondria sites. Cell Death Differ 2011; 18: 1450–1456.
Tuettenberg J, Seiz M, Debatin KM, Hollburg W, von Staden M, Thiemann M et al. Pharmacokinetics, pharmacodynamics, safety and tolerability of APG101, a CD95-Fc fusion protein, in healthy volunteers and two glioma patients. Int Immunopharmacol 2012; 13: 93–100.
Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008; 68: 3421–3428.
Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res 2012; 18: 3163–3169.
Schoenwaelder SM, Jackson SP . Bcl-xL-inhibitory BH3 mimetics (ABT-737 or ABT-263) and the modulation of cytosolic calcium flux and platelet function. Blood 2012; 119: 1320–1321, author reply 1321-1322.
Eno CO, Eckenrode EF, Olberding KE, Zhao G, White C, Li C . Distinct roles of mitochondria- and ER-localized Bcl-xL in apoptosis resistance and Ca2+ homeostasis. Mol Biol Cell 2012; 23: 2605–2618.
Chaigne-Delalande B, Mahfouf W, Daburon S, Moreau JF, Legembre P . CD95 engagement mediates actin-independent and -dependent apoptotic signals. Cell Death Differ 2009; 16: 1654–1664.
Vorndran C, Minta A, Poenie M . New fluorescent calcium indicators designed for cytosolic retention or measuring calcium near membranes. Biophys J 1995; 69: 2112–2124.
Imamura H, Nhat KP, Togawa H, Saito K, Iino R, Kato-Yamada Y et al. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci USA 2009; 106: 15651–15656.
Acknowledgements
We are grateful to Dr R Castellano and Dr Y Collette (TrGET and CRCM animal facilities, Université de la Méditerranée, Marseille). We thank Drs. H Imamura and H Noji for providing the ATeam probes. This work was supported by INCa PLBIO, Ligue Contre le Cancer (Comités d’Ille-et-Vilaine/du Morbihan/des Côtes d’Armor/du Maine et Loire et des Pyrénées Atlantiques), Fondation ARC, Région Bretagne, Rennes Métropole. Biotrial and ANR LabCom. LC was supported by NIH grants (CA106599, CA175003, and GM106386).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by S Nagata
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Fouqué, A., Lepvrier, E., Debure, L. et al. The apoptotic members CD95, BclxL, and Bcl-2 cooperate to promote cell migration by inducing Ca2+ flux from the endoplasmic reticulum to mitochondria. Cell Death Differ 23, 1702–1716 (2016). https://doi.org/10.1038/cdd.2016.61
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.61
- Springer Nature Limited