
Abstract
Background:
Several microRNA (miRNA) molecules have emerged as important post-transcriptional regulators of tumour suppressor and oncogene expression. Ras association domain family member 1 (RASSF1) is a critical tumour suppressor that controls multiple aspects of cell proliferation such as cell cycle, cell division and apoptosis. The expression of RASSF1 is lost in a variety of cancers due to the promoter hypermethylation.
Methods:
miR-193a-3p was identified as a RASSF1-targeting miRNA by a dual screening approach. In cultured human cancer cells, immunoblotting, qRT–PCR, luciferase reporter assays, time-lapse microscopy and immunofluorescence methods were used to study the effects of excess miR-193a-3p on RASSF1 expression and cell division.
Results:
Here, we report a new miRNA-mediated mechanism that regulates RASSF1 expression: miR-193a-3p binds directly to RASSF1-3′UTR and represses the mRNA and protein expression. In human cancer cells, excess of miR-193a-3p causes polyploidy through impairment of the Rassf1-Syntaxin 16 signalling pathway that is needed for completion of cytokinesis. In the next cell cycle the miR-193a-3p-overexpressing cells exhibit multipolar mitotic spindles, mitotic delay and elevated frequency of cell death.
Conclusions:
Our results suggest that besides epigenetic regulation, altered expression of specific miRNAs may contribute to the loss of Rassf1 in cancer cells and cause cell division errors.
Similar content being viewed by others


Main
The tumour suppressor gene Ras association domain family member 1 (RASSF1, (Dammann et al, 2000; Burbee et al, 2001)) encodes eight different transcripts (a–h), of which Rassf1a is the most abundant. The RASSF1 gene is located in a chromosomal locus 3p21.3 in which high frequency of allelic loss is observed in a variety of human cancers (Kok et al, 1987; Zbar et al, 1987; Chen et al, 1994; Wistuba et al, 1997). Indeed, RASSF1 is considered as one of the most frequently inactivated genes in a broad spectrum of human tumour types such as brain, lung, breast, ovarian, bladder, prostate and renal cell carcinomas (Burbee et al, 2001; Lee et al, 2001; Yoon et al, 2001; Liu et al, 2002; Horiguchi et al, 2003). The loss of activity is mostly due to hypermethylation of the gene promoter but also the aforementioned allelic imbalance (Hogg et al, 2002; Ito et al, 2005) and in rare cases inactivating mutations (Kashuba et al, 2009) play a role. Importantly, mice with loss or decline of Rassf1a protein (hereafter Rassf1) are more prone to develop tumours spontaneously and after carcinogen or irradiation exposure (Tommasi et al, 2005; van der Weyden et al, 2005).
Rassf1 carries multiple structural domains (Dammann et al, 2000; Guo et al, 2007; Hamilton et al, 2009) enabling the protein to operate in several key regulatory pathways such as the cell cycle and cell division (Shivakumar et al, 2002; Song et al, 2004), microtubule stability (Liu et al, 2003; Dallol et al, 2004), DNA damage response (Hamilton et al, 2009) and apoptosis signalling networks (Baksh et al, 2005; Matallanas et al, 2007). According to recent evidence, rather the microtubule and cell cycle than the apoptosis-related functions of Rassf1 are pivotal for its tumour suppressor effect; mutation of the Rassf1 microtubule-interacting domain abolishes the protein’s capability to restrain cell cycle progression and suppress cancer cell growth, albeit intact apoptosis induction (Donninger et al, 2014).
One largely unexplored control mechanism of RASSF1 expression is the microRNA (miRNA) pathway. miRNAs regulate gene expression post-transcriptionally by binding to the target gene mRNA, which causes degradation of the mRNA and/or inhibition of protein translation (Guo et al, 2010). Normal function of the miRNA pathway is essential for the maintenance of many physiological processes, such as development, differentiation and apoptosis. Importantly, altered expression of miRNAs have been associated with several human pathologies as cardiovascular diseases (Yang et al, 2007; Thum et al, 2008) and cancer (Cimmino et al, 2005; Johnson et al, 2005). MicroRNAs can be very potent cancer drivers or suppressors, owing to the capability of one miRNA to target several genes in the same signalling cascade or genes with similar function. On the other hand, each gene can be controlled by several miRNAs. (Bueno et al, 2011) To date, few miRNAs have been reported to regulate RASSF1 expression, mainly indirectly via targeting genes that are involved in its epigenetic silencing (Chen et al, 2011; Wang et al, 2011; Li et al, 2012). Moreover, miR-181a/b has been shown to control RASSF1 expression by direct binding to the RASSF1-3′UTR (Meng et al, 2012; Bräuer-Hartmann et al, 2015).
Here, we report a novel Rassf1-regulating miRNA, miR-193a-3p, which directly binds to the 3′UTR of RASSF1 mRNA. Previous reports indicate that miR-193a-3p regulates key metastasis genes, such as ERBB4 (Yu et al, 2015) and RAB27B (Pu et al, 2016). Moreover, the miRNA is also known to promote chemoresistance (Li et al, 2015). Our results reveal a new regulatory function of miR-193a-3p; overexpression of the miRNA in human cancer cells abrogates normal cell division by disturbing Rassf1-Syntaxin16 (Stx16) axis that is needed for faithful cell division.
Materials and methods
Cell culture
HeLa cells (ATCC CCL-2, obtained 2006) were cultured in DMEM (Sigma-Aldrich, St Louis, MO, USA) as described before (Mäki-Jouppila et al, 2015) or in DMEM/F12 (Sigma-Aldrich) supplemented with 10% foetal bovine serum (Gibco, Thermo Fisher Scientific, Carlsbad, CA, USA), 0.1 mM non-essential amino acids (Sigma-Aldrich) and 1% penicillin–streptomycin (Sigma-Aldrich). For H2B-GFP/tubulin-mCherry HeLas, which were obtained in 2012 from Dr Stephan Geley (Medical University, Innsbruck, Austria), G418 (250 μg ml−1) was added to the culture medium. OVCAR-8 cells (DCTD Tumor/Cell Line Repository, NCI) were obtained from Dr Olli Carpén in 2015 and were cultured as previously described (Tambe et al, 2016). HCT116 cells, obtained in 2009 from Dr Lauri Aaltonen (University of Helsinki, Finland), were cultured as previously described (Mäki-Jouppila et al, 2015). All cell lines were grown at 37 °C supplemented with 5% CO2, and tested negative for mycoplasma.
Target prediction screen
The target prediction screen was conducted with a custom miRIDIAN mimic library (v19.0, GE Dharmacon, Lafayette, CO, USA) consisting of 55 miRNAs predicted to target RASSF1. H2B-GFP/tubulin-mCherry HeLas were reverse transfected using HiPerFect (Qiagen, Valencia, CA, USA) with the miRNA mimics (40 nM), printed on a 384-well assay plate in four replicates as described before (Mäki-Jouppila et al, 2015). MicroRNA-transfected cells were then synchronised with a double thymidine block as described in ‘Cell cycle synchronisation’ section. Live-cell imaging with Incucyte (Essen Instruments Ltd, Hertfordshire, UK) was started immediately after cells were released from block. Fluorescent still images were obtained from the transfected cells at the time of the mitotic peak, equalling 72 h after transfection, with Operetta high-content imaging system (PerkinElmer, Waltham, MA,USA), equipped with heating (37 °C) and CO2 supply (5%).
Transient transfections
miRIDIAN miRNA mimics (GE Dharmacon) were reverse transfected into cells at 50 nM concentration using HiPerFect (Qiagen) according to the manufacturer’s protocol. miRIDIAN miRNA Mimic Negative Control #1 served as a non-targeting control miRNA. Anti-miR-193a-3p and corresponding negative control miRNA were purchased from Ambion (Thermo Fisher Scientific, Carlsbad, CA, USA) and used at 50 nM concentration. For luciferase reporter assays, miRNA mimics and reporter plasmids were forward transfected with Lipofectamine 2000 or 3000 (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA) using the manufacturer’s protocol.
Cell cycle synchronisation
For live-cell imaging experiments, cells were synchronised with a double thymidine block. First, cells were plated to 50% confluency and after attachment treated with 2 mM thymidine (Sigma-Aldrich) for 19 h. Cells were released from the first block by washing with fresh culture medium for 2 × 10 min and 1 × 30 min and let recover for 3–4 h before reverse transfection of miRNA mimics. The second thymidine was added 5–6 h after transfection, summing up the total time between the blocks to 8–9 h. After overnight incubation with the second thymidine, cells were released into fresh medium and live-cell imaging started.
RNA isolation and qRT–PCR analysis
For gene expression analyses, RNA isolation from harvested cells was performed with RNeasy Mini Kit (Qiagen) according to manufacturer’s instructions. Complementary DNA was prepared with iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). For TaqMan-based qRT–PCR protocol and GAPDH primers used, please refer to previous publication (Tambe et al, 2016). The primers used for RASSF1 were R1A-Fw 5′-GCTCGTCTGCCTGGACTG-3′ and R1A-Rv 5′-CTCCACAGGCTCGTCCAC-3′. The following primers were used to measure STX16 transcript variant 1 expression: STX16-Fw 5′-CAGCTGTTAGCCGAGCAAGT-3′ and STX16-Rv 5′-CATCAGCAAGCTCGTCCAG-3′. To measure mature miR-193a-3p levels in cells, total RNA was isolated with miRvana miRNA isolation kit (Ambion, Thermo Fisher Scientific) and reverse transcription was performed with TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, Thermo Fisher Scientific, Foster City, CA, USA). miR-193a-3p and RNU6B specific TaqMan MicroRNA assay (Applied Biosystems, Thermo Fisher Scientific) were used to measure the expression of miR-193a-3p and the internal control used for normalisation.
Immunoblotting
The method for cell lysis is described elsewhere (Mäki-Jouppila et al, 2015). Lysed protein samples were run on 4–20% gels (Bio-Rad) and then transferred with semi-dry transfer equipment (Bio-Rad) to nitrocellulose membrane. 5% milk/TBS-T, 5% BSA/TBS-T and Odyssey blocking buffer (LI-COR Biotechnology, Lincoln, NE, USA)/TBS-T (1 : 1) were used as blocking agents. The primary antibodies used were mouse anti-Rassf1a (1 : 500; ab23950, Abcam, Cambridge, UK or SM6017, Acris antibodies GmbH, Herford, Germany), rabbit anti-STX16 (1 : 750; HPA041019, Atlas antibodies, Stockholm, Sweden) and mouse anti-GAPDH (1 : 30 000–50 000; mAb 6C5, Advanced ImmunoChemical Inc., Long Beach, CA, USA, or HyTest Ltd, Turku, Finland). Primary antibodies were diluted into TSB-T or in the case of Rassf1a antibody, into TBS-T/Odyssey blocking buffer (5 : 1) when infrared detection system was used. Secondary antibodies (1 : 5000 in TBS-T) and detection methods used are described in previous publication (Tambe et al, 2016) with the addition of HRP-linked anti-rabbit IgG (Cell Signalling Technology, Danvers, MA, USA) used also as a secondary antibody.
Immunofluorescence labelling
For Stx16 detection, miRNA-transfected cells were grown on coverslips and then fixed with 2% paraformaldehyde in 0.5% Triton-X-100/PHEM (60 mM Pipes, 25 mM Hepes, 10 mM EGTA, 4 mM MgSO4). For microtubule staining, the cells were pre-extracted in PHEM/0.5% Triton-X-100 for 5 min before fixation using the fixative above supplemented with 0.2% glutaraldehyde. After rinse with MBST (10 mM MOPS, 150 mM NaCl and 0.05% Tween 20), coverslips were blocked in 20% boiled normal goat serum (bngs)/MBST for 1 h at RT. Primary antibodies used were rabbit anti-STX16 (1 : 250; HPA041019, Atlas antibodies), mouse anti-CETN3 (1 : 500; H00001070-M01, Abnova), rat anti-α-tubulin (1 : 500; YL1/2, ab6160, Abcam) and rabbit anti-pericentrin (1 : 500; ab4448, Abcam), all diluted in 5% bngs/MBST and incubated 1 h at RT. Secondary Alexa Fluor goat anti-mouse 555, goat anti-rabbit 488, 555 and 647, and chicken anti-rat 488 antibodies (Invitrogen, Thermo Fisher Scientific) were used at 1 : 500 concentration and incubated for 1 h at RT. DNA was stained with DAPI (1 : 10 000 in MQ H2O) and coverslips mounted on slides using Vectashield (Vector laboratories, Burlingame, CA, USA).
Luciferase reporter assay and site-directed mutagenesis
The RASSF1-3′UTR-pMirTarget construct, harbouring the human RASSF1-3′UTR downstream of firefly luciferase sequence, was purchased from OriGene (Rockville, MD, USA). For luciferase reporter assay, cells were seeded on a white 96-well plate with clear bottom, and 1 day later the RASSF1-3′UTR reporter vector (100 ng) was co-transfected with miRIDIAN mimics (50 nM) into cells, along with Renilla luciferase plasmid (25 ng, pRL-SV40, Promega, WI, USA) used for normalisation. Lipofectamine 2000 or 3000 (Invitrogen, Thermo Fisher Scientific) was used as transfection agent, following the manufacturer’s protocol. Luminescence signal was measured 24 h or 48 h post transfection, respectively, with the Dual-Glo Luciferase Assay System (Promega) and EnSight Multimode Plate Reader (PerkinElmer). To mutate the predicted binding site of miR-193a-3p in the RASSF1-3′UTR-pMirTarget, Quick-Change Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA) was used according to manufacturer’s protocol. The following primers were used to generate a 4 nucleotide mutation: Fw 5′-GCCGTGTGAGTGTGACAGGTTACGTGGGGCCTGTGGAATGAG-3′ and Rv 5′-CTCATTCCACAGGCCCCACGTAACCTGTCACACTCACACGGC-3′. The mutation was confirmed by sequencing at the Finnish Microarray and Sequencing Centre (Turku Centre for Biotechnology, Turku, Finland).
Image acquisition and analysis
Immunofluorescence specimens were imaged with a Zeiss inverted 200 M microscope (Zeiss GmbH, Oberkochen, Germany) equipped with Hamamatsu ORCA-ER camera (Hamamatsu Photonics, Hamamatsu City, Japan) and Metamorph software version 6.2r6 (Molecular Devices, Downingtown, PA, USA). Signal intensities were measured from maximum projections of Z-stacks (0.5 μ M step size). The relative area occupied by Stx16 signal was measured from maximum projections of Z-stacks using the thresholding function to include only the Stx16 signal for automated area measurement. Signal area was normalised to cell number for each analysed image. For fluorescent imaging of live cells, Operetta high-content imaging system was used (PerkinElmer) and Incucyte equipment was used for phase-contrast live-cell imaging.
Clinical data analysis
The MicMa breast cancer cohort and the performed analyses are described in more detail in a previous publication (Mäki-Jouppila et al, 2015).
Statistical analysis
Statistical analyses were performed using paired two-tailed Student’s t-test. Statistical significance was defined as *P⩽0.05, **P⩽0.01 and ***P⩽0.001. Values are presented as the average±s.d.
Results
Discovery of novel RASSF1-regulating miRNAs
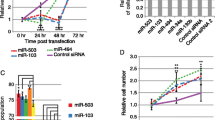
One of the Rassf1 key functions is to control M-phase progression through restriction of APC/C-Cdc20 activity (Song et al, 2004). Importantly, changed amount of Rassf1 protein causes errors both in mitotic timing and in spindle architecture and chromosome alignment (Liu et al, 2003; Song et al, 2004). To identify RASSF1-targeting miRNAs that impair normal mitosis, we selected 55 candidate miRNAs according to miRNA target prediction softwares and tested their ability to induce cell division errors when overexpressed in vitro. Synchronised H2B-GFP/tubulin-mCherry tagged HeLa cells were transiently transfected with individual miRNAs and subjected to live-cell imaging for 3 days. Nine miRNAs were found to induce pronounced mitotic errors; the overexpressing cells exhibited increased frequency of multipolar spindles, lagging chromosomes, chromosome bridges and polyploidy in comparison to miR-control (Figure 1A). Importantly, no significant changes in the progression of cell cycle were detected with any of the tested miRNA. Western blotting assays and qRT–PCR indicated that five out of the nine candidate miRNAs (miR-1271-3p, -323a-3p, -193a-3p, -181a-5p and -506-3p) suppressed Rassf1 mRNA and protein levels by at least 20% in comparison to miR-control (Figure 1A). One of the hit miRNAs, miR-181a-5p, was recently confirmed to target RASSF1 by others (Meng et al, 2012; Bräuer-Hartmann et al, 2015) that provides validation for our screen and hit selection criteria.

Identification of novel Rassf1-regulating miRNAs. Flowchart representations of the two screening approaches utilised. (A) The target prediction screen. Representative micrographs show mitotic anomalies induced by the overexpressed miRNAs. Scale bar is 50 μm. Arrowheads point to examples of detected mitotic defects, such as lagging chromosomes, chromatin bridges and multipolar mitotic spindles. Suppression of Rassf1 mRNA and protein expression by the most potent miRNAs is shown below. (B) The clinical correlation screen. The table shows the Pearson correlations and P-values of the most potent miRNAs (n=19) with RASSF1 mRNA expression in a breast tumour sample set. Results from Rassf1 qRT–PCR and immunoblotting experiments with these miRNAs are shown below. The target prediction screen (A) yielded four and the clinical correlation screen (B) three hit miRNAs that are marked with arrows in the graphs. The data are from one or two experiments (mean±s.d.).
In addition to the target prediction screen, we implemented a second clinical correlation screen based on miRNA-RASSF1 mRNA correlation analysis performed retrospectively from a collection of 101 breast cancer tumour samples profiled for almost 800 miRNAs (Naume et al, 2001; Enerly et al, 2011). The hit miRNAs (n=19) that displayed statistically significant and the most negative Pearson correlation (⩽−0.22) with RASSF1 mRNA expression (Figure 1B), were tested in vitro for suppression of Rassf1 mRNA and protein expression. Western blot analyses and qRT–PCR of HeLa cell populations overexpressing the selected miRNAs separately indicated that three miRNAs (miR-182-3p, -130b-3p and -454) suppressed both the Rassf1 mRNA and protein levels by at least 20%, while six decreased only the mRNA expression (Figure 1B). We conclude that the two screens yielded a total of seven potential RASSF1-targeting miRNAs that efficiently suppressed RASSF1 expression in cultured human cancer cells.
miR-193a-3p regulates Rassf1 expression via direct binding to the RASSF1-3′UTR
To determine if the hit miRNAs bind directly to the RASSF1-3′UTR, we performed a luciferase reporter assay. Out of the seven hit miRNAs, only the co-transfection of miR-193a-3p mimic with firefly luciferase reporter vector harbouring the RASSF1-3′UTR sequence resulted in significant suppression of the luciferase signal in comparison to the negative miR-control (Figure 2A, 0.77±0.06, P=0.02). To validate the luciferase reporter assay result, we mutated the predicted binding site of miR-193a-3p in the RASSF1-3′UTR (Figure 2A) and measured the impact on the luciferase activity. The results show that co-transfection of the miR-193a-3p mimic with the mutated luciferase reporter vector did not suppress the luciferase activity when compared to miR-control, which confirms that miR-193a-3p binds to the predicted binding site in the 3′UTR of the gene (Figure 2A).

miR-193a-3p suppresses Rassf1 expression via direct binding to the RASSF1 -3′UTR. (A) Quantifications from luciferase reporter assays with WT RASSF1-3′UTR and mutated RASSF1-3′UTR, which harbours a 4 nucleotide mutation in the predicted miR-193a-3p binding site. Schematic shows the predicted miR-193a-3p-binding site on RASSF1-3′UTR (bold) and the mutation (underlined). (B) Quantification of miR-193a-3p expression in miR-control or miR-193a-3p mimic-transfected HeLa cells 48 h post transfection. Quantification of Rassf1 (C) mRNA and (D) protein expression in HeLa, OVCAR-8 and HCT116 cell lines 48 h after miR-control or miR-193a-3p transfection, and (E) Rassf1 protein expression in HeLa cells 48 h after miR-control or anti-miR-193a-3p transfection. Representative immunoblots are shown. All data are from two or three independent experiments (mean±s.d.).
To confirm successful miR-193a-3p mimic transfection, we measured the expression of mature miR-193a-3p in miR-control and miR-193a-3p mimic-transfected HeLa cells 48 h post transfection. The miR-193a-3p levels >20 000-fold higher in mimic transfected cells compared to miR-control-transfected cell population (Figure 2B). Next, to exclude the possibility that the impact of miR-193a-3p on Rassf1 expression is a HeLa cell-specific event, we transiently transfected ovarian OVCAR-8 and colon HCT116 carcinoma cells with the miRNA followed by measurement of the Rassf1 mRNA and protein levels 48 h post transfection. The qRT–PCR and Western blotting data show that Rassf1 mRNA and protein levels were equally suppressed in all three cell lines; the mRNA levels were reduced by an average of 39±16%, 31±8% and 49±11%, in HeLa, OVCAR-8 and HCT116 cells, respectively, and the protein levels were declined by an average of 47±11%, 29±2% and 50±17%, respectively, when compared to the miR-controls (Figure 2C and D). Moreover, inhibition of endogenous miR-193a-3p activity in HeLa cells with anti-miRNA resulted in increase of Rassf1 protein expression by an average of 51% (Figure 2E). In conclusion, out of the seven hit miRNAs, only the miR-193a-3p bound to the RASSF1-3′UTR and was found to cause significant downregulation of Rassf1 mRNA and protein expression in various human cancer cell lines.
miR-193a-3p overexpression hampers cytokinesis
Several studies have functionally associated Rassf1 in the regulation of mitosis and cell cycle progression (Shivakumar et al, 2002; Song et al, 2004, 2009; Guo et al, 2007; Tommasi et al, 2011). Rassf1 may control mitosis by perturbing microtubule stability (Liu et al, 2003). However, the protein has also been suggested to control mitotic timing by inhibiting APC/C-Cdc20 activity in early prometaphase (Song et al, 2004). Moreover, Rassf1 localises to the mitotic spindle, centrosomes and later in mitosis to the midzone/midbody, which supports the notion that the protein regulates cell division (Song et al, 2004; Guo et al, 2007). The mitotic defects detected in the miR-193a-3p-overexpressing cells (Figure 1A) prompted us to analyse mitotic processes in more depth. Time-lapse imaging of synchronised HeLa cells with excess miR-193a-3p revealed that a significant portion of the cells failed to separate the daughter cells at the end of M-phase; in average 18.7±3.8% of miR-193a-3p-overexpressing cells failed to form two daughter cells in comparison to 2.7±0.6% observed in miR-control population (P=0.02, Figure 3A). The phenotype was confirmed in OVCAR-8 cells in which excess of miR-193a-3p increased the frequency of cells exhibiting failed cytokinesis from 2.3±0.6% of control cells to 8.0±2.6% (P=0.04, Figure 3A). Also the average duration of mitosis (from nuclear envelope breakdown to anaphase) in miR-193a-3p-transfected cell population was slightly longer compared to miR-control (Figure 3B).

Excess of miR-193a-3p impairs cytokinesis. (A) Representative still images from live-cell films of synchronised HeLa cells transfected with either miR-control or miR-193a-3p. Time is h : min after release from the double thymidine block. Arrowheads point to dividing cells, with either normal (miR-control) or failed cytokinesis (miR-193a-3p). Scale bar equals 25 μm. The bar graph presents quantification for percentage of mitotic cells failing in cytokinesis. The data are from three independent experiments with HeLa and OVCAR-8 cell lines (n=300 cells per category). (B) The scatter plot from one representative live-cell imaging experiment (n=100 cells per group) with miR-control or miR-193a-3p-transfected HeLa cells, showing the duration of mitosis (min; NEBD-to-anaphase, mean±s.d.) and the fate of each recorded cell. Each cell is presented by a diamond shape, and the grey colour indicates a cell with failed cytokinesis. (C) Representative micrographs from miR-control or miR-193a-3p-transfected HeLa cells fixed 48 h post transfection. Arrowheads indicate abnormal binuclear (II), trinuclear (III) and tetranuclear (IV) interphase cells in the miR-193a-3p-transfected population. Quantification from three experiments shows increased frequency of multinuclear interphase cells in the miR-193a-3p-overexpressing HeLa and OVCAR-8 cell populations (mean±s.d.; n>1500 cells per group) in comparison to miR-control. Scale bar is 25 μm.
Failure to segregate the daughter cells often results in formation of progeny cells with two or more nuclei. Quantification of miR-193a-3p or miR-control-overexpressing HeLa cells that were fixed and DAPI-stained 48 h post transfection indicated significantly increased frequency of binuclear cells by miR-193a-3p overexpression (7.8±1.7%) in comparison to miR-control (2.3±0.5%, P=0.03). Moreover, when cells with more than two nuclei were also counted, the fraction of multinuclear cells in miR-193a-3p-overexpressing cell population further increased in comparison to miR-control (9.7±2.3% vs 2.3±0.4%, P=0.03, Figure 3C). Similar observation was also made in OVCAR-8 cells (Figure 3C). We conclude that excess of miR-193a-3p resulted in a failure of cytokinesis that caused formation of multinuclear daughter cells.
Excess of miR-193a-3p disturbs the Rassf1-Stx16 axis needed for cytokinesis
Rassf1 regulates cytokinesis in the spindle midzone by recruiting a t-SNARE protein Stx16 (Song et al, 2009), which is needed for accumulating central abscission factors to the midbody (Neto et al, 2013). Interestingly, Stx16 is also a predicted target of miR-193a-3p. To assess whether excess of miR-193a-3p affects Stx16 expression, we determined Stx16 mRNA and protein levels in the miRNA-transfected HeLa cells. Quantification of the qRT–PCR data showed no difference in Stx16 mRNA levels at 48 h post transfection in comparison to miR-control (Figure 4A). However, immunoblotting data indicated that the amount of Stx16 protein was significantly decreased by 40±0.12% (P=0.03) in the miR-193a-3p-overexpressing cells when compared to control (Figure 4B).

Excess of miR-193a-3p disturbs Stx16 protein expression and localisation. Quantification of Stx16 (A) mRNA and (B) protein expression in HeLa cells 48 h after transfection of miR-control or miR-193a-3p. A representative immunoblot image of Stx16 protein expression is shown. (C) Representative immunofluorescence images of fixed interphase HeLa cells transfected with miR-control or miR-193a-3p and stained with a Stx16 antibody. DAPI depicts the DNA and scale bar is 20 μm. Quantification indicates that the Stx16 signal is dispersed to a larger area in miR-193a-3p-overexpressing cells when compared to miR-control-transfected cells. (D) Representative immunofluorescence images of fixed HeLa cells in late M-phase, transfected with miR-control or miR-193a-3p and stained with antibodies against Stx16 (red) and tubulin (green) 48 h after transfection. DNA is stained with DAPI. Scale bar is 10 μm. Graph shows quantification of Stx16 signal intensity in the midbody of early telophase cells (n>50 cells per group), transfected with miR-control or miR-193a-3p. All quantifications are from three independent experiments (mean±s.d.). A full color version of this figure is available at the British Journal of Cancer journal online.
Earlier studies have shown that Stx16 predominantly localises to Golgi/endosomal compartment in interphase and to spindle midzone and midbody in late M-phase (Neto et al, 2013; Willett et al, 2013). Our immunostainings confirmed that in miR-control-transfected HeLa cells Stx16 resides in these locations (Figure 4C and D). However, in miR-193a-3p-overexpressing HeLa cells, the Stx16 signals were clearly more dispersed at 48 h post transfection (Figure 4C). To quantify the difference, we measured the relative area occupied by Stx16 signals in the miR-control and miR-193a-3p-transfected cells. The analysis indicated a 2.3 times larger areal signal distribution in miR-193a-3p-overexpressing cells compared to miR-control (Figure 4C, P=0.05). Importantly, the Stx16 signal intensity in the midbody of telophase cells was significantly decreased by excess of miR-193a-3p in comparison to miR-control (by 38±13%, P=0.03, Figure 4D). We conclude that excess of miR-193a-3p abrogates cytokinesis due to impairment of Rassf1-Stx16 regulatory axis needed for faithful cytokinesis/abscission.
Overexpression of miR-193a-3p induces multipolarity in mitotic cells
Cytokinesis failure gives rise to polyploid progeny cells with extra centrosomes that can cause errors in spindle organisation and chromosome segregation in the subsequent mitoses (Storchova and Kuffer, 2008). Interestingly, depletion of Rassf1 (Song et al, 2004) or its’ centrosomal recruitment factor MAP1S (Dallol et al, 2007) by RNAi have been reported to cause centrosome defects and multipolar spindles. To investigate if excess of miR-193a-3p induces mitotic spindle anomalies, we fixed and immunostained miR-control and miR-193a-3p-transfected HeLa cells with antibodies against α-tubulin and pericentrin 48 h post transfection. A variety of multipolar mitotic spindles was detected in cells with excess of miR-193a-3p (Figure 5A). According to quantification, an average of 47.3±12.2% of the mitotic cells in the miR-193a-3p-transfected cell population exhibited a multipolar spindle, which was significantly more than in the miR-control showing an average of 13.3±5.0% of multipolar mitoses (P=0.01, Figure 5A). To get more detailed information about the origin of the multipolarity, we performed immunostainings with a centriole marker, centrin-3 antibody, together with pericentrin and α-tubulin. In 40% of the multipolar cells in miR-193a-3p-overexpressing population, all poles were positive for centrin-3 and the rest of the multipolar cells exhibited one or more poles without centrioles (Figure 5B). This suggests that both polyploidisation and pericentriolar matrix fragmentation contribute to the multipolarity induction. In conclusion, excess of miR-193a-3p results in increased frequency of centrosome abnormalities and multipolar spindles in mitotic cells.

Overexpression of miR-193a-3p induces multipolar mitotic spindles and results in accumulation of M-phase cells and cell death. (A) Representative immunofluorescence images of miR-control or miR-193a-3p-transfected mitotic HeLa cells, fixed 48 h after transfection and stained with antibodies against α-tubulin (green) and pericentrin (red). DNA is stained with DAPI. Scale bar equals 10 μm. Quantification shows increased percentage of mitotic cells with multipolar spindle in the miR-193a-3p-overexpressing cell population (n=150 cells per group). (B) Analysis of pericentrin, α-tubulin and centrin-3 immunostained HeLa cells shows the presence of two types of multipolar spindles and their relative frequencies in miR-193a-3p-overexpressing cell population; multipolar mitotic cells where each pole has centrioles and multipolar mitotic cells where one or more poles are negative for centrin-3 staining (n=150 cells per group). (C) Bar graph presents quantification of mitotic and cell death indices from miR-control and miR-193a-3p-transfected HeLa cells 48 h post transfection (n>1500 cells per group). All quantifications are from three independent experiments, and data are mean±s.d. A full color version of this figure is available at the British Journal of Cancer journal online.
Mitotic defects induced by excess miR-193a-3p result in accumulation of M-phase cells and increased cell death
Complete or partial loss of Rassf1 (Guo et al, 2007; Tommasi et al, 2011) or the associated protein MAP1S (Dallol et al, 2007) has been linked to extended mitotic timing. Furthermore, extra chromosomes and/or centrosomes and multipolar spindles have been reported to prolong mitosis per se in mammalian cells (Gisselsson et al, 2008; Yang et al, 2008). To investigate whether excess miR-193a-3p causes accumulation of cells to M-phase, we determined the mitotic index in asynchronous HeLa and OVCAR-8 cells that were fixed and DNA-stained 48 h post transfection. Analysis indicated significantly elevated mitotic index in both cell lines in comparison to miR-control; 9.8±0.2 vs 3.5±0.4 (P=0.002, Figure 5C) and 5.9±0.9 vs 3.3±1.6 (P=0.05), respectively.
Besides the spindle anomalies and delayed mitosis, the cells with excess of miR-193a-3p were found to die more frequently than the miR-control cells. The fraction of dead cells, analysed from fixed and DAPI-stained HeLa cells was increased from the average of 3.5±0.1% in miR-control cells to 14.5±5.2% observed in miR-193a-3p-transfected cells (Figure 5C). To conclude, the mitotic defects induced by overexpression of miR-193a-3p result in a transient mitotic arrest and increased cell death.
Discussion
Our data describe a new molecular mechanism whereby expression of the tumour suppressor gene RASSF1 is controlled in human cells; miR-193a-3p binds directly to the RASSF1-3′UTR, which leads to downregulation of the mRNA and protein levels of the target gene. Phenotypically, overexpression of miR-193a-3p in cultured human cancer cells perturbs normal cell division and causes polyploidy. Moreover, polyploidisation together with de novo centrosome abnormalities induces chromosome alignment problems in the next M-phase, followed by transient mitotic arrest and cell death.
Although Rassf1 is among the most frequently lost tumour suppressor proteins, the regulation of Rassf1 by post-translational mechanisms has not been extensively studied earlier. Among the human miRNAs, only the miR-181a/b cluster has been demonstrated to regulate RASSF1 via direct binding to the 3′UTR of the gene product. This miRNA-mediated regulation of RASSF1 plays a specific role in the pathogenesis and treatment of certain forms of acute promyelocytic leukaemia, in which PML/RAR fusion oncogene can promote proliferation via miR-181a/b upregulation and Rassf1 suppression. (Bräuer-Hartmann et al, 2015) Identification of miR-193a-3p as another miRNA that directly binds to the RASSF1-3′UTR and causes efficient suppression of the gene expression extends the notion that Rassf1 is a target of the miRNA pathway. However, the potential tumorigenic roles of miR-193a-3p and miR-181a/b in vivo remain to be studied further in leukaemia and other neoplasms.
Rassf1 is a tumour suppressor that restrains malignant cell proliferation plausibly via regulating cell cycle progression and microtubule stability (Donninger et al, 2014). Here we show, in line with previous reports (Song et al, 2004, 2009; Guo et al, 2007; Tommasi et al, 2011), that reduction of Rassf1 expression due to overexpression of miR-193a-3p abrogates normal cell division and causes polyploidisation, followed by formation of multipolar mitotic spindles in the next cell cycle. As both tetraploidy and extra centrosomes have been intimately linked to promotion of chromosomal instability and malignant cell growth (Fujiwara et al, 2005; Ganem et al, 2009), we speculate that miR-193a-3p is an oncomiR that fine-tunes the Rassf1 expression to prevent cell division errors that can challenge genomic stability. Previously miR-193a-3p has often been reported to function as a tumour suppressor miRNA that represses the metastatic capability of cancer cells (Yu et al, 2015; Pu et al, 2016) and restricts their proliferation (Seviour et al, 2016). We observed a transient mitotic arrest and increased cell death following the mitotic defects induced by excess of miR-193a-3p. However, also the other putative target genes of miR-193a-3p, such as the anti-apoptotic protein Mcl-1 (Kwon et al, 2013) may contribute to the cell fate.
To conclude, here we demonstrate that Rassf1 expression is under post-transcriptional regulation by a novel miRNA, miR-193a-3p. Moreover, our data indicate a new, potentially tumorigenic function of miR-193a-3p; overexpression of the miRNA disturbs normal cell division, leading to polyploidisation and accumulation of mitotic defects. The potential tumorigenic function of the miR-193a-3p-Rassf1-regulatory pathway in vivo remains as a subject for further studies.
Change history
23 May 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Baksh S, Tommasi S, Fenton S, Yu VC, Martins LM, Pfeifer GP, Latif F, Downward J, Neel BG (2005) The tumor suppressor RASSF1A and MAP-1 link death receptor signaling to bax conformational change and cell death. Mol Cell 18: 637–650.
Bräuer-Hartmann D, Hartmann JU, Wurm AA, Gerloff D, Katzerke C, Falzacappa MVV, Pelicci PG, Müller-Tidow C, Tenen DG, Niederwieser D, Behre G (2015) PML/RARα-regulated miR-181a/b cluster targets the tumor suppressor RASSF1A in acute promyelocytic leukemia. Cancer Res 75: 3411–3424.
Bueno MJ, de Cedrón MG, Gómez-López G, de Castro IP, Di Lisio L, Montes-Moreno S, Martínez N, Guerrero M, Sánchez-Martínez R, Santos J, Pisano DG, Piris MA, Fernández-Piqueras J, Malumbres M (2011) Combinatorial effects of microRNAs to suppress the Myc oncogenic pathway. Blood 117: 6255–6266.
Burbee DG, Forgacs E, Zöchbauer-Müller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, Sekido Y, Latif F, Milchgrub S, Toyooka S, Gazdar AF, Lerman MI, Zabarovsky E, White M, Minna JD (2001) Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst 93: 691–699.
Chen LC, Matsumura K, Deng G, Kurisu W, Ljung BM, Lerman MI, Waldman FM, Smith HS (1994) Deletion of two separate regions on chromosome 3p in breast cancers. Cancer Res 54: 3021–3024.
Chen Y, Luo J, Tian R, Sun H, Zou S (2011) miR-373 negatively regulates methyl-CpG-binding domain protein 2 (MBD2) in hilar cholangiocarcinoma. Dig Dis Sci 56: 1693–1701.
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu C-G, Kipps TJ, Negrini M, Croce CM (2005) miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 102: 13944–13949.
Dallol A, Agathanggelou A, Fenton SL, Ahmed-choudhury J, Hesson L, Vos MD, Clark GJ, Downward J, Maher ER, Latif F (2004) RASSF1A interacts with microtubule-associated proteins and modulates microtubule dynamics. Cancer Res 64: 4112–4116.
Dallol A, Cooper WN, Al-Mulla F, Agathanggelou A, Maher ER, Latif F (2007) Depletion of the Ras association domain family 1, isoform A-associated novel microtubule-associated protein, C19ORF5/MAP1S, causes mitotic abnormalities. Cancer Res 67: 492–500.
Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP (2000) Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 25: 315–319.
Donninger H, Clark JA, Monaghan MK, Lee Schmidt M, Vos M, Clark GJ (2014) Cell cycle restriction is more important than apoptosis induction for RASSF1A Protein tumor suppression. J Biol Chem 289: 31287–31295.
Enerly E, Steinfeld I, Kleivi K, Leivonen S-K, Aure MR, Russnes HG, Rønneberg JA, Johnsen H, Navon R, Rødland E, Mäkelä R, Naume B, Perälä M, Kallioniemi O, Kristensen VN, Yakhini Z, Børresen-Dale A-L (2011) miRNA-mRNA integrated analysis reveals roles for miRNAs in primary breast tumors. PLoS ONE 6: e16915.
Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D (2005) Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437: 1043–1047.
Ganem NJ, Godinho Sa, Pellman D (2009) A mechanism linking extra centrosomes to chromosomal instability. Nature 460: 278–282.
Gisselsson D, Håkanson U, Stoller P, Marti D, Jin Y, Rosengren AH, Stewénius Y, Kahl F, Panagopoulos I (2008) When the genome plays dice: circumvention of the spindle assembly checkpoint and near-random chromosome segregation in multipolar cancer cell mitoses. PLoS One 3: e1871.
Guo C, Tommasi S, Liu L, Yee JK, Dammann R, Pfeifer G (2007) RASSF1A is part of a complex similar to the Drosophila hippo/salvador/lats tumor-suppressor network. Curr Biol 17: 700–705.
Guo H, Ingolia NT, Weissman JS, Bartel DP (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840.
Hamilton G, Yee KS, Scrace S, O’Neill E (2009) ATM regulates a RASSF1A-dependent DNA damage response. Curr Biol 19: 2020–2025.
Hogg RP, Honorio S, Martinez A, Agathanggelou A, Dallol A, Fullwood P, Weichselbaum R, Kuo MJ, Maher ER, Latif F (2002) Frequent 3p allele loss and epigenetic inactivation of the RASSF1A tumour suppressor gene from region 3p21.3 in head and neck squamous cell carcinoma. Eur J Cancer 38: 1585–1592.
Horiguchi K, Tomizawa Y, Tosaka M, Ishiuchi S, Kurihara H, Mori M, Saito N (2003) Epigenetic inactivation of RASSF1A candidate tumor suppressor gene at 3p21.3 in brain tumors. Oncogene 22: 7862–7865.
Ito M, Ito G, Kondo M, Uchiyama M, Fukui T, Mori S, Yoshioka H, Ueda Y, Shimokata K, Sekido Y (2005) Frequent inactivation of RASSF1A, BLU, and SEMA3B on 3p21.3 by promoter hypermethylation and allele loss in non-small cell lung cancer. Cancer Lett 225: 131–139.
Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ (2005) RAS is regulated by the let-7 microRNA family. Cell 120: 635–647.
Kashuba VI, Pavlova TV, Grigorieva EV, Kutsenko A, Yenamandra SP, Li J, Wang F, Protopopov AI, Zabarovska VI, Senchenko V, Haraldson K, Eshchenko T, Kobliakova J, Vorontsova O, Kuzmin I, Braga E, Blinov VM, Kisselev LL, Zeng YX, Ernberg I, Lerman MI, Klein G, Zabarovsky ER (2009) High mutability of the tumor suppressor genes RASSF1 and RBSP3 (CTDSPL) in cancer. PLoS ONE 4: e5231.
Kok K, Osinga J, Carritt B, Davis MB, van der Hout AH, van der Veen AY, Landsvater RM, de Leij LF, Berendsen HH, Postmus PE (1987) Deletion of a DNA sequence at the chromosomal region 3p21 in all major types of lung cancer. Nature 330: 578–581.
Kwon JE, Kim BY, Kwak SY, Bae IH, Han YH (2013) Ionizing radiation-inducible microRNA miR-193a-3p induces apoptosis by directly targeting Mcl-1. Apoptosis 18: 896–909.
Lee MG, Kim HY, Byun DS, Lee SJ, Lee CH, Kim JI, Chang SG, Chi SG (2001) Frequent epigenetic inactivation of RASSF1A in human bladder carcinoma. Cancer Res 61: 6688–6692.
Li Q, Zhu F, Chen P (2012) MiR-7 and miR-218 epigenetically control tumor suppressor genes RASSF1A and Claudin-6 by targeting HoxB3 in breast cancer. Biochem Biophys Res Commun 424: 28–33.
Li Y, Deng H, Lv L, Zhang C, Qian L, Xiao J, Zhao W, Liu Q, Zhang D, Wang Y, Yan J, Zhang H, He Y, Zhu J (2015) The miR-193a-3p-regulated ING5 gene activates the DNA damage response pathway and inhibits multi-chemoresistance in bladder cancer. Oncotarget 6: 10195–10206.
Liu L, Tommasi S, Lee D-H, Dammann R, Pfeifer GP (2003) Control of microtubule stability by the RASSF1A tumor suppressor. Oncogene 22: 8125–8136.
Liu L, Yoon J-H, Dammann R, Pfeifer GP (2002) Frequent hypermethylation of the RASSF1A gene in prostate cancer. Oncogene 21: 6835–6840.
Matallanas D, Romano D, Yee K, Meissl K, Kucerova L, Piazzolla D, Baccarini M, Vass JK, Kolch W, O’Neill E (2007) RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol Cell 27: 962–975.
Meng F, Glaser SS, Francis H, Demorrow S, Passarini JD, Stokes A, Cleary JP, Liu X, Venter J, Kumar P, Priester S, Hubble L, Stoloch D, Sharma J (2012) Functional analysis of microRNAs in human hepatocellular cancer stem cells. J Cell Mol Med 16: 160–173.
Mäki-Jouppila JHE, Pruikkonen S, Tambe MB, Aure MR, Halonen T, Salmela AL, Laine L, Børresen-Dale AL, Kallio MJ (2015) MicroRNA let-7b regulates genomic balance by targeting Aurora B kinase. Mol Oncol 9: 1056–1070.
Naume B, Borgen E, Kvalheim G, Kåresen R, Qvist H, Sauer T, Kumar T, Nesland JM (2001) Detection of isolated tumor cells in bone marrow in early-stage breast carcinoma patients: comparison with preoperative clinical parameters and primary tumor characteristics. Clin Cancer Res 7: 4122–4129.
Neto H, Kaupisch A, Collins LL, Gould GW (2013) Syntaxin 16 is a master recruitment factor for cytokinesis. Mol Biol Cell 24: 3663–3674.
Pu Y, Zhao F, Cai W, Meng X, Li Y, Cai S (2016) MiR-193a-3p and miR-193a-5p suppress the metastasis of human osteosarcoma cells by down-regulating Rab27B and SRR, respectively. Clin Exp Metastasis 33: 359–372.
Seviour EG, Sehgal V, Mishra D, Rupaimoole R, Rodriguez-Aguayo C, Lopez-Berestein G, Lee J-S, Sood AK, Kim MP, Mills GB, Ram PT (2016) Targeting KRas-dependent tumour growth, circulating tumour cells and metastasis in vivo by clinically significant miR-193a-3p. Oncogene 36: 1–12.
Shivakumar L, Minna J, Sakamaki T, Pestell R, White MA (2002) The RASSF1A tumor suppressor blocks cell cycle progression and inhibits cyclin D1 accumulation. Mol Cell Biol 22: 4309–4318.
Song MS, Song SJ, Ayad NG, Chang JS, Lee JH, Hong HK, Lee H, Choi N, Kim J, Kim H, Kim JW, Choi E-J, Kirschner MW, Lim D-S (2004) The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat Cell Biol 6: 129–137.
Song SJ, Kim SJ, Song MS, Lim DS (2009) Aurora B-mediated phosphorylation of RASSF1A maintains proper cytokinesis by recruiting syntaxin16 to the midzone and midbody. Cancer Res 69: 8540–8544.
Storchova Z, Kuffer C (2008) The consequences of tetraploidy and aneuploidy. J Cell Sci 121: 3859–3866.
Tambe M, Pruikkonen S, Mäki-Jouppila J, Chen P, Elgaaen BV, Straume AH, Huhtinen K, Cárpen O, Lønning PE, Davidson B, Hautaniemi S, Kallio MJ (2016) Novel Mad2-targeting miR-493-3p controls mitotic fidelity and cancer cells’ sensitivity to paclitaxel. Oncotarget 7: 12267–12285.
Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JTR, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S (2008) MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456: 980–984.
Tommasi S, Besaratinia A, Wilczynski SP, Pfeifer GP (2011) Loss of Rassf1a enhances p53-mediated tumor predisposition and accelerates progression to aneuploidy. Oncogene 30: 690–700.
Tommasi S, Dammann R, Zhang Z, Wang Y, Liu L, Tsark WM, Wilczynski SP, Li J, You M, Pfeifer GP (2005) Tumor susceptibility of Rassf1a knockout mice. Cancer Res 65: 92–98.
Wang H, Wu J, Meng X, Ying X, Zuo Y, Liu R, Pan Z, Kang T, Huang W (2011) MicroRNA-342 inhibits colorectal cancer cell proliferation and invasion by directly targeting dna methyltransferase 1. Carcinogenesis 32: 1033–1042.
van der Weyden L, Tachibana KK, Gonzalez MA, Adams DJ, Ng BL, Petty R, Venkitaraman AR, Arends MJ, Bradley A (2005) The RASSF1A isoform of RASSF1 promotes microtubule stability and suppresses tumorigenesis. Mol Cell Biol 25: 8356–8367.
Willett R, Kudlyk T, Pokrovskaya I, Schönherr R, Ungar D, Duden R, Lupashin V (2013) COG complexes form spatial landmarks for distinct SNARE complexes. Nat Commun 4: 1553.
Wistuba, II, Montellano FD, Milchgrub S, Virmani AK, Behrens C, Chen H, Ahmadian M, Nowak JA, Muller C, Minna JD, Gazdar AF (1997) Deletions of chromosome 3p are frequent and early events in the pathogenesis of uterine cervical carcinoma. Cancer Res 57: 3154–3158.
Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, Wang H, Chen G, Wang Z (2007) The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med 13: 486–491.
Yang Z, Loncarek J, Khodjakov A, Rieder CL (2008) Extra centrosomes and/or chromosomes prolong mitosis in human cells. Nat Cell Biol 10: 748–751.
Yoon JH, Dammann R, Pfeifer GP (2001) Hypermethylation of the CpG island of the RASSF1A gene in ovarian and renal cell carcinomas. Int J cancer 94: 212–217.
Yu T, Li J, Yan M, Liu L, Lin H, Zhao F, Sun L, Zhang Y, Cui Y, Zhang F, Li J, He X, Yao M (2015) MicroRNA-193a-3p and -5p suppress the metastasis of human non-small-cell lung cancer by downregulating the ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Oncogene 34: 413–423.
Zbar B, Brauch H, Talmadge C, Linehan M (1987) Loss of alleles of loci on the short arm of chromosome 3 in renal\ncell carcinoma. Nature 327: 721–724.
Acknowledgements
We acknowledge Dr Miriam Ragle Aure and Dr Anne-Lise Børresen-Dale (Oslo University Hospital and University of Oslo) for the provided in vivo data. Rami Mäkelä and Johannes Hattara are acknowledged for technical assistance in the cell-based screen. The authors thank Dr Lauri Aaltonen, Dr Olli Carpén and Dr Stephen Gelay for providing cell lines used in this study, and Dr Jeroen Pouwels for providing secondary antibodies for immunoblotting. This study was supported by a grant from Academy of Finland (268360), a Finnish Cancer Organisations grant to MJK, a Finnish Cultural Foundation grant to SP and Turku Doctoral Programme of Molecular Medicine funding for SP. MJK is K. Albin Johansson Senior Cancer Researcher for the Finnish Cancer Institute.
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Pruikkonen, S., Kallio, M. Excess of a Rassf1-targeting microRNA, miR-193a-3p, perturbs cell division fidelity. Br J Cancer 116, 1451–1461 (2017). https://doi.org/10.1038/bjc.2017.110
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.110
- Springer Nature Limited
Keywords
This article is cited by
-
Long non-coding RNA MIAT serves as a biomarker of fragility fracture and promotes fracture healing
Journal of Orthopaedic Surgery and Research (2024)