
Abstract
In the context of solid tumours, the evolution of cancer therapies to more targeted and nuanced approaches has led to the impetus for personalised medicine. The targets for these therapies are largely based on the driving genetic mutations of the tumours. To track these multiple driving mutations the use of next generation sequencing (NGS) coupled with a morphomolecular approach to tumours, has the potential to deliver on the promises of personalised medicine. A review of NGS and its application in a universal healthcare (UHC) setting is undertaken as the technology has a wide appeal and utility in diagnostic, clinical trial and research paradigms. Furthermore, we suggest that these can be accommodated with a unified integromic approach. Challenges remain in bringing NGS to routine clinical use and these include validation, handling of the large amounts of information flow and production of a clinically useful report. These challenges are particularly acute in the setting of UHC where tests are not reimbursed and there are finite resources available. It is our opinion that the challenges faced in applying NGS in a UHC setting are surmountable and we outline our approach for its routine application in diagnostic, clinical trial and research paradigms.
Similar content being viewed by others

Main
The future of modern medicine will be dictated by new discoveries in the molecular basis of disease, new technological advances and the accessibility to these developments that healthcare systems provide for individual patients. Indeed, the question of next generation sequencing (NGS) in routine diagnostics epitomises these aspects; a new technology delivering novel DNA-based discoveries in routine diagnostics, which can be applied within those healthcare systems making it affordable for routine use. There are excellent examples of NGS validations in the literature (Frampton et al, 2013; Cheng et al, 2015) coming from well-resourced, reimbursement-based healthcare systems. However, many countries have at least a partly or wholly universal healthcare (UHC) system (Figure 1) and the application of NGS technology has some common challenges but also further distinct considerations in this environment.

Global expenditure on health related to GDP and the efficacy of the global UHC environment. We have highlighted countries with a private healthcare spend of <40% as an index for UHC (WHO and Bloomberg).
An all-encompassing definition of UHC can be difficult. A reasonable working definition is all required healthcare which is free at the point of care. However, due to the rapid nature of progress particularly in the area of molecular-based medicine the line between what is necessary and what is required is obscured. The proliferation of genetic technology and in particular sequencing technology means that many patients are familiar with and proactive in seeking genetic information regarding their disease. Many oncologists will have the experience of patients requesting genomic sequencing of their tumour. This can be accomplished in private healthcare or if ones insurance coverage allows for reimbursement. In contrast, in a UHC environment, resources are finite and therefore it is important to ensure that cancer patients are provided with at the very least all the molecular information regarding their tumour that can affect its management. It has also been shown that prognostic signatures can provide confidence for the patient in management and be of economic benefit such as oncotype Dx (Holt et al, 2013).
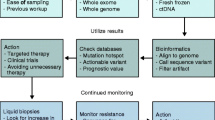
At present the molecular information required for standard-of-care management can be achieved predominantly through single-gene tests. However, we are at a tipping point with more targeted therapies coming online and immune-checkpoint modulation a real option in combatting solid tumours. These interventions require more detailed information than ever regarding the molecular composition of tumours and it is arguable that this information can be provided in a more efficient and timely fashion utilising NGS technology as a ‘one-stop-shop’ for all the currently targetable driver mutations in a tumour (Souilmi et al, 2015). In addition, given that tumour heterogeneity drives the evolution of a cancer the identification of potential targets which may become relevant following late recurrences or first line treatment failure are important for future management of the patient.
It is clear that the age of personalised medicine is here, with many cancer care pathways requiring both histological-based diagnosis and molecular pathology mutational analysis of the tumour (Friedman et al, 2015). Frequent allelic variants of actionable target mutations are reliably detected by clinically applied low-throughput technologies. However, analysis of 4742 tumour-normal matched controls across 21 tumours types revealed over 200 cancer-related genes many having clinically relevant frequencies of 2–20% (Lawrence et al, 2014). Furthermore, down-sampling analysis has estimated that increasing the sample size will reveal more clinically relevant mutations. Clearly, attempting to describe these mutations using single-gene technology is not feasible (Lawrence et al, 2014).
There are a number of paradigms where NGS can be applied (Table 1). The most significant is its role in clinical diagnostics where it can examine all standard of care, actionable mutations simultaneously from low-quality formalin-fixed paraffin-embedded (FFPE)-derived DNA. These specimens can include different types including fine needle aspirations (smears and/or cell blocks), biopsies and resections with many differences in their pre-analytical processing. The second paradigm is a clinical trial where it can provide comprehensive data regarding potential actionable mutations. Finally, NGS can be used in the research paradigm by rapidly identifying candidate biomarkers. We would argue that these paradigms need a holistic, patient-centred and affordable approach for the appropriate healthcare system, ideally in an integrated molecular pathology environment (Salto-Tellez et al, 2014).
Models for application of NGS in a UHC setting
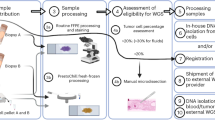
It may seem self evident that centralisation of molecular pathology services is required to make them viable in a UHC setting, however, there are still many hospitals currently carrying out molecular tests, usually with single-gene technology, on small numbers of samples in UHC settings which obviously raises costs. Given the current capital expense of NGS platforms, specialised personnel required and bioinformatics infrastructure needed, these costs could only be justified in a centralised laboratory (Figure 2). In this model there are logistical issues regarding sample transport and ensuring standardisation of pre-analytical factors. Moreover, if the hospital is a cancer centre and does not have molecular diagnostics there may be a disconnect with the MDTs where feedback on results may be required. This can be easily addressed through teleconferencing.

Diagrammatic version of our suggested models for the evolution of NGS in a UHC environment.
The expense of NGS platforms and capital equipment will likely come down in the future. With the proliferation of this technology a centralised model may evolve over time to a more decentralised model with several cancer centres serving regional populations carrying out the tests while sending their results to a central bioinformatics core. One might suggest that such bioinformatics cores could be superseded by cloud-based computing (Souilmi et al, 2015). However, this raises significant issues regarding sensitive health data travelling across jurisdictions for storage or analysis.
Future directions for the evolution of NGS in solid tumour testing include its use to analyse so-called ‘liquid biopsies’ which involves the analysis of cell free DNA from the patient’s serum. The use of serial liquid biopsies has been shown to be useful in monitoring disease progression/treatment failure. In particular this has been shown in lung cancer studies where new driver mutations were identified in patients with a previous T790M mutation (Chia et al, 2016) Patient preference for less invasive procedures and the difficulties in obtaining serial tissue biopsies means that the increased volume of liquid biopsies may contribute to making the scalable NGS more cost-effective for UHC-based institutions.
NGS clinical applications
The chief considerations as to whether NGS will be successfully applied to routine diagnostics are clinical utility and cost (Aronson, 2015). Even in these early days of genomic-based oncology, where numbers of actionable mutations are still relatively modest, the test result must relate to an evidence-based management decision. The majority of management decisions for cancer patients in a UHC environment are made at vital MDTs, with oncology, pathology, radiology and surgery input amongst others. At present the binary form of information regarding mutations (mutant/wild type) makes the addition of molecular information to the MDT environment equivalent to prognostic immunohistochemistry such as HER2 receptor analysis. However, as molecular pathologists we are aware that such information is rarely binary and requires setting of thresholds and other elements which may be obscured in the bioinformatics that produce a result for the oncologist. Therefore, it is vital to curate the data in the reports using up-to-date clinical trial information and to be able to interpret these results at MDTs.
Indeed, the curation of the NGS data into a meaningful diagnostic report is challenging, particularly for large gene panels, due to several degrees of ‘clinical relevance’ within the information provided. When crafting these reports, it can be unclear whether a specific mutation activates or inactivates the gene, or if it engenders sensitivity to specific therapies, or how therapies should be prioritised where there are multiple genomic alterations. Consequently, mutations are identified and actioned as part of (a) standard-of-care pathways, (b) clinical trial enrolment and (c) ‘no option’ patients without significant evidence for a targeted therapy. Indeed, one could argue that in these early days of genomic diagnostics reporting we are ‘managing levels of genomic (un)certainty’. Therefore, communicating the level of evidence/confidence in these results needs to be illuminated for clinicians. This approach is epitomised in the model of Dienstmann et al (2014), where the genomic results are organised, into levels of clinical applicability, as ‘treatment recommendation’, ‘clinical trial eligibility’ or ‘observe /talk to an expert’.
Within a UHC system it is clear that a centralised database for annotation and curation of reports is vitally important. Such a system allows finite resources to be used economically including information technology infrastructure and bioinformatics as well as molecular pathology expertise. Furthermore, it would allow standard reports with clear recommendations to be provided to clinicians. Similarly to our radiology colleagues' change to digital pathology, such a change in histopathology would require up-front investment in IT and personnel by a UHC system.
An individual institution’s capacity to engage in personalised medicine depends on the defined standard of care in a given country, the institutional access of patients to clinical trials, and the openness of institutional ethical review boards to allow therapeutic intervention, based solely on biological observation. This is accentuated in the 58 countries, as of 2009, with UHC (Stuckler et al, 2010). In such systems, there is frequently a contrast between the approach of healthcare administrators managing finite resources, who must apply strict criteria regarding the ‘rules of engagement’ for therapeutics and consequently for diagnostics, and academic healthcare professionals who wish to offer a patient-specific best option and derive as much as possible for future patients. Micro-costing has shown that for lung cancer in a reimbursement model the cost of a 5–50 gene panel is $578 to $908 (Sabatini et al, 2016). Interestingly, there is an estimated decrease in costs compared with single-gene testing when clinical trial participation is factored in, where investigational drugs do not cost much. When this is factored in there is a modest cost increase due to increased use of targeted therapies overall (Sabatini et al, 2016). Never before has the gap between what is possible and what is affordable in patient diagnostics and treatment been so obvious in modern medicine.
However, exceptions to this dynamic exist. In Canada, a credible private pathway is available which provides reimbursable testing at a national level led by global experts in the field such as Dr. Samuel Aparicio (http://contextualgenomics.com). In contrast, the German Network for Genomic Medicine (NGM) in lung cancer is a healthcare network providing NGS-based multiplex genotyping for all inoperable lung cancer patients (Kostenko et al, 2015). Since German reimbursable procedures do not include NGS-based multiplex genotyping, NGM has successfully started a nationwide, pilot-project establishing a flat rate reimbursement model for NGS in Germany, a model transferrable to other tumour types. In the UK, the 100 000 genome project run by Genomics England (http://www.genomicsengland.co.uk/the-100000-genomes-project/) aims to generate substantial whole genome sequencing data from different cancer types, including rarer forms and, at the same time, shape the testing landscape where no model for diagnostic testing currently exists. The longevity of these initiatives remains to be seen.
Pre-analytical factors for the clinical use of NGS
Many of the pre-analytical factors which affect the performance of NGS are common regardless of the health system encountered due to the commonality of FFPE. Within a UHC system the use of consistent guidelines is important to provide high-quality DNA from such specimens for testing (Lindeman et al, 2013; Gargis et al, 2016). Moreover, within UHC systems the use of such standardised guidelines and tissue pathways, fixation times and a morphomolecular approach would all contribute to high-quality DNA. Moreover the use of cytological samples can provide higher concentrations of tumour DNA. In histological specimens we advocate the use of a guiding H&E slide along with a morphomolecular approach (Salto-Tellez et al, 2014). Many laboratories will be familiar with providing scrolls of FFPE tissue for molecular analysis to private laboratories for analysis. However, without expert review the relative amount of tumour or, indeed whether tumour is present at all, cannot be guaranteed. Low or no tumour concentration in a sample increases the chance of test failure and misinterpretation of wild-type results, that is, too low a concentration of tumour may result in too weak a mutation signal to be picked up above the wild type ‘noise’. One caveat regarding all tests and tumour sampling is tumour molecular heterogeneity which may reflect the site of the tumour sampling. The use of a morphomolecular approach, allows tumour morphology to inform sampling and indeed clonal areas may be identified on histology which can be sampled and tested separately to help guide treatment.
Where appropriate cytological samples may be recommended to clinicians regarding molecular testing of a tumour and this provides useful genetic material for testing once the tumour concentration is at least 20% and the specimen is sufficiently diagnostically advanced (Salto-Tellez, 2015). For example lung cytology cases which are equivocal for squamous or adenocarcinoma would skew EGFR results if mistyped and cause unnecessary expense. Indeed, the combination of rapid on site assessment, the lack of fixation effect and the potential use of direct smears in addition to cell blocks makes cytology specimens very attractive for NGS testing as described and utilised by the MD Andersen group (Roy-Chowdhuri and Stewart, 2016).
Choice of gene panel for NGS
Presuming that most laboratories will adopt a pragmatic NGS-targeted approach, detecting hotspot mutations, there are several variables that determine the choice of panel and its associated costs:
1. Design of the gene panel: a number of possible designs are outlined below (Figure 3).

Choice of gene panel based on the paradigm being investigated by NGS.
Class 1 design: basic actionable mutations, up to 10–15 genes hotspots and larger gene regions
This will hold hotspot mutations considered routine, for genes such as KRAS (colorectal and lung), NRAS (colorectal and melanoma), BRAF (melanoma and colorectal), EGFR (lung), KIT (gastrointestinal stromal tumour (GIST) and melanoma), PDGFRA (GIST and melanoma) and larger gene regions such as BRCA1 and 2 (ovarian). PIK3CA and TP53 could be added to this hotspot list, as well as some of the myeloproliferative related mutations such as JAK2, CALR and MPL.
Class 2 design: broad, clinically relevant, up to 50 genes
This design will include those in Class 1 and, in addition, mutations that allow oncologists to enrol patients in clinical trials supported by the individual institution. Therefore, these are very much hospital-dependent. They may include mutations of AKT1 or ESR1 (breast cancer), PTEN (prostate cancer) or MET in several cancer types (Dietel et al, 2015).
Class 3 design: tumour comprehensive, up to 150 genes
This is a cancer-specific design for translational research, comprising all relevant mutations relating to the cancer’s biology.
Class 4 design: human cancer comprehensive, up to 400 genes
This includes all cancer-relevant genes.
2. The volume of testing: presuming a TAT of 1–2 weeks and a minimum of 1 run per week, an accurate assessment of the laboratory’s average working volume throughout the year is vital for allocation of resources and choice of testing platform. Clinical factors impacting on this such as introduction of new surgical services must be anticipated through a management structure.
3. The size of the runs: tightly related to the previous point. A chip with capacity for 20 cases may be more affordable per case than a chip of 12 samples. However, if a lab has 9–12 cases per week, and runs a chip per week for TAT purposes, then a 12 sample chip may be more cost-effective (Figure 4).

Estimation of costs associated with NGS runs linked to testing volume.
4. The technology: different platforms may need different up-front costs, and many have different running costs.
5. The minimum DNA requirement: Diagnostic practices with resection specimens are likely to yield significant DNA content per sample, and thus the minimum DNA concentration threshold necessary to run the test is easily achieved. However, in small biopsy or cytology samples total DNA may be limiting, with larger numbers of cases deemed inadequate. This is an important consideration for overall laboratory budgeting, because the number of fails and need to maintain accreditation for single-gene orthogonal tests can make the overall cost of NGS testing significantly more expensive.
6. The availability of a clinical reporting bioinformatics system: discussed with validation below.
Validation
The validation of NGS includes pre-analytical, nucleic acid preparation, sequencing and bioinformatics. Validation of NGS needs to examine analytical sensitivity, specificity, accuracy and precision as well as validating for minimum sequence coverage and downstream validation of bioinformatics pipelines. With the levels of validation required it is important to set standard parameters in advance. In particular, choices of instrument and bioinformatics approach are important considerations. Crucially pre-analytics must be considered as discussed above. New embedding materials and processing of specimens particularly for auto-embedding units need to be validated to ensure no loss of mutant signal. Interestingly, the authors have personal experience of gel-based embedding material interfering with downstream molecular analysis.
Accreditation for molecular tests should be achievable by a professional and competent laboratory (Aziz et al, 2015). Low-throughput assays have a straightforward validation/verification process with the precise variants having matched, characterised reference material. For NGS, however, validating every variant within the target DNA is not feasible (Salto-Tellez et al, 2014). No such reference material exists to accommodate all the variants that could be examined using NGS. Reference materials, in the form of pooled cell lines, have proven helpful in providing strength to NGS validation although are imperfect (Frampton et al, 2013). A pragmatic approach is to validate the main clinically relevant targets initially and then validate the less frequent mutations in an ongoing continuous manner as they are detected. This requires the use of orthogonal reference standard methods in addition to the NGS testing which may make costs prohibitive. Moreover, the gold-standard method, Sanger sequencing, has a lower sensitivity than NGS and will miss lower frequency mutations.
The most successful validations of diagnostic NGS thus far have been carried out using single-platform approaches with tumour samples or pooled cell lines which have well-characterised molecular aberrations. From our own experience there has been a tendency to have local variations in sample type (scrolls vs morphomolecular slides vs fresh samples), platform type and bioinformatics tools which inhibits collaborative validations. We propose that cross centre collaborative validations represent the best model for carrying out efficient NGS validations within a UHC system (Figure 5). These would take the form of specific molecular pathology centres having strict provisions regarding agreed instrumentation and protocols, including bioinformatics, to carry out benchmarking validations of various mutations and mutation types. This would subsequently be followed by validations of other mutations carried out separately in the different centres and then extrapolated between the centres. Regular internal ‘calibration’ sets of tumours would be routinely analysed to ensure adequate quality control.

A model for NGS collaborative validations as suggested for a UHC environment. We have proposed to use this system within the UK NHS for collaborative validations between centres. This is a non-centralised, non-hierarchical system across centres of excellence.
Some of the limitations of NGS include its ability to detect different mutation types, in particular large insertions/deletions, which may be suboptimally recognised in this setting and these would need careful validation to ensure adequate sensitivity. Practical solutions have been described with validations of NGS in the literature (Frampton et al, 2013; McCourt et al, 2013; Cottrell et al, 2014; Cheng et al, 2015; Wang et al, 2016). It is interesting to note that the most detailed validations have occurred in the US where the health system is predominantly a private concern. Frameworks in this health setting have been published from several sources including the Association for Molecular Pathology, and the College of American Pathologists. To date, the ability to demonstrate validation of NGS in a UHC setting has been limited and may require the innovations described above to be successful.
The most comprehensive attempts to validate NGS in clinical practice include the Foundation Medicine Group validation (Frampton et al, 2013) and the Memorial Sloan Kettering integrated mutation profiling of actionable cancer targets (MSK-IMPACT) validation (Cheng et al, 2015). Foundation examined 287 cancer-related gene exons and introns from 19 genes frequently rearranged in solid tumours. They validated their process with cell lines mimicking a range of mutant allele frequency percentages (Frampton et al, 2013). A similar cell line approach was used to validate for single-nucleotide variants (SNVs) in the Washington University cancer mutation profiling NGS assay (McCourt et al, 2013). In addition, within the Foundation validation, indels were validated by spiking tumour pools with known somatic indel variants. Copy number alterations were validated using tumours with various amplifications and homozygous deletions matched with normal cell lines in a range of tumour concentrations (Frampton et al, 2013). The MSK-IMPACT approach analysed 341 cancer genes (Cheng et al, 2015). Validation in this case was carried out using 284 tumour DNA samples and 75 matched controls. The tumour samples had previously been genotyped/sequenced using alternative methods and had SNVs or indels in 47 exons of 19 genes with 393 known variants detected.
Outside of an environment where there is insurance reimbursement for tests and significant endowment/direct investment, the cost of validation is also a sensible concern. For NGS with modest scope (<20 genes), validation of all possible variations including all clinical hotspots, extraction protocols, sample types and a bioinformatics pipeline could cost close to £60 000 in the UK setting for consumables alone. Furthermore, in the UK, where cost effectiveness of new standards of care is driven by organisations like the National Institute for health and Care Effectiveness (NICE), the dialectic between what is necessary to test, what is desirable to test in the clinical trial and academic context, and what would be useful to inform future therapeutic decisions without current clinical trial evidence, is difficult to manage. However, it is important to consider that current analysis for point mutations of clinical relevance may already cost £200–300 per case to the National Health Service, and hence NGS solutions able to provide a test within that boundary would be cost neutral to UHC systems.
Information flow with NGS
All three aspects of information flow for NGS must be validated, these include (1) the bioinformatics pipeline, (2) the compiling and communication of the clinical report, and (3) the integration of (1) and (2) with the laboratory information system (LIS). Increasing amounts of information at each level raises a number of issues for service delivery including adequately skilled personnel and infrastructure issues with associated costs of storage and processing power. A hybrid facility, as suggested by the ‘Belfast model’ (Salto-Tellez et al, 2014), combines the skill sets of an academic bioinformatics department within a clinical environment to produce a cost-effective bioinformatics pipeline with clinical utility and appropriate TATs for application in a UHC setting (Oliver et al, 2015). It is clear from the ISO15189 standard for accreditation that the same rigorous validation is required for the bioinformatics pipeline and any change results in re-validation, although the raw FASTQ data can be used to condense this process considerably.
Data generated following the initial reads, sequence alignment and variant calling within the pipeline must be assimilated into a meaningful clinical report requiring interpretation of the bioinformatics by the reporting team. The role of clinical scientist within this context is key to bridge the bioinformatics to the clinical report. In departments with an integrated model consisting of molecular diagnostics, clinical trial work and biomarker discovery, a dedicated bioinformatician is required. Communicating these results to clinicians is a critical role for the reporting team who need to incorporate this information into the care pathway in a clinically useful TAT. They will rely on annotation databases which are accurate and constantly updated/curated (Dienstmann et al, 2014). However, as highlighted in a germline setting, evidence-based annotations can be inaccurate in a third of cases (Bell et al, 2011). To ensure optimal interpretation of the tests, consistent bottom line diagnoses must be used and it may be argued that the reporting team members should partake in multidisciplinary team meetings to aid dissemination of the results.
Workflow management for single-gene assays can be achieved using spreadsheet databases. However, this is not scalable for NGS testing. It will be a necessity in the future that the (LIS) will require integration with the bioinformatics pipeline. Furthermore, consolidation of an LIS within a UHC setting must take into account molecular testing from pathology, clinical genetics, microbiology and haematology.
Clinical trials and NGS
Perhaps the most obvious application for NGS is in clinical trials. Tumour heterogeneity gives cancer the ability to evolve and be driven by resistance clones developing in response to the selective pressures of treatment (Enriquez-Navas et al, 2015). Tracking this complex biology cannot be done with low-throughput technologies and NGS is necessary to allow for full tumour taxonomy within the timeframe of a clinical trial.
Trial designs which aim to incorporate molecular stratification of patients include umbrella, basket and adaptive. Umbrella trials are merely an extension of the current dogma in clinical trials with solid tumours characterised in the first instance by histology and then with additional limited mutation parameters which allow substratification of tumours. Although only an incremental step, it has proven fruitful in lung cancer with EGFR analysis, colorectal cancer with N/KRAS and melanoma with BRAF. Basket trial design allows mutation characteristics of a tumour, independent of site and other histological factors to be used to select patients. However, this must be done with a biological rationale (Sleijfer et al, 2013), as highlighted by KIT mutations in GISTs (Nakahara et al, 1998).
In our opinion the clinical trial design which has best incorporated molecular pathology and which would be ideal for use in an NGS environment is the adaptive design. An example of the use of this design in a UHC system has been the Biomarker-integrated Approach of Targeted Therapy for Lung Cancer Elimination (BATTLE) trials which utilised a statistical adaptive design (Kim et al, 2011) allowing the trial to be more ethical and effective. The National Lung Matrix Trial being carried out within the NHS in the UK seeks to enhance the number of molecular targets which may used in non-small cell lung cancer (Middleton et al, 2015). The aim is to identify known molecular targets found at low frequencies. Similar to the BATTLE design this study utilises a Bayesian adaptive design which allows for more power for low frequency mutations where recruiting sufficient patients in a traditional clinical trial would not be possible. It may be anticipated that single-gene testing will have a limited role in future clinical trials and by combining NGS diagnostics and clinical trial patients overall costs may be reduced in UHC as already described in a reimbursement model (Sabatini et al, 2016).
Conclusions
In these early days of genomic medicine, how cancer practitioners learn to generate, manage and act upon genomic information may dictate the speed with which we help our cancer patients live longer and better lives. Thus, even at a time in which cancer healthcare costs are consistently increasing, this is not the moment to have a ‘common denominator’ approach to genomic testing. NGS technology is bringing high-quality analysis at an affordable cost, and it may be strategically sound for UHC systems to accept that an increase in budgets associated with genomics testing, whilst we learn to apply it clinically, will be a great long-term investment.
The ideal model for provision of molecular diagnostics in UHC systems remains controversial but is at the heart of how we wish to deliver NGS for diagnostics. The Canadian private model, the unified model for NGS lung cancer testing in Germany, or the lack of a defined model for commissioning of molecular diagnostics currently favoured in most of the UK, are currently available. Significant centralisation, ensuring concentration of technical skills, resources and economy of scale will make for the most cost-efficient framework for NGS diagnostics in the future.
Genes and HUGO Nomenclature:
- NRAS :
-
neuroblastoma RAS viral (v-ras) oncogene homolog
- BRAF :
-
B-Raf proto-oncogene, serine/threonine kinase
- EGFR :
-
epidermal growth factor receptor
- KIT :
-
KIT proto-oncogene receptor tyrosine kinase
- PDGFRA :
-
platelet-derived growth factor receptor alpha
- BRCA :
-
breast cancer 1
- BRCA2 :
-
breast cancer 2
- PIK3CA :
-
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- TP53 :
-
tumour protein p53
- JAK2 :
-
Janus kinase 2
- CALR :
-
calreticulin
- MPL :
-
MPL proto-oncogene, thrombopoietin receptor
- AKT1:
-
v-akt murine thymoma viral oncogene homolog 1
- ESR1 :
-
oestrogen receptor 1
- PTEN :
-
phosphatase and tensin homolog
- MET :
-
MET proto-oncogene, receptor tyrosine kinase
- FAT4 :
-
FAT atypical cadherin 4
References
Aronson N (2015) Making personalized medicine more affordable. Ann N Y Acad Sci 1346: 81–89.
Aziz N, Zhao Q, Bry L, Driscoll DK, Funke B, Gibson JS, Grody WW, Hegde MR, Hoeltge GA, Leonard DG, Merker JD, Nagarajan R, Palicki LA, Robetorye RS, Schrijver I, Weck KE, Voelkerding KV (2015) College of American Pathologists' laboratory standards for next-generation sequencing clinical tests. Arch Pathol Lab Med 139: 481–493.
Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, Langley RJ, Zhang L, Lee CC, Schilkey FD, Sheth V, Woodward JE, Peckham HE, Schroth GP, Kim RW, Kingsmore SF (2011) Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med 3: 65ra4.
Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, Brannon AR, O'Reilly C, Sadowska J, Casanova J, Yannes A, Hechtman JF, Yao J, Song W, Ross DS, Oultache A, Dogan S, Borsu L, Hameed M, Nafa K, Arcila ME, Ladanyi M, Berger MF (2015) Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 17: 251–264.
Chia PL, Do H, Morey A, Mitchell P, Dobrovic A, John T (2016) Temporal changes of EGFR mutations and T790M levels in tumour and plasma DNA following AZD9291 treatment. Lung Cancer 98: 29–32.
Cottrell CE, Al-Kateb H, Bredemeyer AJ, Duncavage EJ, Spencer DH, Abel HJ, Lockwood CM, Hagemann IS, O'Guin SM, Burcea LC, Sawyer CS, Oschwald DM, Stratman JL, Sher DA, Johnson MR, Brown JT, Cliften PF, George B, McIntosh LD, Shrivastava S, Nguyen TT, Payton JE, Watson MA, Crosby SD, Head RD, Mitra RD, Nagarajan R, Kulkarni S, Seibert K, Virgin HW 4th, Milbrandt J, Pfeifer JD (2014) Validation of a next-generation sequencing assay for clinical molecular oncology. J Mol Diagn 16: 89–105.
Dienstmann R, Dong F, Borger D, Dias-Santagata D, Ellisen LW, Le LP, Iafrate AJ (2014) Standardized decision support in next generation sequencing reports of somatic cancer variants. Mol Oncol 8: 859–873.
Dietel M, Jöhrens K, Laffert MV, Hummel M, Bläker H, Pfitzner BM, Lehmann A, Denkert C, Darb-Esfahani S, Lenze D, Heppner FL, Koch A, Sers C, Klauschen F, Anagnostopoulos I (2015) A 2015 update on predictive molecular pathology and its role in targeted cancer therapy: a review focussing on clinical relevance. Cancer Gene Ther 22: 417–430.
Enriquez-Navas PM, Wojtkowiak JW, Gatenby RA (2015) Application of evolutionary principles to cancer therapy. Cancer Res 75: 4675–4680.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, Sun J, Juhn F, Brennan K, Iwanik K, Maillet A, Buell J, White E, Zhao M, Balasubramanian S, Terzic S, Richards T, Banning V, Garcia L, Mahoney K, Zwirko Z, Donahue A, Beltran H, Mosquera JM, Rubin MA, Dogan S, Hedvat CV, Berger MF, Pusztai L, Lechner M, Boshoff C, Jarosz M, Vietz C, Parker A, Miller VA, Ross JS, Curran J, Cronin MT, Stephens PJ, Lipson D, Yelensky R (2013) Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31: 1023–1031.
Friedman AA, Letai A, Fisher DE, Flaherty KT (2015) Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer 15: 747–756.
Gargis AS, Kalman L, Lubin IM (2016) Assuring the quality of next-generation sequencing in clinical microbiology and public health laboratories. J Clin Microbiol 54: 2857–2865.
Holt S, Bertelli G, Humphreys I, Valentine W, Durrani S, Pudney D, Rolles M, Moe M, Khawaja S, Sharaiha Y, Brinkworth E, Whelan S, Jones S, Bennett H, Phillips CJ (2013) A decision impact, decision conflict and economic assessment of routine Oncotype DX testing of 146 women with node-negative or pNImi, ER-positive breast cancer in the UK. Br J Cancer 108 (11): 2250–2258.
Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR Jr, Tsao A, Stewart DJ, Hicks ME, Erasmus J Jr, Gupta S, Alden CM, Liu S, Tang X, Khuri FR, Tran HT, Johnson BE, Heymach JV, Mao L, Fossella F, Kies MS, Papadimitrakopoulou V, Davis SE, Lippman SM, Hong WK (2011) The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov 1: 44–53.
Network Genomic Medicine Lung Cancer, Kostenko A, Glossmann JP, Michels SYF, Sueptitz J, Scheffler M, Fischer R, Markiefka B, Scheel A, De Mary P, Kron F, Buettner R, Wolf J (2015) The network genomic medicine cost reimbursement model for implementation of comprehensive lung cancer genotyping in clinical routine. J Clin Oncol 33 (suppl): abstract e12556.
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G (2014) Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505: 495–501.
Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, Jenkins RB, Kwiatkowski DJ, Saldivar JS, Squire J, Thunnissen E, Ladanyi M (2013) Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. Arch Pathol Lab Med 137: 828–860.
McCourt CM, McArt DG, Mills K, Catherwood MA, Maxwell P, Waugh DJ, Hamilton P, O'Sullivan JM, Salto-Tellez M (2013) Validation of next generation sequencing technologies in comparison to current diagnostic gold standards for BRAF, EGFR and KRAS mutational analysis. PLoS One 8: e69604.
Middleton G, Crack LR, Popat S, Swanton C, Hollingsworth SJ, Buller R, Walker I, Carr TH, Wherton D, Billingham LJ (2015) The national lung matrix trial: translating the biology of stratification in advanced non-small-cell lung cancer. Ann Oncol 26: 2464–2469.
Nakahara M, Isozaki K, Hirota S, Miyagawa J, Hase-Sawada N, Taniguchi M, Nishida T, Kanayama S, Kitamura Y, Shinomura Y, Matsuzawa Y (1998) A novel gain-of-function mutation of c-kit gene in gastrointestinal stromal tumors. Gastroenterology 115: 1090–1095.
Oliver GR, Hart SN, Klee EW (2015) Bioinformatics for clinical next generation sequencing. Clin Chem 61: 124–135.
Roy-Chowdhuri S, Stewart J (2016) Preanalytic variables in cytology: lessons learned from next-generation sequencing-the MD Anderson experience. Arch Pathol Lab Med Available at: https://www.ncbi.nlm.nih.gov/pubmed/27333361 (accessed 2 January 2017).
Sabatini LM, Mathews C, Ptak D, Doshi S, Tynan K, Hegde MR, Burke TL, Bossler AD (2016) Genomic sequencing procedure microcosting analysis and health economic cost-impact analysis: a report of the association for molecular pathology. J Mol Diagn 18: 319–328.
Salto-Tellez M (2015) Diagnostic molecular cytopathology - a further decade of progress. Cytopathology 26: 269–270.
Salto-Tellez M, Gonzalez de Castro D (2014) Next-generation sequencing: a change of paradigm in molecular diagnostic validation. J Pathol 234: 5–10.
Salto-Tellez M, James JA, Hamilton PW (2014) Molecular pathology - the value of an integrative approach. Mol Oncol 8: 1163–1168.
Sleijfer S, Bogaerts J, Siu LL (2013) Designing transformative clinical trials in the cancer genome era. J Clin Oncol 31: 1834–1841.
Souilmi Y, Lancaster AK, Jung JY, Rizzo E, Hawkins JB, Powles R, Amzazi S, Ghazal H, Tonellato PJ, Wall DP (2015) Scalable and cost-effective NGS genotyping in the cloud. BMC Med Genomics 8: 64.
Stuckler D, Feigl AB, Basu S, McKee M (2010) The political economy of universal health coverage. First Global Symposium on Health Systems Research; 16–19 November 2010; Montreaux, Switzerland.
Wang SR, Malik S, Tan IB, Chan YS, Hoi Q, Ow JL, He CZ, Ching CE, Poh DY, Seah HM, Cheung KH, Perumal D, Devasia AG, Pan L, Ang S, Lee SE, Ten R, Chua C, Tan DS, Qu JZ, Bylstra YM, Lim L, Lezhava A, Ng PC, Wong CW, Lim T, Tan P (2016) Technical validation of a next-generation sequencing assay for detecting actionable mutations in patients with gastrointestinal cancer. J Mol Diagn 18: 416–424.
Acknowledgements
Dr Seán Hynes is supported by a Dr Richard Steevens Scholarship from the Health Service Executive, Ireland. No specific funding was utilised to prepare this commentary. In general, the Northern Ireland Molecular Pathology Laboratory is supported by Cancer Research UK, Experimental Cancer Medicine Centre Network, the NI Health and Social Care Research and Development Division, the Sean Crummey Memorial Fund, the Tom Simms Memorial Fund and the Friends of the Cancer Centre.
Author contributions
All authors have contributed to the intellectual content of this paper and have made significant contributions to the conception and design, drafting or revising the article for intellectual content; and final approval of the published article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Hynes, S., Pang, B., James, J. et al. Tissue-based next generation sequencing: application in a universal healthcare system. Br J Cancer 116, 553–560 (2017). https://doi.org/10.1038/bjc.2016.452
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.452
- Springer Nature Limited
Keywords
This article is cited by
-
Invited review—next-generation sequencing: a modern tool in cytopathology
Virchows Archiv (2019)