
Abstract
Background:
Heat-shock protein 990 (HSP990) is a potent and selective synthetic small-molecule HSP90 inhibitor. The primary objectives of this phase I first-in-human study were to determine dose-limiting toxicities (DLTs), maximum-tolerated dose (MTD) and recommended phase II dose (RP2D). Secondary objectives included characterisation of the safety profile, pharmacokinetics (PKs) and pharmacodynamics (PDs).
Methods:
Heat-shock protein 990 was administered orally once or two times weekly on a 28-day cycle schedule in patients with advanced solid tumours. Dose escalation was guided by a Bayesian logistic regression model with overdose control.
Results:
A total of 64 patients were enrolled. Fifty-three patients received HSP990 once weekly at 2.5, 5, 10, 20, 30, 50 or 60 mg, whereas 11 patients received HSP990 two times weekly at 25 mg. Median duration of exposure was 8 weeks (range 1–116 weeks) and 12 patients remained on treatment for >16 weeks. Dose-limiting toxicities occurred in seven patients and included diarrhoea, QTc prolongation, ALT/AST elevations and central neurological toxicities. The most common drug-related adverse events were diarrhoea, fatigue and decreased appetite. Further dose escalation beyond 60 mg once weekly was not possible owing to neurological toxicity. Rapid absorption, no drug accumulation and large interpatient variability in PK exposures were observed. No objective responses were seen; 25 patients had a best overall response of stable disease.
Conclusions:
Heat-shock protein 990 is relatively well tolerated, with neurological toxicity being the most relevant DLT. The single agent MTD/RP2D of HSP990 was declared at 50 mg once weekly.
Similar content being viewed by others

Main
Heat-shock proteins (HSPs) are molecular chaperones present in almost every cellular compartment, stimulated by environmental stress conditions such as infection, inflammation, starvation, hypoxia and cancer (Macario and Conway de Macario, 2005). Through correct protein folding and functional conformation, HSPs protect cells from stress damage, leading to protein stability (Macario and Conway de Macario, 2005). They are classified based on their molecular weight into high-molecular-weight HSPs (⩾100 kDa), in contrast to small HSPs (<34 kDa), as well as those that fall in between these two categories with molecular weights between 99 and 35 kDa (Macario and Conway de Macario, 2005). Within HSPs, HSP90 and HSP70 are two functionally related ATP-dependent chaperones that, despite their cooperation to prevent protein aggregation through heat-shock factor 1, are inversely regulated (i.e. HPS90 inhibits the expression of HSP70), leading to feedback loop that limits the response to stress (Erlichman, 2009). Overexpression of HSPs, in particular HSP90, with consequent oncogenic protein stabilisation, stimulation of cell proliferation, survival and angiogenesis has been shown to occur commonly in cancer cells (Whitesell and Lindquist, 2005; Powers and Workman, 2007; Neckers and Workman, 2012). For instance, HSP90 is known to stabilise several growth factor receptors and signal-transduction pathway regulators such as the epidermal growth factor receptor (EGFR) (Shimamura et al, 2005). In clinicopathological studies, tumour progression and poor prognosis are associated with HSP overexpression in several cancer types (Li et al, 2008; Simpson et al, 2010). Therefore, HSPs are an attractive cancer target and their inhibitors have been developed in preclinical and clinical settings.
Heat-shock protein 90 inhibitors demonstrate antitumour activity in multiple tumour types both as single agents and in combination with targeted therapies, standard chemotherapy or radiotherapy in preclinical models (Sawai et al, 2008; Kim et al, 2009; Menezes et al, 2012). In addition, their role in HER-2-positive breast cancer alone or in combination with anti-HER2 and hormonal therapy has been described previously (De Mattos-Arruda and Cortes, 2012; Scaltriti et al, 2012). An additional area of interest has been lung cancer. In lung cancer cell lines, radioresistance can be induced through hypoxia-inducible factor-1 (HIF-1) upregulation, enhanced HSP90–HIF-1 interaction and PI3K/Akt/mTOR pathway activation. This process can be reversed by the natural HSP90 inhibitor deregulin (Kim et al, 2009). Recent studies have described potent in vitro and in vivo ability of HSP90 inhibition in restoring drug responsiveness in crizotinib-resistant anaplastic lymphoma kinase (ALK)-driven non-small-cell lung cancer (NSCLC) models, via the loss of echinoderm microtubule-associated protein-like 4 (ELM4)-ALK fusion gene expression and oncogenic protein depletion (Chen et al, 2013; Sang et al, 2013). In the clinical setting, HSP90 inhibitors have been evaluated in multiple phase I and II clinical studies as single agent and in combination with chemotherapy, hormonal therapy or targeted agents (Goldman et al, 2013; Johnson et al, 2013; Sessa et al, 2013; Socinski et al, 2013). Promising clinical activity has been observed in patients with ALK-translocated crizotinib-resistant NSCLC and in those with acquired EGFR T790M mutations, which confer resistance to EGFR tyrosine kinase inhibitors (Johnson et al, 2013; Socinski et al, 2013).
Heat-shock protein 990 is an orally bioavailable synthetic small molecule, which inhibits HSP90 via an ATP-binding site, resulting in the degradation of ubiquitin proteasome pathway-driven client proteins and inhibition of multiple oncoproteins. Its antitumour effects have been demonstrated in preclinical models of different malignancies that are dependent on HSP90 client proteins (Lamottke et al, 2012; Menezes et al, 2012; Stuhmer et al, 2012; Fu et al, 2013; Zitzmann et al, 2013). In this first-in-human, open-label, phase I dose-escalation study, HSP990 was administered as a single agent orally in two different dosing schedules of once weekly and two times weekly. The primary objective of this study was to establish the maximum-tolerated dose (MTD) and recommended phase II dose (RP2D) of HSP990 in patients with advanced solid malignancies based on these schedules. Secondary objectives included the assessment of safety, tolerability and preliminary efficacy, as well as the evaluation of the pharmacokinetic (PK) profile and the PK–pharmacodynamic (PD) relationship of HSP990.
Patients and methods
Heat-shock protein 990 (Novartis, Basel, Switzerland) was administered orally either once a week or two times a week on a 28-day cycle schedule. Regulatory and independent ethics committee approvals were obtained in all participating sites of this multicentre study. The National Cancer Institute Common Toxicity Criteria for Adverse Events (NCI CTCAE) version 3.0 were used to evaluate toxicity.
Patient selection
Eligible patients were those with histologically confirmed, advanced malignant solid tumours whose disease had either progressed on standard therapy or for whom no standard therapy was available. Inclusion criteria included: age 18 years or older; World Health Organisation performance status (WHO PS) of 0–2; at least one measurable lesion as defined by Response Evaluation Criteria in Solid Tumours (RECIST) version 1.0; documented disease progression before study entry, predicted life expectancy of at least 12 weeks; ability to swallow capsules; adequate haematopoietic, hepatic, and renal function and normal electrolytes. Exclusion criteria included: pregnant or lactating women; another primary malignancy; presence or history of central nervous system metastases; acute or chronic hepatic or renal disease; gastrointestinal impairment that may affect absorption of oral medications; significant cardiac comorbidities; HIV infection; other severe or uncontrolled medical conditions; prior treatment with HSP90 or histone deacetylase inhibitors; poor or intermediate CYP2C9 metabolisers based on genotyping performed at screening (homozygous for the CYP2C9*2 allele, or either heterozygous or homozygous for the CYP2C9*3 allele); concomitant use of CYP2C9 inhibitors; active therapeutic doses of sodium warfarin or acenocumarol; and unresolved side effects (⩾grade 2) from previous treatments. Patients required a washout period of at least 4 weeks from prior systemic anticancer treatment and radiation and 2 weeks from prior major surgery before starting study drug.
Study design and dosing
The study design comprised a dose-escalation part followed by a dose expansion cohort. In the dose-escalation part, a minimum of three patients per cohort were enrolled, although two patients without any treatment-related adverse events (AEs >grade 1 were sufficient for dose-escalation decision. An adaptive Bayesian Logistic Regression Model (BLRM) guided by the Escalation with Overdose Control principle was used in the dose-escalation part (Rogatko et al, 2007). The BLRM with overdose control was used to model the relationship between dose and the probability of a patient experiencing a dose-limiting toxicity (Babb et al, 1998). The dose-escalation part initially planned to only include a once-weekly dose regimen of HSP990 starting from 2.5 mg given orally, derived based on preclinical studies. Owing to the potential for dropout during the first cycle of treatment, cohort expansion was allowed provided enrolment occurred within 14 days of treatment start of the third patient in the same cohort. If either of the two patients in the cohort experienced a DLT before the enrolment of the third patient, the model was to be re-evaluated before enrolment of any additional patients to that cohort. The dose expansion part of the once-weekly schedule was performed at the MTD. To assess whether the frequency and severity of neurologic toxicities could be attenuated by splitting HSP990 dosing as a twice-weekly regimen, this alternative schedule was introduced after expansion of the once-weekly dose regimen. No dose expansion was conducted for the twice-weekly dose regimen due to slow accrual and early termination of the study. Intrapatient dose escalation was permitted only after the first four cycles of treatment utilising the same dosing schedule patients were initially assigned. A DLT was defined as any clinically relevant AE or abnormal laboratory values occurring within the first 28 days of HSP990 treatment as specified in Supplementary Table 1. The MTD/RP2D was defined based on recommendations of the Bayesian model, plus other available safety and tolerability information. All investigators reviewed safety data collected at each dose level before selection of the next dose level. At the completion of dose escalation, all reviewers evaluated safety and DLT data, as well as recommendations by the Bayesian model, to select the MTD/RP2D.
HSP990 formulation and administration
Heat-shock protein 990 was administered orally once or two times weekly on a flat dosing scale and treatment cycles were of 28 days duration. Heat-shock protein 990 was supplied as 1, 2.5, 20 and 50 mg hard gelatin capsules. Fasting conditions were required (study drug was taken 30 min after a light breakfast, followed by a 3-h fasting period). The twice-weekly dosing schedule required at least 72 h between the two doses, with both doses taken within a 7-day period. Administration of HSP990 was allowed until disease progression, unacceptable toxicity, investigator’s decision or patient withdrawal of consent.
Safety and efficacy assessments
Safety assessments consisted of physical examination including neurological examination, vital signs, weight, performance status assessment and documentation of AEs and serious AEs (SAEs). In addition, laboratory investigations such as haematology, coagulation, biochemistry, urinalysis and pregnancy test, if applicable, were conducted at baseline, on day 1 of every treatment cycle, on the last visit and at any time when clinically indicated. Electrocardiograms were performed on day 1 of each cycle and multiple times on specific days throughout the study treatment. Cardiac evaluations by ECHO or MUGA scans were performed at baseline, at the end of cycles 2–4 and every other month thereafter until the end of study or when clinically indicated. Imaging with fluorodeoxyglucose positron emission tomography (FDG-PET) was performed at baseline, on day 10 of cycle 1 and on day 2 of cycle 3. Tumour assessments based on radiological evaluations occurred at baseline, the end of every two cycles and the end of treatment. Response assessment was performed according to RECIST version 1.0.
PK assessments
Pharmacokinetic samples of HSP990 were obtained and evaluated in all patients at all dose levels to characterise the disposition of HSP990 and its metabolite(s) (when appropriate) after oral administration of HSP990. Whole blood samples (2.0 ml) were obtained via venipuncture into an ethylene diamine tetraacetic acid containing tube. Immediately after each sample collection, the blood tubes were centrifuged to separate out plasma. The plasma samples from all patients were assayed centrally for HSP990 concentrations using a validated liquid chromatography-tandem mass spectrometry assay.
Blood samples were collected at prescheduled timepoints after the oral doses on days 1 and 22 of cycle 1 and on day 1 of cycle 2. Owing to the food effect noted in dogs, a record of the meal and related timing to HSP990 drug dosing was required. Non-compartmental analysis using WinNonlin Pro (version 5.2; Pharsight, Mountain View, CA, USA) was performed to calculate the relevant PK parameters.
PD assessments
As HSP70 may be a novel biomarker to assess the pharmacological effects of HSP90 inhibitors (Dakappagari et al, 2010), pre- and posttreatment peripheral blood mononuclear cell (PBMC) samples were analysed to compare the levels of HSP70 measured by ELISA (Enzo Life Sciences, Farmingdale, NY, USA; cat. no. ADI-EKS-700B). One 8 ml sample of blood was collected at pre- and posttreatment time points on days 1, 2, 3, 4 and 22 of cycle 1 and on days 1, 2 and 3 of cycle 2.
Exploratory functional imaging
Exploratory efficacy assessment was performed in all patients enrolled in the study utilising FDG-PET to explore antitumour response by analysing changes in FDG avidity. Scans were performed at screening on day 10 of cycle 1and on day 2 of cycle 3. A reduction in maximum standardised uptake value (SUV) summed across all target lesions (sSUVmax) of ⩾25% was considered evidence of objective metabolic response. An increase in sSUVmax of ⩾25% was considered evidence of progression. Any appearance of new PET lesions was categorised as metabolic progression regardless of changes observed in target lesion SUV assessment.
Statistical methods
Data analysis was performed using SAS version 9.3 (Cary, NC, USA), with the exception of the BLRM, which was performed using R version 2.8.1 and WinBUGS version 1.4.1. Data were summarised for demographic and baseline characteristics, safety and efficacy measurements, and all relevant PK and PD evaluations using descriptive statistics. For PK data, an analysis of covariance was performed on log-transformed AUC and Cmax (day 1 of cycle 1 and day 22 of cycle 1) using a linear mixed-effect model to assess day effect. Summary statistics for HSP70 induction in PBMCs were tabulated. Estimation of the MTD in the dose-escalation part of the study was based on the estimation of the probability of DLT in cycle 1 for patients in the dose-determining set. For the once-weekly regimen, a BLRM was used to model the dose–toxicity relationship and to compute the posterior probability of a DLT at each dose level.
Results
Patient characteristics
A total of 64 patients from three investigational sites were enrolled in this study. Sixty-three patients (98%) were eligible for dose-determining set given that one patient, because of progression of disease, did not receive ⩾75% of dose without a DLT and 50 patients were evaluable for the biomarker analysis subset. Fifty-four patients (84%) discontinued treatment for disease progression, nine (14%) for AEs of any causality, of which 78% (7 out of 9 patients) were considered drug-related, and one patient (2%) withdrew consent. As summarised in Table 1, colorectal cancer was the most common tumour type (45%), followed by non-colorectal gastrointestinal malignancies (16%) and breast cancer (14%). All patients previously received systemic treatments, with the majority of them (83%) having received three or more lines of therapy. Nearly all (98%) patients had WHO PS 0 or 1 at baseline.
Dose escalation and MTD
During the dose-escalation part for the once-weekly schedule, seven dose levels ranging from 2.5 to 60 mg were evaluated. A total of seven patients experienced DLTs. At 60 mg once weekly, two patients experienced DLTs (40%). One patient developed grade 1 decreased appetite, dysdiadochokinesia, fatigue, hyperreflexia and tremor. Of note, this patient experienced drug-related AEs that caused an inability to administer 75% of the planned dose of HSP990, even though the reported events were not in the grade range defined as a DLT. The second patient experienced drug-related grade 3 tremor without any confounding factor. In one patient, the dose of HSP990 was reduced, whereas the other patient discontinued the study treatment. The MTD in this study was declared as 50 mg administered once weekly; therefore, 16 additional patients were recruited at this dose in the expansion phase. Of the 22 patients who received HSP990 at 50 mg once a week, 5 DLTs were encountered in 4 patients (grade 3 bilateral myoclonic hand movement, grade 3 diarrhoea, grade 3 prolonged QTc seen in 2 patients and grade 4 ALT and AST increase), with diarrhoea and QTc prolongation occurring in the same patient. The frequency of DLT at the 50 mg once-weekly dose level was 18% (4 out of 22 patients).
Dose escalation for the twice-weekly dose regimen started with 25 mg two times a week and enrolled 11 patients. One of them experienced DLT in the form of grade 1 extremity tremors, superior limb extrapyramidal hypertropia and dysmetria; grade 2 ataxia, confusion and visual hallucination. Further dose escalation beyond 25 mg two times a week was not pursued, the study stopped due to high interpatient PK variability and the lack of an efficacy trend across all dose groups. Table 2 provides a summary of the occurrences of DLT by dose level.
Safety
The most frequent drug-related AEs on this study were: diarrhoea in 47 patients (73%), asthenia in 26 patients (41%), anorexia in 16 patients (25%), insomnia in 16 patients (25%), nausea in 14 patients (22%), dizziness in 12 patients (19%), tremor in 12 patients (19%), abdominal pain in 10 patients (16%) and vomiting in 9 patients (14%).
Study drug-related grade 3 or 4 AEs occurred in 15 patients (23%). Grade 3 AEs were: diarrhoea (13%), increased ALT and/or AST, asthenia, QTc prolongation and hyponatremia, each occurring in two patients (3%). Grade 4 ALT and AST elevations were each experienced in one patient in the 50 mg weekly group. Adverse events requiring dose adjustment or study drug interruption were reported in 19 patients (30%). Out of six deaths reported on study and up to 28 days after the last dose, none of them were considered treatment-related. At least one SAE was experienced in 26 patients (41%). Heat-shock protein 990-related SAEs were: grade 1, ataxia, dizziness and tremor; grade 2, confusional state and visual hallucination; grade 3, gastrointestinal disorder, myoclonic hand movements and QTc prolongation; and grade 4, AST/ALT elevation. Neurotoxicities leading to study drug discontinuation, during and beyond cycle 1, were reported in nine patients (14%) who presented with tremor, cerebellar syndrome, ataxia, extrapyramidal disorder, hyperreflexia, presyncope and syncope. Seven patients (11%) overall reported a drug-related AE that led to study discontinuation as shown in Table 2. No drug-related deaths occurred in this study. All drug-related toxicities occurring in at least 5% of all patients are summarised in Table 3 and grade 3 and 4 AEs suspected to be study drug related are reported in Table 4.
Treatment exposure, dose delay and dose modifications
The median treatment duration for all patients was 8.0 weeks (range 1.0–116.0 weeks). The highest median treatment duration (12.0 weeks) was reported in the 25 mg twice-weekly group. Forty-six patients (72%) received up to 12 weeks of treatment, with six patients (9%) receiving 12 to 16 weeks of treatment, and the remaining 12 patients (19%) receiving >16 weeks of treatment. The majority of patients (89%) had a relative dose intensity of 0.9 to <1.1, with an overall median relative dose intensity of 1.0. Overall, dose reductions, observed primarily in the cohorts dosed at ⩾50 mg weekly, occurred in six patients, with only one patient requiring more than one dose reductions. Main reasons for dose reduction were neurological toxicities, QTc prolongation and diarrhoea. Dose delays, mostly due to AEs, were observed in 22 patients, with five patients (8%) requiring more than one dose delays.
PK analysis
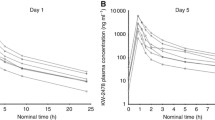
In both dosing schedules, HSP990 was well absorbed, with time to peak concentrations achieved ∼3 h postdose and then declined mono- or biexponentially with a t1/2 of ∼20 h (Figures 1A and B). Similar absorption rates (Tmax) and comparable PK profiles were observed on days 1 and 22 of cycle 1 and day 1 of cycle 2 after multiple oral doses in individual patients, which suggested low intrapatient PK variability. At the 50 and 60 mg dose levels, a distribution phase after the peak was observed in the concentration–time profiles, indicating rapid oral absorption. On day 1, Cmax values were 496 ng ml−1 in the 50 mg once-weekly and 270 ng ml−1 in the 25 mg twice-weekly schedules, and 552 to 232 ng ml−1 at steady states, respectively. From days 1 to 22, related AUClast values changed from 8408 ng.h ml−1 to 7959 ng ml−1 in the 50 mg once-weekly schedule and from 12 580 to 14 195 ng.h ml−1 in the 25 mg twice-weekly schedule. The dose splitting from 50 mg once weekly to 25 mg twice weekly decreased the Cmax by half, while the weekly AUC was increased. The increase in AUC was generally dose proportional in the dose range from 2.5 to 60 mg following the weekly oral doses on days 1 or 22 of cycle 1. The Cmax values increased slightly greater than dose proportionally in the dose range from 2.5 to 60 mg. Intersubject variability in Cmax and AUC differed at each dose level but the differences were relatively large at higher doses. Apparent clearance (∼6.4 l h−1) and volume of distribution (∼200 l) suggest that HSP990 has low systemic clearance and a large tissue distribution. Similar PK profiles were observed on days 1 and 22 of cycle 1, indicating no drug accumulation after multiple- or twice-weekly doses. The HSP990 PK parameters after the oral doses on day 1 of cycle 1 are summarised in Table 5.

Median concentration–time profiles for plasma HSP990 on day 1 of cycle 1 (A) and day 22 of cycle 1 (B).
PD analysis
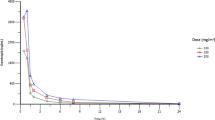
To assess the PD effects of HSP990, HSP70 analysis in PBMCs was performed by ELISA on a total of 50 patients. Supplementary Figure 1 shows the highest HSP70 changes by treatment group. For once-weekly administration, at dose ranges from 2.5 to 60 mg, HSP70 induction tended to increase with dose across the 5 mg weekly to 30 mg weekly dose groups. Dose groups above 20 mg appeared to plateau, indicating a saturation effect. There were a wide range of HSP70 values at each dose level, suggesting interpatient PD variability. For twice-weekly administration at a dose of 25 mg, the level of HSP990 induction achieved was greater than that seen in once weekly 30 mg to 60 mg dose levels, indicating no saturation effect in the limit of our clinical findings in a small group of patients.
Response evaluation
No partial or complete responses were observed. Twenty-five (39%) patients showed stable disease as best overall response, with a trend (7 out of 11 patients, 64%) towards higher rates of stable disease in the twice-weekly dosing schedule. Seven (11%) patients demonstrated a prolonged stable disease (⩾6 months), of which 71% (5 out of 7) were seen in either 50 or 60 mg once-weekly or 25 mg twice-weekly regimens.
FDG-PET imaging
An overall decrease in FDG-PET uptake was observed in the higher dose levels (50 mg weekly and 25 mg twice-weekly groups) compared with the lower dose levels (2.5–30 mg weekly groups) as shown in Supplementary Figure 2. No complete metabolic response was seen in any of the patients treated. A total of 11 patients (17%) had a ⩾25% reduction in sSUVmax of target lesions from baseline. These 11 patients received HSP990 at 5 mg weekly (1 patient), 30 mg weekly (1 patient), 50 mg weekly (6 patients) and 25 mg twice weekly (3 patients). A summary of tumour response as per RECIST version 1.0 is summarised in Supplementary Figure 3.
Discussion
This first-in-human, multicentre, phase 1 study of HSP990 established the MTD/RP2D at 50 mg once weekly. The twice-weekly schedule was evaluated to determine if neurological toxicities encountered at the higher dose levels in the once-weekly schedule could be minimised by reducing the peak drug concentrations without compromising drug AUC exposures. However, the alternate schedule of twice-weekly dosing did not conclusively show improved tolerability. Therefore, in light of neurological toxicities that persisted despite schedule change, and the substantial interpatient PK variability observed, this study was terminated without reaching MTD in the twice-weekly schedule.
Neurological toxicities occurring during phase I studies often limit the development of novel compounds despite evidence of preliminary antitumour activity (Cavaliere and Schiff, 2006). Diaz-Rubio et al (1994) reported severe unexpected central nervous system (CNS) toxicities of the cytostatic agent mitonafide, whose development was later abandoned despite evidence of antitumour activity (Diaz-Rubio et al, 1994). More recently, both central and peripheral neurological toxicities of the JAK2 inhibitor XL019 led to early termination of the phase I study (Verstovsek et al, 2014). For many agents, even with a broad spectrum of activity, the presence of neurological events likely result in suboptimal dose and inappropriate schedule recommendation, therefore limiting potential activity. Pivotal examples are represented by interleukin-2 and interferon, where the severity of CNS neurological toxicity events has been associated with higher doses leading to the modification of treatment schedules in different tumour types (Chiarion-Sileni et al, 2006; Alwan et al, 2013).
Heat-shock protein 990 was found to be highly distributed in the brain tissues in the rat model. Preclinical toxicology studies are relevant to determine a reasonable expectation of safety for all new molecules. Evaluation of neurological toxicity caused by anticancer agents is challenging in animal models and interspecies differences can lead to false predictions of a drug’s neurological toxicity profile. Preclinical pharmacology studies of HSP990 showed no effects on CNS in rats, but tremors and ataxia in dogs were observed at doses of ⩾2 mg kg−1 (Novartis, 2012). This finding corroborates previous reports, which suggests that non-rodent models might be more predictive of neurological toxicities in humans (Olson et al, 2000), highlighting the role of non-rodent large animal model toxicology studies to identify appropriately potential drug-related non-haematologic toxicities.
Although the majority of AEs reported in this phase I study of HSP990 were mild or moderate in intensity, temporary and reversible in nature and consistent with those typically encountered by patients with advanced cancers, the occurrence of dose-dependent neurological toxicities led to frequent treatment delays or drug discontinuation. The attempt to split the daily dose of HSP990 into two to reduce peak drug concentrations did not attenuate its neurological toxicity, suggesting that this event is likely related to drug exposure. Previous findings have shown toxic effects of HSP90 inhibitors on oligodendrocyte precursor cells, with consequent reduction of oligodendrocyte population and prevention of remyelination during HSP90 inhibitors therapy in cell cultures (Alcazar and Cid, 2009). In addition, in vitro and in vivo data have shown the dual ability of HSP90 inhibitors to protect murine neural progenitor cells from their natural apoptosis at low doses and increase their death at high doses (Wang et al, 2011). These findings may explain the neurological toxicities seen in our study, particularly at higher dose levels of HSP990, and reflect the toxicity profile seen with other molecules that belong to the same class of agents (Dickson et al, 2013; Saif et al, 2014).
Despite several challenges, including the identification of potential therapeutic targets and exploitable therapeutic index, lack of predictive biomarker and occurrence of severe toxicities, the development of HSP90 inhibitors has gained increasing interest in the cancer field, given the molecular chaperones regulation on several vital proteins. Phase II and III trials with AUY922 and ganetespib (STA-9090) are ongoing in prostate, gastric, pancreatic, breast and lung cancers. These agents have shown modest clinical benefit in both monotherapy or combination with chemotherapy or targeted agents, with the exception of NSCLC and triple-negative breast cancer where activity appears promising (Awada et al, 2013; Johnson et al, 2013; Ramalingam et al, 2013; Thota et al, 2014).
In contrast to the major classes of molecular chaperones, HSP90 uses repeated cycles of client protein binding, ATP hydrolysis as well as interaction with cochaperones, such as HSP70, to stabilise and activate ∼200 client proteins, several of which represent oncoproteins such as HER2, EGFR, AKT and RAF kinase (Zhang and Burrows, 2004; Chandarlapaty et al, 2010). Interesting preclinical and clinical results have supported the role of these agents in NSCLC, particularly in patients with tumours resistant to ALK inhibitors or EGFR inhibitors. (Johnson et al, 2013; Sang et al, 2013; Socinski et al, 2013). In contrast to ganetespib and AUY922, which are available in intravenous formulation only, our study investigated HSP990 that has the advantage of oral availability (Goldman et al, 2013; Sessa et al, 2013). Disappointingly in this study, the narrow therapeutic index, interpatient PK variability and neurological toxicities limited the development of HSP990.
The induction of HSP70 and HSP27 through the heat-shock transcription factor 1 frequently occurs as a result of HSP90 inhibitor effect (Erlichman, 2009). In normal tissue, the increased expression of these proteins leads to protection from some toxicities related to HSP90 inhibition. The upregulation of these molecular chaperones may also protect cancer cells, and thus may potentially result in resistance to HSP90 blockade (Guo et al, 2005; McCollum et al, 2006). Previous data have shown that silencing of HSP70 and/or HSP27 markedly increases cancer cell sensitivity to HSP90 inhibitors (McCollum et al, 2006; Powers et al, 2008). In our study, the HSP70 was evaluated as a measure of target inhibition in PBMCs. Heat-shock protein 70 induction range appeared wide, increasing with HSP990 doses of up to 30 mg once weekly, but plateaued at higher dose levels, indicating a saturation effect. Surrogate tissues such as PBMCs are not always reflective of tumour effects, and in this case, while HSP70 induction may reflect on-target HSP90 inhibition, this PD biomarker may actually reflect the emergence of escape mechanisms in the tumour cells. The evaluation of pharmacological inhibitors of HSP70 and HSP27 is under current development (Evans et al, 2010), and it would be of interest to combine such drugs with HSP90 inhibitors to reduce compensatory escapes.
Several trials have tested the role of FDG-PET as an important early marker of efficacy in drug development (Kraeber-Bodere et al, 2012; McArthur et al, 2012). In the current study, no complete metabolic responses were seen, whereas 17% of patients had partial metabolic responses, mostly at higher HSP990 dose levels. The metabolic responses seen in this study did not correlate with conventional objective tumour response. These results may reflect the unclear role of FDG-PET as an indicator of drug-induced modulation of tumour metabolism (O'Brien et al, 2012; Piessen et al, 2013). In our study, no complete or partial responses were observed, and stable disease as per RECIST was achieved in 39% of patients. Although not a primary end point in phase I studies, preliminary efficacy data often influence go-no-go decisions on the continued development of a new agent. Substantial biases such as differences in tumour biology, tumour measurement errors and timing of radiological assessments can affect the reliability of disease stabilisation as a true measure of antitumour activity (Le Tourneau et al, 2014). Despite the development of alternative methods such as tumour growth rate (Ferte et al, 2014), at present no consensus has been reached and RECIST still remains the conventional approach to evaluate tumour response in clinical trials.
Many molecularly targeted agents, which have successfully attained regulatory approval, are linked to specific predictive biomarkers that help identify sensitive or resistant patient populations. However, this characteristic may not be applicable to HSP90 inhibitors as their antitumour activity is mediated by downstream effects via multiple pathways. Given the pleiotropic spectrum of targets of this class of agents that may preclude the utility of specific predictive biomarkers, a major challenge is the identification of specific client proteins that may ultimately serve as predictive or surrogate biomarkers for those cancers likely to respond to HSP90 inhibitors. Nevertheless, many compounds such as ipilimumab or bevacizumab are used as standard of care despite the absence of predictive biomarkers (Hurwitz et al, 2004; Hodi et al, 2010).
The relevant questions remain as to whether drugs such as HSP90 inhibitors with their wide range of downstream targets can exert sufficient antitumour activity to translate into clinical impact in one or more malignancies, and whether their therapeutic indices are dampened by off-target effects and compensatory mechanisms that induce drug resistance. Based on the promising results in NSCLC and the activity of HSP90 inhibitors on the EGFR pathway, new studies have been developed to evaluate the combination of EGFR and HSP90 inhibitors (Johnson et al, 2013). In addition, given their effects on cell cycle arrest and their ability to enhance DNA damage and apoptosis, combination studies with taxanes or with radiotherapy (Sain et al, 2006; Milanovic et al, 2013) may be of interest for further development of HSP90 inhibitors.
Change history
17 February 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Alcazar A, Cid C (2009) High cytotoxic sensitivity of the oligodendrocyte precursor cells to HSP90 inhibitors in cell cultures. Exp Neurol 216 (2): 511–514.
Alwan LM, Grossmann K, Sageser D, Van Atta J, Agarwal N, Gilreath JA (2013) Comparison of acute toxicity and mortality after two different dosing regimens of high-dose interleukin-2 for patients with metastatic melanoma. Target Oncol 9 (1): 63–71.
Awada A, Rodriguez AA, Kong A, Erban JK, Cortes J, Mano MS, Perez EA, El Hariry A, Vukovic VM, Teofilovici F, Yalcin I, Bradley R, Cameron DA (2013) The ENCHANT-1 trial (NCT01677455): An open label multicenter phase II proof of concept study evaluating first-line ganetespib monotherapy in women with metastatic HER2-positive or triple-negative breast cancer (TNBC). J Clin Oncol 31 (suppl): abstr TPS1136.
Babb J, Rogatko A, Zacks S (1998) Cancer phase I clinical trials: efficient dose escalation with overdose control. Stat Med 17 (10): 1103–1120.
Cavaliere R, Schiff D (2006) Neurologic toxicities of cancer therapies. Curr Neurol Neurosci Rep 6 (3): 218–226.
Chandarlapaty S, Scaltriti M, Angelini P, Ye Q, Guzman M, Hudis CA, Norton L, Solit DB, Arribas J, Baselga J, Rosen N (2010) Inhibitors of HSP90 block p95-HER2 signaling in Trastuzumab-resistant tumors and suppress their growth. Oncogene 29 (3): 325–334.
Chen Z, Akbay EA, Mikse OR, Tupper T, Cheng K, Wang Y, Tan X, Altabef A, Woo SA, Chen L, Reibel J, Janne PA, Engelman JA, Sharpless NE, Kung AL, Shapiro GI, Wong KK (2013) Co-clinical trials demonstrate superiority of crizotinib to chemotherapy in ALK-rearranged non-small cell lung cancer and predict strategies to overcome resistance. Clin CancerRes 20 (5): 1204–1211.
Chiarion-Sileni V, Del Bianco P, Romanini A, Guida M, Paccagnella A, Dalla Palma M, Naglieri E, Ridolfi R, Silvestri B, Michiara M, De Salvo GL (2006) Tolerability of intensified intravenous interferon alfa-2b versus the ECOG 1684 schedule as adjuvant therapy for stage III melanoma: a randomized phase III Italian Melanoma Inter-group trial (IMI – Mel.A.) [ISRCTN75125874]. BMC Cancer 6: 44.
Dakappagari N, Neely L, Tangri S, Lundgren K, Hipolito L, Estrellado A, Burrows F, Zhang H (2010) An investigation into the potential use of serum Hsp70 as a novel tumour biomarker for Hsp90 inhibitors. Biomarkers 15 (1): 31–38.
De Mattos-Arruda L, Cortes J (2012) Breast cancer and HSP90 inhibitors: is there a role beyond the HER2-positive subtype? Breast 21 (4): 604–607.
Diaz-Rubio E, Martin M, Lopez-Vega JM, Casado A, Benavides A (1994) Phase I study of mitonafide with a 3-day administration schedule: early interruption due to severe central nervous system toxicity. Invest N Drugs 12 (4): 277–281.
Dickson MA, Okuno SH, Keohan ML, Maki RG, D'Adamo DR, Akhurst TJ, Antonescu CR, Schwartz GK (2013) Phase II study of the HSP90-inhibitor BIIB021 in gastrointestinal stromal tumors. Ann Oncol 24 (1): 252–257.
Erlichman C (2009) Tanespimycin: the opportunities and challenges of targeting heat shock protein 90. Expert Opin Invest Drugs 18 (6): 861–868.
Evans CG, Chang L, Gestwicki JE (2010) Heat shock protein 70 (hsp70) as an emerging drug target. J Med Chem 53 (12): 4585–4602.
Ferte C, Fernandez M, Hollebecque A, Koscielny S, Levy A, Massard C, Balheda R, Bot B, Gomez-Roca C, Dromain C, Ammari S, Soria JC (2014) Tumor growth rate is an early indicator of antitumor drug activity in phase I clinical trials. Clin CancerRes 20 (1): 246–252.
Fu J, Koul D, Yao J, Wang S, Yuan Y, Colman H, Sulman EP, Lang FF, Yung WK (2013) Novel HSP90 inhibitor NVP-HSP990 targets cell-cycle regulators to ablate Olig2-positive glioma tumor-initiating cells. Cancer Res 73 (10): 3062–3074.
Goldman JW, Raju RN, Gordon GA, El-Hariry I, Teofilivici F, Vukovic VM, Bradley R, Karol MD, Chen Y, Guo W, Inoue T, Rosen LS (2013) A first in human, safety, pharmacokinetics, and clinical activity phase I study of once weekly administration of the Hsp90 inhibitor ganetespib (STA-9090) in patients with solid malignancies. BMC Cancer 13: 152.
Guo F, Rocha K, Bali P, Pranpat M, Fiskus W, Boyapalle S, Kumaraswamy S, Balasis M, Greedy B, Armitage ES, Lawrence N, Bhalla K (2005) Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. CancerRes 65 (22): 10536–10544.
Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363 (8): 711–723.
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350 (23): 2335–2342.
Johnson ML, Hart EM, Rademarker A, Weitner BB, Urman A, SImm HD, Fountas LM, Worden R, Patel JD, Miller VA, Riely GJ (2013) A phase II study of HSP90 inhibitor AUY922 and erlotinib (E) for patients (pts) with EGFR-mutant lung cancer and acquired resistance (AR) to EGRFR tyrosine kinase inhibitors (EGFR TKIs). J Clin Oncol 31 (Suppl): abstr 8036.
Kim WY, Oh SH, Woo JK, Hong WK, Lee HY (2009) Targeting heat shock protein 90 overrides the resistance of lung cancer cells by blocking radiation-induced stabilization of hypoxia-inducible factor-1alpha. CancerRes 69 (4): 1624–1632.
Kraeber-Bodere F, Carlier T, Naegelen VM, Shochat E, Lumbroso J, Trampal C, Nagarajah J, Chua S, Hugonnet F, Stokkel M, Gleeson F, Tessier J (2012) Differences in the biologic activity of 2 novel MEK inhibitors revealed by 18F-FDG PET: analysis of imaging data from 2 phase I trials. J Nucl Med 53 (12): 1836–1846.
Lamottke B, Kaiser M, Mieth M, Heider U, Gao Z, Nikolova Z, Jensen MR, Sterz J, von Metzler I, Sezer O (2012) The novel, orally bioavailable HSP90 inhibitor NVP-HSP990 induces cell cycle arrest and apoptosis in multiple myeloma cells and acts synergistically with melphalan by increased cleavage of caspases. Eur J Haematol 88 (5): 406–415.
Le Tourneau C, Paoletti X, Coquan E, Sablin MP, Zoubir M, Tannock IF (2014) Critical evaluation of disease stabilization as a measure of activity of systemic therapy: lessons from trials with arms in which patients do not receive active treatment. J Clin Oncol 32 (3): 260–263.
Li CF, Huang WW, Wu JM, Yu SC, Hu TH, Uen YH, Tian YF, Lin CN, Lu D, Fang FM, Huang HY (2008) Heat shock protein 90 overexpression independently predicts inferior disease-free survival with differential expression of the alpha and beta isoforms in gastrointestinal stromal tumors. Clin CancerRes 14 (23): 7822–7831.
Macario AJ, Conway de Macario E (2005) Sick chaperones, cellular stress, and disease. N Engl J Med 353 (14): 1489–1501.
McArthur GA, Puzanov I, Amaravadi R, Ribas A, Chapman P, Kim KB, Sosman JA, Lee RJ, Nolop K, Flaherty KT, Callahan J, Hicks RJ (2012) Marked, homogeneous, and early [18F]fluorodeoxyglucose-positron emission tomography responses to vemurafenib in BRAF-mutant advanced melanoma. J Clin Oncol 30 (14): 1628–1634.
McCollum AK, Teneyck CJ, Sauer BM, Toft DO, Erlichman C (2006) Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer Res 66 (22): 10967–10975.
Menezes DL, Taverna P, Jensen MR, Abrams T, Stuart D, Yu GK, Duhl D, Machajewski T, Sellers WR, Pryer NK, Gao Z (2012) The novel oral Hsp90 inhibitor NVP-HSP990 exhibits potent and broad-spectrum antitumor activities in vitro and in vivo. Mol Cancer Ther 11 (3): 730–739.
Milanovic D, Firat E, Grosu AL, Niedermann G (2013) Increased radiosensitivity and radiothermosensitivity of human pancreatic MIA PaCa-2 and U251 glioblastoma cell lines treated with the novel Hsp90 inhibitor NVP-HSP990. Radiat Oncol 8: 42.
Neckers L, Workman P (2012) Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 18 (1): 64–76.
Novartis (2012) HSP990A Investigator's Brochure, Vol. 4. Novartis: Basel, Switzerland.
O'Brien ME, Myerson JS, Coward JI, Puglisi M, Trani L, Wotherspoon A, Sharma B, Cook G, Ashley S, Gunapala R, Chua S, Popat S (2012) A phase II study of (1)(8)F-fluorodeoxyglucose PET-CT in non-small cell lung cancer patients receiving erlotinib (Tarceva); objective and symptomatic responses at 6 and 12 weeks. Eur J Cancer 48 (1): 68–74.
Olson H, Betton G, Robinson D, Thomas K, Monro A, Kolaja G, Lilly P, Sanders J, Sipes G, Bracken W, Dorato M, Van Deun K, Smith P, Berger B, Heller A (2000) Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul Toxicol Pharmacol 32 (1): 56–67.
Piessen G, Petyt G, Duhamel A, Mirabel X, Huglo D, Mariette C (2013) Ineffectiveness of (1)(8)F-fluorodeoxyglucose positron emission tomography in the evaluation of tumor response after completion of neoadjuvant chemoradiation in esophageal cancer. Ann Surg 258 (1): 66–76.
Powers MV, Clarke PA, Workman P (2008) Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 14 (3): 250–262.
Powers MV, Workman P (2007) Inhibitors of the heat shock response: biology and pharmacology. FEBS Lett 581 (19): 3758–3769.
Ramalingam SS, Goss GD, Andric ZG, Bondarenko I, Zaric B, Ceric T, Poddubskaya EV, Ciuleanu T-E, Spicer JF, Felip E, Hirsh V, Manegold C, Rosell R, Khuri FR, Vukovic VM, Teofilovici F, El-Hariry I, Guo W, Bahcall SR, Fennell D (2013) A randomized study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel versus docetaxel alone for second-line therapy of lung adenocarcinoma (GALAXY-1). J Clin Oncol 31 (Suppl): abstr CRA8007.
Rogatko A, Schoeneck D, Jonas W, Tighiouart M, Khuri FR, Porter A (2007) Translation of innovative designs into phase I trials. J Clin Oncol 25 (31): 4982–4986.
Saif MW, Takimoto C, Mita M, Banerji U, Lamanna N, Castro J, O'Brien S, Stogard C, Von Hoff D (2014) A phase 1, dose-escalation, pharmacokinetic and pharmacodynamic study of BIIB021 administered orally in patients with advanced solid tumors. Clin CancerRes 20 (2): 445–455.
Sain N, Krishnan B, Ormerod MG, De Rienzo A, Liu WM, Kaye SB, Workman P, Jackman AL (2006) Potentiation of paclitaxel activity by the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin in human ovarian carcinoma cell lines with high levels of activated AKT. Mol Cancer Ther 5 (5): 1197–1208.
Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, Zhang C, Jiang Q, Xue L, Lovly CM, Jimenez JP, Shaw AT, Doebele RC, He S, Bates RC, Camidge DR, Morris SW, El-Hariry I, Proia DA (2013) Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov 3 (4): 430–443.
Sawai A, Chandarlapaty S, Greulich H, Gonen M, Ye Q, Arteaga CL, Sellers W, Rosen N, Solit DB (2008) Inhibition of Hsp90 down-regulates mutant epidermal growth factor receptor (EGFR) expression and sensitizes EGFR mutant tumors to paclitaxel. Cancer Res 68 (2): 589–596.
Scaltriti M, Dawood S, Cortes J (2012) Molecular pathways: targeting hsp90 – who benefits and who does not. Clin Cancer Res 18 (17): 4508–4513.
Sessa C, Shapiro GI, Bhalla KN, Britten C, Jacks KS, Mita M, Papadimitrakopoulou V, Pluard T, Samuel TA, Akimov M, Quadt C, Fernandez-Ibarra C, Lu H, Bailey S, Chica S, Banerji U (2013) First-in-human phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin Cancer Res 19 (13): 3671–3680.
Shimamura T, Lowell AM, Engelman JA, Shapiro GI (2005) Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res 65 (14): 6401–6408.
Simpson NE, Lambert WM, Watkins R, Giashuddin S, Huang SJ, Oxelmark E, Arju R, Hochman T, Goldberg JD, Schneider RJ, Reiz LF, Soares FA, Logan SK, Garabedian MJ (2010) High levels of Hsp90 cochaperone p23 promote tumor progression and poor prognosis in breast cancer by increasing lymph node metastases and drug resistance. Cancer Res 70 (21): 8446–8456.
Socinski MA, Goldman J, El-Hariry I, Koczywas M, Vukovic V, Horn L, Paschold E, Salgia R, West H, Sequist LV, Bonomi P, Brahmer J, Chen LC, Sandler A, Belani CP, Webb T, Harper H, Huberman M, Ramalingam S, Wong KK, Teofilovici F, Guo W, Shapiro GI (2013) A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin Cancer Res 19 (11): 3068–3077.
Stuhmer T, Iskandarov K, Gao Z, Bumm T, Grella E, Jensen MR, Einsele H, Chatterjee M, Bargou RC (2012) Preclinical activity of the novel orally bioavailable HSP90 inhibitor NVP-HSP990 against multiple myeloma cells. Anticancer Res 32 (2): 453–462.
Thota R, Williams Goff L, Chan E, Berlin J, Jones CM, McClanahan P, Ayers GD, Backlund Cardin D (2014) A phase II study of ganetespib (G) as second- or third-line therapy for metastatic pancreatic cancer (MPC). J Clin Oncol 32 (suppl 3): abstr 297.
Verstovsek S, Tam CS, Wadleigh M, Sokol L, Smith CC, Bui LA, Song C, Clary DO, Olszynski P, Cortes J, Kantarjian H, Shah NP (2014) Phase I evaluation of XL019, an oral, potent, and selective JAK2 inhibitor. Leuk Res 38 (3): 316–322.
Wang G, Krishnamurthy K, Tangpisuthipongsa D (2011) Protection of murine neural progenitor cells by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin in the low nanomolar concentration range. J Neurochem 117 (4): 703–711.
Whitesell L, Lindquist SL (2005) HSP90 and the chaperoning of cancer. Nat Rev Cancer 5 (10): 761–772.
Zhang H, Burrows F (2004) Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med (Berl) 82 (8): 488–499.
Zitzmann K, Ailer G, Vlotides G, Spoettl G, Maurer J, Goke B, Beuschlein F, Auernhammer CJ (2013) Potent antitumor activity of the novel HSP90 inhibitors AUY922 and HSP990 in neuroendocrine carcinoid cells. Int J Oncol 43 (6): 1824–1832.
Acknowledgements
This study was supported by Novartis Pharmaceutical.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
AS, J-PD, LDeM-A: none. and YB: none. JR: honoraria for advisory board Novartis; EC: none; PLB: commercial research grant, Novartis; MA: Novartis Employee, Ownership interest Novartis; HL: Novartis Employee; SP: Novartis Employee; AK: Novartis Employee; LLS: commercial research grant, Novartis; JC: honoraria for speaker lectures from Novartis.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Spreafico, A., Delord, JP., De Mattos-Arruda, L. et al. A first-in-human phase I, dose-escalation, multicentre study of HSP990 administered orally in adult patients with advanced solid malignancies. Br J Cancer 112, 650–659 (2015). https://doi.org/10.1038/bjc.2014.653
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.653
- Springer Nature Limited
Keywords
This article is cited by
-
A phase I trial of ganetespib in combination with paclitaxel and trastuzumab in patients with human epidermal growth factor receptor-2 (HER2)-positive metastatic breast cancer
Breast Cancer Research (2017)
-
Hyponatremia and V2 vasopressin receptor upregulation: a result of HSP90 inhibition
Cancer Chemotherapy and Pharmacology (2017)
-
Advances in HSP27 and HSP90-targeting strategies for glioblastoma
Journal of Neuro-Oncology (2016)