
Abstract
In recent years, melanoma has become a poster-child for the development of oncogene-directed targeted therapies. This approach, which has been exemplified by the development of small-molecule BRAF inhibitors and the BRAF/MEK inhibitor combination for BRAF-mutant melanoma, has brought new hope to patients. Despite these successes, treatment failure seems near inevitable in the majority of cases—even in individuals treated with the BRAF/MEK inhibitor doublet. In the current review, we discuss the future of combination strategies for patients with BRAF-mutant melanoma as well as the emerging therapeutic options for patients with NRAS-mutant and BRAF/NRAS-wild-type melanoma. We also outline some of the newest developments in the in-depth personalisation of therapy that should allow melanoma treatment to continue shaping the field precision cancer medicine.
Similar content being viewed by others

Beyond BRAF: developing combination strategies that abrogate resistance
The era of targeted therapy in melanoma began with the identification of driver mutations in the serine threonine kinase BRAF (Davies et al, 2002). It is now known that 40–60% of all cutaneous melanoma patients harbour position 600 mutations in BRAF, with the majority of these (∼80%) being V600E mutations (Dhomen and Marais, 2009). The next most frequent BRAF mutation is V600K, which occurs in ∼20% of BRAF-mutant patients (Long et al, 2011) and is associated with advancing age—with chronic sun damage being a risk factor (El-Osta et al, 2011). It has also been suggested that BRAF V600K-mutant melanoma shows an increased propensity to metastasise to the brain and lungs (El-Osta et al, 2011). The BRAF V600R mutation is the third most prevalent position 600 BRAF mutation and occurs in 5–7% of patients (Lovly et al, 2012).
In the clinical setting, the BRAF inhibitors vemurafenib and dabrafenib lead to significant levels of tumour shrinkage (according to RECIST criteria) in the majority of patients whose melanomas harbour position 600 BRAF mutations (Table 1) (Chapman et al, 2011; Hauschild et al, 2012). The three most common BRAF mutations (V600E/K/R) all exhibit BRAF inhibitor sensitivity, with response rates in the BRAF V600E group being ∼50% (Chapman et al, 2011; Klein et al, 2013). Regardless of these encouraging results, most patients have responses that are relatively short-lived (progression-free survival (PFS) for vemurafenib and dabrafenib was 5.3 and 5.1 months, respectively in phase III trials) with resistance eventually occurring in most cases (Chapman et al, 2011; Hauschild et al, 2012). Despite this and preclinical data suggesting that some resistant melanoma patient xenografts as well as cell lines with BRAF splice mutants may depend upon continuous BRAF inhibition for their fitness, there seems to be benefit to keeping patients on therapy beyond disease progression (Das Thakur et al, 2013; Azer et al, 2014; Hartsough et al, 2014). A recent study of 114 patients treated with either dabrafenib or vemurafenib showed an increase in OS when patients were kept on therapy despite progressing, compared with those who stopped drug (17.8 vs 7.0 months, P<0.001) (Azer et al, 2014). Studies are currently ongoing to determine whether resistance is better forestalled through continuous or discontinuous dosing schedules.
The efficacy of these inhibitors is not restricted to extra-cranial sites, with vemurafenib and dabrafenib found to cross the blood–brain barrier and to have efficacy against melanoma brain metastases (Long et al, 2012). At the same time, a subset of BRAF-mutant melanoma patients—∼10%—have been identified who did not meet the RECIST criteria for a response to vemurafenib or dabrafenib, suggesting the existence of complex genetic profiles that may convey intrinsic (or pre-existing) resistance (Salama and Flaherty, 2013).
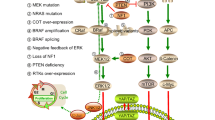
Resistance to BRAF inhibitors in melanoma is complex and mediated through multiple mechanisms with heterogeneous patterns of progression observed (for extensive reviews, see Solit and Rosen, 2011 and Holderfield et al, 2014). A recent analysis of a small cohort of patients showed the majority to progress at new metastatic sites (50%), with a slightly small percentage showing disease progression at existing sites (Menzies et al, 2014). In the majority of cases (>70%), acquired BRAF inhibitor resistance (here defined as the diminishment of drug response that that occurs following chronic BRAF inhibitor treatment) is characterised by the reactivation of MAPK signalling that can be mediated through multiple mechanisms including NRAS (Q61K) mutations, BRAF amplification, activating MEK1 (C121S and P124L) mutations, MEK2 (Q60P) mutations concurrent with BRAF amplification and BRAF splice-form mutants (Emery et al, 2009; Wagle et al, 2011; Villanueva et al, 2013; Shi et al, 2014). In vitro, MAPK signalling recovers rapidly following BRAF inhibition, in part through the relief of feedback inhibition in the pathway and an increased sensitivity to growth factors such as epidermal growth factor (EGF), neuregulin (NRG-1), hepatocyte growth factor (HGF) and fibroblasts growth factor (FGF) (Lito et al, 2012). In this context, reactivation of MAPK signalling following BRAF inhibition is important for therapeutic escape with increased levels of cell death and tumour regression being seen when BRAF and MEK are co-targeted (Paraiso et al, 2010; Lito et al, 2012). Clinical trials have confirmed these preclinical observations with the BRAF/MEK inhibitor combination (dabrafenib plus trametinib) showing an increased PFS compared with BRAF inhibitor alone (Table 1). A later trial—the phase III COMBI-D regimen—further showed an increase in the overall response rate to the combination compared with monotherapy and an improvement in a range of clinical end points. Another BRAF/MEK inhibitor combination—vemurafenib+cobimetinib—also appears promising, with newly released data from the BRIM-7 trial demonstrating an 87% confirmed response rate by RECIST and a median PFS of 13.7 months (Ribas et al, 2014).
Despite hopes that vertical MAPK pathway targeting would limit resistance, treatment failure still occurs with the resistance mechanisms observed to the BRAF/MEK inhibitor combination being analogous to those seen in patients on BRAF inhibitor monotherapy. A preliminary genetic analysis of five patients failing dabrafenib+trametinib revealed similar mechanisms of resistance to those seen in patients on BRAF inhibitor monotherapy and highlighted the role of MEK2 Q60P mutations, BRAF-splice mutants and BRAF amplification (Wagle et al, 2014). It is therefore perhaps not surprising that failure on BRAF inhibitor therapy also confers resistance to MEK inhibition, with minimal clinical activity being seen to trametinib in patients failing dabrafenib and a 15% response rate (median PFS 2.8 months) observed in patients failing BRAF inhibitor following treatment with vemurafenib+cobimetinib (Ribas et al, 2014).
Attention is now being turned to further targeted agents that can be added to the BRAF/MEK inhibitor backbone. There is already preclinical evidence that ERK inhibitors, such as SCH772984, can overcome acquired resistance to single agent BRAF and MEK inhibition (Morris et al, 2013). Although clinical trials of ERK inhibitors in the monotherapy setting have been initiated, little information has been yet presented on their efficacy or toxicity. Another surprise recent finding was the dependency of the MAPK signalling pathway upon copper ions, with the copper-MEK1 interaction being required for efficient ERK phosphorylation (Brady et al, 2014). Chelation of copper or the knockdown of the copper transporter CRT1 had anti-proliferative activity against BRAF-mutant melanoma cells and could overcome vemurafenib resistance mediated through the C121S MEK1 mutations (Brady et al, 2014).
Many components of the MAPK pathway, including mutant BRAF, CRAF and COT proteins, are clients of the chaperone protein HSP90 (da Rocha Dias et al, 2005; Grbovic et al, 2006; Paraiso et al, 2012). This, along with the requirement for HSP90 in multiple BRAF inhibitor resistance mechanisms, has raised interest in co-targeting the HSP90 ‘clientome’ along with mutant BRAF. This concept has already been validated in other cancers, with studies showing that HSP90 inhibitors reverse trastuzamab resistance in HER2-positive breast cancer as well as bortezomib resistance in multiple myeloma (Modi et al, 2011; Richardson et al, 2011). At least three preclinical studies have now demonstrated that HSP90 inhibitors (including XL888 and ganetespib) can overcome or abrogate the onset of BRAF inhibitor resistance (Paraiso et al, 2012; Wu et al, 2013; Acquaviva et al, 2014). At this time, a phase I dose escalation study of vemurafenib+XL888 in patients with metastatic BRAF V600-mutant melanoma is ongoing (Table 1) (NCT01657591).
Although reactivation of MAPK signalling is frequently associated with BRAF and BRAF/MEK inhibitor failure, other pathways such as the PI3K/AKT pathway have also been implicated in resistance. In melanoma cell lines, constitutive PI3K/AKT signalling is commonly observed and can result from multiple mechanisms, including the loss/mutation of the tumour suppressors PTEN or neurofibromin (NF1) or increased expression of AKT3 (Stahl et al, 2004; Tsao et al, 2004; Maertens et al, 2012). In vitro studies have suggested that BRAF and MEK inhibition may sometimes lead to rebound PI3K/AKT signalling, resulting in therapeutic escape mediated through the suppression of apoptosis (Gopal et al, 2010; Paraiso et al, 2011). These lab-based findings have also been supported by the observation that increased PI3K/AKT signalling occurs in some early (4–25 days) on-therapy specimens from patients treated with a BRAF inhibitor (Shi et al, 2014). One study showed that 22% of tumours from patients failing therapy exhibited increased AKT activity (Shi et al, 2014). In the majority of cases (81%) increased AKT signalling overlapped with recovery of signalling in the MAPK pathway, with the AKT pathway representing the primary resistance pathway in only a minority of cases (Shi et al, 2014). A number of potential mechanisms of PI3K/AKT activation were identified including mutations in PI3KCA (D350G and E544G), PI3KCG (V983E), PTEN (134M_ and fs.40), PI3KR2 (N561D), AKT1 (Q79K), AKT3 (E17K) and PHLPP1 (K596E) (Shi et al, 2014). Of these, loss of PTEN function, either through mutation or loss of expression, has been the most extensively investigated with somewhat conflicting results about its relevancy. Although there is preclinical evidence suggesting that loss of PTEN expression predicts for a reduced cytotoxic response to BRAF inhibition, some BRAF V600E/PTEN-null melanoma cell lines have also been identified with sensitivity to vemurafenib (Atefi et al, 2011; Paraiso et al, 2011). In addition, good levels of tumour regression have been observed in BRAF V600E/PTEN-null GEMM models of melanoma following BRAF inhibitor treatment (Marsh Durban et al, 2013). A similarly nuanced picture also emerged when patients receiving dabrafenib were stratified according to their PTEN status, with a trend being seen towards a lower PFS in individuals whose tumours lacked PTEN function (Nathanson et al, 2013).
The identification of PI3K/AKT signalling as a core pathway for melanoma development and therapeutic escape suggested the possibility of co-targeting MAPK and PI3K/AKT signalling in BRAF-mutant melanoma (Table 1). A number of early preclinical studies demonstrated the utility of concurrently targeting the MAPK and PI3K/AKT pathways across multiple melanoma cell lines and xenografts (Bedogni et al, 2006; Smalley et al, 2006). In BRAF V600E/PTEN-null GEMM models, the combination of the BRAF inhibitor LGX818 with the PI3K inhibitor BKM-120 was associated with a more rapid and durable pattern of tumour regression compared with LGX818 alone (Marsh Durban et al, 2013). With the clinical availability of PI3K, AKT and mTOR inhibitors, multiple trials are now underway with combined inhibition of the MAPK and PI3K pathways (Tables 1 and 2).
Melanomas have the highest mutational loads of all cancers (Alexandrov et al, 2013). One central question that has arisen is whether resistance is mediated through pre-existing clones that are BRAF wild type or occurs through drug-induced selection pressure that drives the mutational landscape. Evidence in favour of drug-induced selection pressure comes from a recent whole-exome sequencing study of multiple progressing lesions from one patient failing dabrafenib therapy after 383 days (Shi et al, 2014). Of the nine distinct progressing lesions analysed, at least five co-existent mechanisms of resistance were identified, including an acquired KRAS mutation, a BRAF splice-mutant, BRAF amplification, a PTEN indel and one mechanism that remains unknown (Shi et al, 2014). At the same time, the mutational spectra of the progressing tumours significantly altered on BRAF inhibitor therapy, with a reduction in the frequency of C>T transition mutations being observed in the resistant tumours compared with the pre-treatment tumours (Shi et al, 2014).
NRAS-mutant and BRAF/NRAS-wild-type melanoma
Although the majority of therapeutic advancements in the past few years have been largely focused on patients with mutations in BRAF, NRAS was actually the first oncogene identified in melanoma (Albino et al, 1984). NRAS is part of family of low-molecular weight GTP-binding proteins that are associated with the plasma membrane. Ras proteins control a wide array of cellular functions, including growth, survival and invasion, by relaying signals from activated RTKs at the cell surface to downstream effectors in the nucleus, including cell-cycle proteins and transcription factors (Malumbres and Barbacid, 2003; Cully and Downward, 2008). Activated Ras can trigger a number of intracellular signalling pathways such as the Raf/MEK/ERK mitogen-activated protein kinase (MAPK) pathway and the PI3K/AKT pathway (Downward, 2003). Mutated Ras can mediate cellular transformation through a network of signal-transduction pathways independent of upstream RTK activation (Malumbres and Barbacid, 2003). The role of NRAS in driving growth of melanoma cells was confirmed through knockdown of NRAS in melanoma cell lines using small-interfering RNA, which showed a marked reduction in cell growth and with decreased expression of cyclins D1 and E2 (Eskandarpour et al, 2009). It is currently known that NRAS, KRAS and HRAS mutations are present in 20%, 2% and 1% of all melanomas, respectively, with the most common NRAS mutation occurring at position Q61 (Milagre et al, 2010).
NRAS-mutant melanomas differ from BRAF-mutant melanomas in clinical presentation and prognostic features (Devitt et al, 2011; Ellerhorst et al, 2011). Patients who present with NRAS-mutant melanomas tend to be older and have a history of chronic UV exposure (Devitt et al, 2011). These individuals tend to have thicker primary tumours that are located on the extremities and have higher rates of mitosis (Devitt et al, 2011). While MAPK signalling in melanocytes is typically driven through BRAF, BRAF activity is not required for MAPK activation in NRAS-mutant melanomas, which alternatively rely on CRAF signalling (Dumaz et al, 2006). In NRAS-mutant melanoma, the switch to CRAF signalling is dependent on both the phosphorylation and inactivation of BRAF at S151, T401, S750 and T753 and the deregulation of protein kinase A (PKA) activity (which serves to prevent CRAF from being phosphorylated at inhibitory sites) (Dumaz et al, 2006; Marquette et al, 2011). The role of PKA in the regulation of CRAF suggests the possibility for therapeutic intervention, with studies showing selective phosphodiesterase IV inhibitors to be growth inhibitory and pro-apoptotic in NRAS-mutant cell lines (Marquette et al, 2011). Similarly, PI3K/AKT pathway regulation in NRAS-mutant melanoma cells proceeds differently than in those harbouring a BRAF mutation, and occurs directly through the Ras-mediated recruitment of PI3K, rather than the concurrent loss of PTEN or NF1 function (Tsao et al, 2004; Maertens et al, 2012).
So far, the direct targeting of NRAS has proven to be a challenge. Several approaches have been explored for targeting Ras directly by designing drugs that prevent the post-translational modifications required for the insertion of Ras into the plasma membrane. Farnesyl transferase inhibitors initially showed great preclinical potential, but have ultimately been disappointing in the clinical setting (Konstantinopoulos et al, 2007). The recent years have seen renewed interest in the development of small-molecule RAS inhibitors that bind to domains unique to the mutant protein. One approach has utilised the binding of drug to the cysteine in the G12-mutant form of KRAS to achieve selectivity over the wild-type protein (Ostrem et al, 2013). In preclinical studies, compounds directed against KRAS-G12 had anti-proliferative and pro-apoptotic activity against KRAS-mutant lung cancer cell lines in the low micromolar range. Another RAS targeting approach is to inhibit the interaction between KRAS and the prenyl binding protein PDEδ, which in turn prevents KRAS signalling by altering its localisation to endomembranes (Zimmermann et al, 2013). Although these new inhibitors have been developed for mutant KRAS, it is likely that a similar concept can be applied to NRAS-mutant tumours in the near future.
One other strategy being actively explored is the inhibition of the downstream mediators of Ras signalling, including MEK, ERK, PI3K and CDK4. The greatest focus of preclinical studies for NRAS-mutant melanoma has been upon MAPK pathway inhibition in combination with inhibitors of other pathways activated by Ras. Preclinical work using shRNA to knock down Ras targets demonstrated that good levels of tumour regression could be achieved in vivo following the ablation of either BRAF+CRAF or BRAF+PI3K (Jaiswal et al, 2009). Other studies, in which small-molecule inhibitors of the MAPK and PI3K/AKT pathway were evaluated in NRAS-mutant melanoma xenograft models, demonstrated the combination of MEK/PI3K inhibitors to be superior to a combination targeting MEK+mTOR (Posch et al, 2013). It was further found that the combination of a MEK inhibitor with a PI3K/mTOR inhibitor was synergistic and was associated with profound levels of tumour regression (Posch et al, 2013). Further support for this combination comes from studies of BRAF-mutant melanoma cell lines in which resistance was mediated through the acquisition of an NRAS mutation(Greger et al, 2012). There is also evidence from unbiased bioinformatic studies of synergy between MEK and CDK4/6 inhibitors in NRAS (Q61K)/CDKN2ANull mouse melanoma models, and this combination is currently being evaluated clinically in patients with NRAS-mutant melanoma as well as those with BRAF-mutant melanoma (Table 2) (NCT01781572, NCT01777776) (Kwong et al, 2012).
Despite the focus upon the co-targeting of the MAPK and PI3K/AKT pathways in NRAS-mutant melanoma, other Ras-driven pathways may also be important for tumour initiation and maintenance—and significantly less is known about these. A recent study in which ARF-null immortalised melanocytes were transformed with active forms of PI3K, MEK or Ral-GDS revealed distinct roles for all of these signal transduction mediators in tumour initiation, with Ral-GDS being a prerequisite for anchorage-independent growth (Mishra et al, 2010). Other work showed the RAL-GEF effector TANK-binding kinase 1 (TBK1) to be a key mediator of RAL activity in NRAS-mutant melanoma, with its siRNA-mediated knockdown found to prevent tumour invasion and migration (Vu and Aplin, 2014).
In the same vein, the HSP inhibitor XL888 demonstrated efficacy against a panel of NRAS-mutant cell lines through simultaneous suppression of a number of targets crucial for NRAS-mutant melanoma cell survival, including CDK4, AKT and WEE1 (Haarberg et al, 2013). The potential role of WEE1 in Ras-mutant tumours has also been confirmed in other systems, with positive interactions seen between the WEE1 inhibitor MK-1775 and PI3K/AKT/mTOR inhibitors in various models including acute leukaemia (Weisberg et al, 2014).
Despite strong preclinical evidence for treating NRAS-mutant melanoma using inhibitors of MEK, clinical trials so far have demonstrated only modest activity in patients. At first, early generation MEK inhibitors exhibited only a 10% objective response rate. However, excitement at the idea of targeting MEK resurfaced with the development of potent third-generation MEK inhibitors trametinib (GSK1120212) and MEK162 (ARRY-438162) (Table 2). In the recent phase II clinical trial of MEK162, 20% of NRAS-mutant patients exhibited objective responses while a further 43% showed stable disease; however, median PFS was 3.7 months (Ascierto et al, 2013). A phase III trial is currently ongoing comparing MEK162 with dacarbazine specifically in NRAS-mutant metastatic melanoma patients (Table 2) (NCT01763164). The mechanisms by which NRAS-mutant melanomas escape from MEK inhibition are starting to be elucidated. Treatment of NRAS-mutant melanoma cell lines with the MEK inhibitors AZD6244 and trametinib is associated with recovery of MAPK signalling, an effect that can be overcome through dual MEK/ERK inhibition. Another potential mechanism of escape from MEK inhibition in NRAS-mutant melanoma is adaptive (here defined as a change in signalling that occurs following the administration of a first drug) RTK signalling, with increased PDGFR-B signalling being observed in some cell lines following treatment with the MEK inhibitor AZD6244 (Rebecca et al, 2014b). In these instances, therapeutic escape was abrogated and cytotoxicity was enhanced when MEK inhibitors were combined with the PDGFR inhibitor crenolanib (Rebecca et al, 2014b).
NRAS-mutant melanoma cell lines are known to have constitutive activity in many RTKs and there is growing preclinical evidence that multi-RTK targeted inhibitors have efficacy against subsets of NRAS-mutant melanoma (Tworkoski et al, 2011; Fedorenko et al, 2014). Studies have already shown RAF-265 (which inhibits CRAF, BRAF, VEGFR and FLT-3 among other things) to have good anti-tumour activity in some BRAF-wild-type melanoma patient-derived xenografts and that amuvatinib (inhibits c-KIT, c-MET, Axl and RAD51) is effective in some NRAS-mutant melanoma cell lines (Su et al, 2012; Fedorenko et al, 2014). In the clinical setting, NRAS-mutant melanoma patients also showed better responses to the combination of sorafenib with carboplatin+paclitaxel than their BRAF-mutant counterparts (Wilson et al, 2014).
Strategies to target melanomas that are BRAF/NRAS/KIT-wild type (so-called ‘triple-negative’ or ‘pan-negative’ melanomas) have proven even more elusive. The recent whole-exome sequencing of 21 melanomas that were BRAF/NRAS-wild type painted a complex picture involving mutations in NF1 as well as rare CRAF and MAP2K1 mutations (Hodis et al, 2012). Some of the BRAF/NRAS-wild-type melanomas in this study showed focal amplification in potential oncogenes such as cyclin D1 and CDK4, although it was not determined whether this conveyed sensitivity to CDK4 inhibition (Hodis et al, 2012). In a second more recent study, the next-generation sequencing of 623 cancer-related genes across 241 melanoma samples included 69 tumours that were ‘triple-negative’. In this cohort, a number of potential driver mutations were identified including ALK (3.5%), RAC1 (2.9%), STK31 (8.7%), DGK1 (4.7%), NF1 (7.6%), KDR (6.4%) and ERBB4 (11.6%) (Xia et al, 2014). Although both these sequencing studies identified mutations in NF1 (which is a negative regulator of Ras signalling) as a potential driver of BRAF/NRAS-wild-type melanoma, its loss seems to also play a role in BRAF-mutant melanoma (Maertens et al, 2012; Whittaker et al, 2012; Nissan et al, 2014). In the BRAF-mutant context, loss of NF1 function leads to increased CRAF-mediated MAPK signalling and activation of the PI3K/AKT pathway, and it is implicated in both BRAF inhibitor resistance and melanoma development (Maertens et al, 2012; Whittaker et al, 2012; Nissan et al, 2014). It is likely that melanoma patients lacking NF1 function (regardless of other co-operating oncogenes) will require combination therapy treatment; with preclinical studies suggesting the utility of the MEK+mTOR inhibitor combination, a pan-RAF inhibitor or an ERK inhibitor (Maertens et al, 2012; Whittaker et al, 2012; Nissan et al, 2014).
RAC1 is a small GTPase that has been linked to cancer cell motility (Sanz-Moreno et al, 2008). Recurrent P29S mutations in RAC1 were recently reported in 3.3–9.2% of cutaneous melanomas, with mutations occurring at a greater frequency in male patients (Krauthammer et al, 2012; Mar et al, 2014). Although an association was reported between RAC1 mutations and BRAF/NRAS-wild-type mutational status, this was not found in a second, larger study (Krauthammer et al, 2012; Mar et al, 2014). From a functional standpoint, the presence of a RAC1 mutation was associated with a greater risk of nodal metastasis and it was suggested that the acquisition of a Rac1 mutation led to a greater risk of early disease dissemination (Mar et al, 2014).
Although triple-negative melanomas may lack BRAF mutations, they may still be dependent upon BRAF signalling, with two recent reports identifying the potential role of BRAF fusion proteins (Botton et al, 2013; Hutchinson et al, 2013). These fusion events, which typically involve the fusion of the BRAF kinase domain to other N-terminal binding partners including PASSP1, CDC27, TAX1BP1 and TRIM24, occur in 4–8% of cases of triple-negative melanomas (Hutchinson et al, 2013). Preliminary evidence suggests that these fusion proteins activate the MAPK pathway in melanoma cell lines and convey sensitivity to either sorafenib or MEK inhibition (Botton et al, 2013; Hutchinson et al, 2013). At the same time there have also been reports of melanomas with non-position 600 mutations in BRAF such as K601, L597R and L597Q showing sensitivity to MEK inhibition (Dahlman et al, 2012; Bowyer et al, 2014).
Inhibition of the MAPK and PI3K/AKT signalling pathways leads to adaptive RTK signalling in multiple cancer types (Chandarlapaty et al, 2011; Duncan et al, 2012; Lito et al, 2012). BRAF/NRAS-wild-type melanoma is no exception and there is evidence that inhibition of MEK signalling leads to increased endothelin-B receptor (EDNRB) expression that limits therapeutic efficacy (Asundi et al, 2014). In an in vivo model of BRAF/NRAS-wild-type melanoma, the co-targeting of MEK with an antibody drug conjugate targeted against EDNRB was more efficacious than either agent alone and was associated with good levels of tumour suppression (Asundi et al, 2014). Similarly, inhibition of AKT in combination with paclitaxel and carboplatin suppressed the long-term growth of BRAF/NRAS-wild-type melanoma cell lines in vitro, and was associated with stable disease (>10 months) in two cases of BRAF-wild-type melanoma (Rebecca et al, 2014a).
Another potential therapeutic target that is frequently either amplified or overexpressed in BRAF-wild-type and BRAF/NRAS-wild-type melanoma is p21-activated kinase (PAK)-1 (Ong et al, 2013). This kinase, which is downstream of both RAC1 and CDC42, stimulates the MAPK pathway by directly phosphorylating CRAF at S338 and MEK1 at S298. In NRAS-mutant and BRAF/NRAS-wild-type melanomas, inhibition of PAK1 through either siRNA knockdown or the PAK1 inhibitor PF-3758309 suppresses ERK phosphorylation and was associated with the reduction growth in a BRAF/NRAS-wild-type melanoma xenograft model (Ong et al, 2013). In this instance, the effect seemed to be more cytostatic than cytotoxic: suggesting that other drugs may need to be combined with PF-3758309 to achieve cytoreduction and durable responses.
Conclusion
Tremendous progress has been made in developing oncogene-directed therapies for treating BRAF-mutant melanoma. Resistance remains a major problem that limits the long-term responsiveness of the majority of the patients to these drugs. Current strategies to improve the durability of response are now focused on the development of personalised combination therapy strategies, the majority of which centre upon the suppression of adaptive MAPK and PI3K/AKT signalling (Table 1). Given the current success of the BRAF/MEK inhibitor doublet, future combinations will likely be based upon this backbone. At this time, the relationship between the genetic prolife of the tumour and patterns of adaptive signalling are not well understood. Better assays and biomarkers will be needed to interrogate the early treatment responses so that combinations can be rapidly personalised before resistance ensues. Strategies being explored in this realm include the analysis of circulating tumour cells, circulating tumour DNA and proteomic methods. Considerably less progress has been made in the development of precision medicine strategies for BRAF-wild-type melanoma. Although there are hints that MEK inhibitors may be effective in NRAS-mutant melanoma, responses have been sub-optimal and combinations (likely highly personalised ones) will be needed.
References
Acquaviva J, Smith DL, Jimenez JP, Zhang C, Sequeira M, He S, Sang J, Bates RC, Proia DA (2014) Overcoming acquired BRAF inhibitor resistance in melanoma via targeted inhibition of Hsp90 with ganetespib. Mol Cancer Ther 13 (2): 353–363.
Albino AP, Le Strange R, Oliff AI, Furth ME, Old LJ (1984) Transforming ras genes from human melanoma: a manifestation of tumour heterogeneity? Nature 308 (5954): 69–72.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, Lopez-Otin C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdes-Mas R, van Buuren MM, van 't Veer L, Vincent-Salomon A, Waddell N, Yates LR, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR (2013) Signatures of mutational processes in human cancer. Nature 500 (7463): 415–421.
Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, Blank CU, Hauschild A, Beck JT, St-Pierre A, Niazi F, Wandel S, Peters M, Zubel A, Dummer R (2013) MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol 14 (3): 249–256.
Asundi J, Lacap JA, Clark S, Nannini M, Roth L, Polakis P (2014) MAPK pathway inhibition enhances the efficacy of an anti-endothelin B receptor drug conjugate by inducing target expression in melanoma. Mol Cancer Ther 13 (6): 1599–1610.
Atefi M, von Euw E, Attar N, Ng C, Chu C, Guo D, Nazarian R, Chmielowski B, Glaspy JA, Comin-Anduix B, Mischel PS, Lo RS, Ribas A (2011) Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS One 6 (12): e28973.
Azer MWF, Menzies AM, Haydu LE, Kefford RF, Long GV (2014) Patterns of response and progression in patients with BRAF-mutant melanoma metastatic to the brain who were treated with dabrafenib. Cancer 120 (4): 530–536.
Bedogni B, Welford SM, Kwan AC, Ranger-Moore J, Saboda K, Powell MB (2006) Inhibition of phosphatidylinositol-3-kinase and mitogen-activated protein kinase kinase 1/2 prevents melanoma development and promotes melanoma regression in the transgenic TPRas mouse model. Mol Cancer Ther 5 (12): 3071–3077.
Botton T, Yeh I, Nelson T, Vemula SS, Sparatta A, Garrido MC, Allegra M, Rocchi S, Bahadoran P, McCalmont TH, LeBoit PE, Burton EA, Bollag G, Ballotti R, Bastian BC (2013) Recurrent BRAF kinase fusions in melanocytic tumors offer an opportunity for targeted therapy. Pigment Cell Melanoma Res 26 (6): 845–851.
Bowyer SE, Rao AD, Lyle M, Sandhu S, Long GV, McArthur GA, Raleigh JM, Hicks RJ, Millward M (2014) Activity of trametinib in K601E and L597Q BRAF mutation-positive metastatic melanoma. Melanoma Res; e-pub ahead of print 13 June 2014.
Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, Knapp S, Xiao K, Campbell SL, Thiele DJ, Counter CM (2014) Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 509 (7501): 492–496.
Catalanotti F, Solit DB, Pulitzer MP, Berger MF, Scott SN, Iyriboz T, Lacouture ME, Panageas KS, Wolchok JD, Carvajal RD, Schwartz GK, Rosen N, Chapman PB (2013) Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K-mutated melanoma. Clin Cancer Res 19 (8): 2257–2264.
Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N (2011) AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 19 (1): 58–71.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O'Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. New Engl J Med 364 (26): 2507–2516.
Cully M, Downward J (2008) SnapShot: Ras signaling. Cell 133 (7): 1292–1292 e1.
da Rocha Dias S, Friedlos F, Light Y, Springer C, Workman P, Marais R (2005) Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res 65 (23): 10686–10691.
Dahlman KB, Xia JF, Hutchinson K, Ng C, Hucks D, Jia PL, Atefi M, Su ZL, Branch S, Lyle PL, Hicks DJ, Bozon V, Glaspy JA, Rosen N, Solit DB, Netterville JL, Vnencak-Jones CL, Sosman JA, Ribas A, Zhao ZM, Pao W (2012) BRAF(L597) Mutations in Melanoma Are Associated with Sensitivity to MEK Inhibitors. Cancer Discov 2 (9): 791–797.
Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD (2013) Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 494 (7436): 251–255.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA (2002) Mutations of the BRAF gene in human cancer. Nature 417 (6892): 949–954.
Devitt B, Liu W, Salemi R, Wolfe R, Kelly J, Tzen CY, Dobrovic A, McArthur G (2011) Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res 24 (4): 666–672.
Dhomen N, Marais R (2009) BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 23 (3): 529–545, ix.
Downward J (2003) Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3 (1): 11–22.
Dumaz N, Hayward R, Martin J, Ogilvie L, Hedley D, Curtin JA, Bastian BC, Springer C, Marais R (2006) In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res 66 (19): 9483–9491.
Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, Usary J, Kuan PF, Smalley DM, Major B, He X, Hoadley KA, Zhou B, Sharpless NE, Perou CM, Kim WY, Gomez SM, Chen X, Jin J, Frye SV, Earp HS, Graves LM, Johnson GL (2012) Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149 (2): 307–321.
El-Osta H, Falchook G, Tsimberidou A, Hong D, Naing A, Kim K, Wen S, Janku F, Kurzrock R (2011) BRAF mutations in advanced cancers: clinical characteristics and outcomes. PLoS One 6 (10): e25806.
Ellerhorst JA, Greene VR, Ekmekcioglu S, Warneke CL, Johnson MM, Cooke CP, Wang LE, Prieto VG, Gershenwald JE, Wei Q, Grimm EA (2011) Clinical correlates of NRAS and BRAF mutations in primary human melanoma. Clin Cancer Res 17 (2): 229–235.
Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB, Macconaill LE, Zhang J, Gray NS, Sellers WR, Dummer R, Garraway LA (2009) MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci USA 106 (48): 20411–20416.
Eskandarpour M, Huang F, Reeves KA, Clark E, Hansson J (2009) Oncogenic NRAS has multiple effects on the malignant phenotype of human melanoma cells cultured in vitro. Int J Cancer 124 (1): 16–26.
Fedorenko IV, Fang B, Koomen JM, Gibney GT, Smalley KS (2014) Amuvatinib has cytotoxic effects against NRAS-mutant melanoma but not BRAF-mutant melanoma. Melanoma Res; e-pub ahead of print 19 June 2014.
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA 3rd, Falchook G, Algazi A, Lewis K, Long GV, Puzanov I, Lebowitz P, Singh A, Little S, Sun P, Allred A, Ouellet D, Kim KB, Patel K, Weber J (2012a) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. New Engl J Med 367 (18): 1694–1703.
Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, Dummer R, Trefzer U, Larkin JM, Utikal J, Dreno B, Nyakas M, Middleton MR, Becker JC, Casey M, Sherman LJ, Wu FS, Ouellet D, Martin AM, Patel K, Schadendorf D (2012b) Improved survival with MEK inhibition in BRAF-mutated melanoma. New Engl J Med 367 (2): 107–114.
Gopal YN, Deng W, Woodman SE, Komurov K, Ram P, Smith PD, Davies MA (2010) Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res 70 (21): 8736–8747.
Grbovic OM, Basso AD, Sawai A, Ye Q, Friedlander P, Solit D, Rosen N (2006) V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc Natl Acad Sci US A 103 (1): 57–62.
Greger JG, Eastman SD, Zhang VV, Bleam MR, Hughes AM, Smitheman KN, Dickerson SH, Laquerre SG, Liu L, Gilmer TM (2012) Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther 11 (4): 909–920.
Haarberg HE, Paraiso KH, Wood E, Rebecca VW, Sondak VK, Koomen JM, Smalley KS (2013) Inhibition of Wee1, AKT, and CDK4 underlies the efficacy of the HSP90 inhibitor XL888 in an in vivo model of NRAS-mutant melanoma. Mol Cancer Ther 12 (6): 901–912.
Hainsworth JD, Infante JR, Spigel DR, Peyton JD, Thompson DS, Lane CM, Clark BL, Rubin MS, Trent DF, Burris HA 3rd (2010) Bevacizumab and everolimus in the treatment of patients with metastatic melanoma: a phase 2 trial of the Sarah Cannon Oncology Research Consortium. Cancer 116 (17): 4122–4129.
Hartsough EJ, Basile KJ, Aplin AE (2014) Beneficial effects of RAF inhibitor in mutant BRAF splice variant-expressing melanoma. Mol Cancer Res 12 (5): 795–802.
Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E, Martin-Algarra S, Karaszewska B, Mauch C, Chiarion-Sileni V, Martin AM, Swann S, Haney P, Mirakhur B, Guckert ME, Goodman V, Chapman PB (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380 (9839): 358–365.
Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li LR, Place C, DiCara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DSB, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L (2012) A landscape of driver mutations in melanoma. Cell 150 (2): 251–263.
Holderfield M, Deuker MM, McCormick F, McMahon M (2014) Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 14 (7): 455–467.
Hutchinson KE, Lipson D, Stephens PJ, Otto G, Lehmann BD, Lyle PL, Vnencak-Jones CL, Ross JS, Pietenpol JA, Sosman JA, Puzanov I, Miller VA, Pao W (2013) BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin Cancer Res 19 (24): 6696–6702.
Jaiswal BS, Janakiraman V, Kljavin NM, Eastham-Anderson J, Cupp JE, Liang Y, Davis DP, Hoeflich KP, Seshagiri S (2009) Combined targeting of BRAF and CRAF or BRAF and PI3K effector pathways is required for efficacy in NRAS mutant tumors. PLoS One 4 (5): e5717.
Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P, Middleton M, Cantarini M, Zazulina V, Kemsley K, Dummer R (2012) Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 18 (2): 555–567.
Klein O, Clements A, Menzies AM, O'Toole S, Kefford RF, Long GV (2013) BRAF inhibitor activity in V600R metastatic melanoma. Eur J Cancer 49 (5): 1073–1079.
Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG (2007) Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov 6 (7): 541–555.
Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K, Capatana A, Holman EC, Bosenberg M, Sznol M, Kluger HM, Brash DE, Stern DF, Materin MA, Lo RS, Mane S, Ma S, Kidd KK, Hayward NK, Lifton RP, Schlessinger J, Boggon TJ, Halaban R (2012) Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 44 (9): 1006–1014.
Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, Jakubosky D, Genovese G, Muller FL, Jeong JH, Bender RP, Chu GC, Flaherty KT, Wargo JA, Collins JJ, Chin L (2012) Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med 18 (10): 1503–1510.
Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, Wolchok JD, de Stanchina E, Chandarlapaty S, Poulikakos PI, Fagin JA, Rosen N (2012) Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 22 (5): 668–682.
Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, Hughes TM, Thompson JF, Scolyer RA, Kefford RF (2011) Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol 29 (10): 1239–1246.
Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB, Puzanov I, Hauschild A, Robert C, Algazi A, Mortier L, Tawbi H, Wilhelm T, Zimmer L, Switzky J, Swann S, Martin AM, Guckert M, Goodman V, Streit M, Kirkwood JM, Schadendorf D (2012) Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol 13 (11): 1087–1095.
Lovly CM, Dahlman KB, Fohn LE, Su Z, Dias-Santagata D, Hicks DJ, Hucks D, Berry E, Terry C, Duke M, Su Y, Sobolik-Delmaire T, Richmond A, Kelley MC, Vnencak-Jones CL, Iafrate AJ, Sosman J, Pao W (2012) Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS One 7 (4): e35309.
Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messaien L, Bronson RT, McMahon M, Granter S, Flaherty KT, Wargo JA, Marais R, Cichowski K (2012) Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov 3 (3): 338–349.
Malumbres M, Barbacid M (2003) RAS oncogenes: the first 30 years. Nat Rev Cancer 3 (6): 459–465.
Mar VJ, Wong SQ, Logan A, Nguyen T, Cebon J, Kelly J, Wolfe R, Dobrovic A, McLean C, McArthur GA (2014) Clinical and pathological associations of the activating RAC1 P29S mutation in primary cutaneous melanoma. Pigment Cell Melanoma Res; e-pub ahead of print 17 July 2014; doi:10.1111/pcmr.12295.
Marquette A, Andre J, Bagot M, Bensussan A, Dumaz N (2011) ERK and PDE4 cooperate to induce RAF isoform switching in melanoma. Nat Struct Mol Biol 18 (5): 584–591.
Marsh Durban V, Deuker MM, Bosenberg MW, Phillips W, McMahon M (2013) Differential AKT dependency displayed by mouse models of BRAFV600E-initiated melanoma. J Clin Invest 123 (12): 5104–5118.
Means-Powell JA, Adjei AA, Puzanov I, Dy GK, Goff LW, Ma WW, Fetterly GJ, Michael SA, Chai F, Lamar M, Schwartz BE, Sosman JA (2012) Safety and efficacy of MET inhibitor tivantinib (ARQ 197) combined with sorafenib in patients (pts) with NRAS wild-type or mutant melanoma from a phase I study. J Clin Oncol 30 (15_suppl): 8519.
Menzies AM, Haydu LE, Carlino MS, Azer MW, Carr PJ, Kefford RF, Long GV (2014) Inter- and intra-patient heterogeneity of response and progression to targeted therapy in metastatic melanoma. PLoS ONE 9 (1): e85004.
Milagre C, Dhomen N, Geyer FC, Hayward R, Lambros M, Reis-Filho JS, Marais R (2010) A mouse model of melanoma driven by oncogenic KRAS. Cancer Res 70 (13): 5549–5557.
Minor DR, Kashani-Sabet M, Garrido M, O'Day SJ, Hamid O, Bastian BC (2012) Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res 18 (5): 1457–1463.
Mishra PJ, Ha L, Rieker J, Sviderskaya EV, Bennett DC, Oberst MD, Kelly K, Merlino G (2010) Dissection of RAS downstream pathways in melanomagenesis: a role for Ral in transformation. Oncogene 29 (16): 2449–2456.
Modi S, Stopeck A, Linden H, Solit D, Chandarlapaty S, Rosen N, D'Andrea G, Dickler M, Moynahan ME, Sugarman S, Ma W, Patil S, Norton L, Hannah AL, Hudis C (2011) HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res 17 (15): 5132–5139.
Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, Long B, Liu J, Dinunzio E, Windsor W, Zhang R, Zhao S, Angagaw MH, Pinheiro EM, Desai J, Xiao L, Shipps G, Hruza A, Wang J, Kelly J, Paliwal S, Gao X, Babu BS, Zhu L, Daublain P, Zhang L, Lutterbach BA, Pelletier MR, Philippar U, Siliphaivanh P, Witter D, Kirschmeier P, Bishop WR, Hicklin D, Gilliland DG, Jayaraman L, Zawel L, Fawell S, Samatar AA (2013) Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov 3 (7): 742–750.
Nathanson KL, Martin AM, Wubbenhorst B, Greshock J, Letrero R, D'Andrea K, O'Day S, Infante JR, Falchook GS, Arkenau HT, Millward M, Brown MP, Pavlick A, Davies MA, Ma B, Gagnon R, Curtis M, Lebowitz PF, Kefford R, Long GV (2013) Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin Cancer Res 19 (17): 4868–4878.
Nissan MH, Pratilas CA, Jones AM, Ramirez R, Won H, Liu C, Tiwari S, Kong L, Hanrahan AJ, Yao Z, Merghoub T, Ribas A, Chapman PB, Yaeger R, Taylor BS, Schultz N, Berger MF, Rosen N, Solit DB (2014) Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 74 (8): 2340–2350.
Ong CC, Jubb AM, Jakubiak D, Zhou W, Rudolph J, Haverty PM, Kowanetz M, Yan Y, Tremayne J, Lisle R, Harris AL, Freidman LS, Belvin M, Middleton MR, Blackwood EM, Koeppen H, Hoeflich KP (2013) P21-activated kinase 1 (PAK1) is a therapeutic target in BRAF wild-type melanoma. J Natl Cancer Inst 105 (9): 606–607.
Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503 (7477): 548–551.
Paraiso KH, Fedorenko IV, Cantini LP, Munko AC, Hall M, Sondak VK, Messina JL, Flaherty KT, Smalley KS (2010) Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer 102 (12): 1724–1730.
Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson AR, Ribas A, Palma MD, Nathanson KL, Koomen JM, Messina JL, Smalley KS (2011) PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res 71 (7): 2750–2760.
Paraiso KHT, Haarberg HE, Wood E, Rebecca VW, Chen YA, Xiang Y, Ribas A, Lo RS, Weber JS, Sondak VK, John JK, Sarnaik AA, Koomen JM, Smalley KSM (2012) The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clin Cancer Res 18 (9): 2502–2514.
Posch C, Moslehi H, Feeney L, Green GA, Ebaee A, Feichtenschlager V, Chong K, Peng L, Dimon MT, Phillips T, Daud AI, McCalmont TH, LeBoit PE, Ortiz-Urda S (2013) Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci USA 110 (10): 4015–4020.
Rebecca VW, Massaro RR, Fedorenko IV, Sondak VK, Anderson AR, Kim E, Amaravadi RK, Maria-Engler SS, Messina JL, Gibney GT, Kudchadkar RR, Smalley KS (2014a) Inhibition of autophagy enhances the effects of the AKT inhibitor MK-2206 when combined with paclitaxel and carboplatin in BRAF wild-type melanoma. Pigment Cell Melanoma Res 27 (3): 465–478.
Rebecca VW, Wood ER, Fedorenko IV, Paraiso KH, Haarberg HE, Chen Y, Xiang Y, Sarnaik A, Gibney GT, Sondak VK, Koomen JM, Smalley KS (2014b) Evaluating melanoma drug response and therapeutic escape with quantitative proteomics. Mol Cell ProteomicsMCP 13 (7): 1844–1854.
Ribas A, Gonzalez R, Pavlick A, Hamid O, Gajewski TF, Daud A, Flaherty L, Logan T, Chmielowski B, Lewis K, Kee D, Boasberg P, Yin M, Chan I, Musib L, Choong N, Puzanov I, McArthur GA (2014) Combination of vemurafenib and cobimetinib in patients with advanced BRAF-mutated melanoma: a phase 1b study. Lancet Oncol 15 (9): 954–965.
Richardson PG, Chanan-Khan AA, Lonial S, Krishnan AY, Carroll MP, Alsina M, Albitar M, Berman D, Messina M, Anderson KC (2011) Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: results of a phase 1/2 study. Br J Haematol 153 (6): 729–740.
Robert C, Dummer R, Gutzmer R, Lorigan P, Kim KB, Nyakas M, Arance A, Liszkay G, Schadendorf D, Cantarini M, Spencer S, Middleton MR (2013) Selumetinib plus dacarbazine versus placebo plus dacarbazine as first-line treatment for BRAF-mutant metastatic melanoma: a phase 2 double-blind randomised study. Lancet Oncol 14 (8): 733–740.
Salama AK, Flaherty KT (2013) BRAF in melanoma: current strategies and future directions. Clin Cancer Res 19 (16): 4326–4334.
Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ (2008) Rac activation and inactivation control plasticity of tumor cell movement. Cell 135 (3): 510–523.
Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, Kelley MC, Kefford RF, Chmielowski B, Glaspy JA, Sosman JA, van Baren N, Long GV, Ribas A, Lo RS (2014) Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 4 (1): 80–93.
Smalley KS, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M (2006) Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther 5 (5): 1136–1144.
Solit DB, Rosen N (2011) Resistance to BRAF inhibition in melanomas. New Engl J Med 364 (8): 772–774.
Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, Kester M, Sandirasegarane L, Robertson GP (2004) Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res 64 (19): 7002–7010.
Su Y, Vilgelm AE, Kelley MC, Hawkins OE, Liu Y, Boyd KL, Kantrow S, Splittgerber RC, Short SP, Sobolik T, Zaja-Milatovic S, Dahlman KB, Amiri KI, Jiang A, Lu P, Shyr Y, Stuart DD, Levy S, Sosman JA, Richmond A (2012) RAF265 inhibits the growth of advanced human melanoma tumors. Clin Cancer Res 18 (8): 2184–2198.
Tsao H, Goel V, Wu H, Yang G, Haluska FG (2004) Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol 122 (2): 337–341.
Tworkoski K, Singhal G, Szpakowski S, Zito CI, Bacchiocchi A, Muthusamy V, Bosenberg M, Krauthammer M, Halaban R, Stern DF (2011) Phosphoproteomic screen identifies potential therapeutic targets in melanoma. Mol Cancer Res 9 (6): 801–812.
Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Che H-Y, Swoboda R, Wilson M, Vultur A, Fukunaga-Kalabis M, Wubbenhorst B, Chen TY, Liu Q, Sproesser K, DeMarini DJ, Gilmer TM, Martin A, Marmorstein R, Schultz DC, Speicher D, Karakousis GC, Xu W, Amavaradi RK, Xu X, Schuchter L, Herlyn M, Nathanson KL (2013) Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep 4 (6): 1090–1099.
Vu HL, Aplin AE (2014) Targeting TBK1 inhibits migration and resistance to MEK inhibitors in mutant NRAS melanoma. Mol Cancer Res; e-pub ahead of print 24 June 2014.
Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC, Meyerson M, Garraway LA (2011) Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol 29 (22): 3085–3096.
Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, Friedrich DC, Anderka K, Perrin D, Johannessen CM, McKenna A, Cibulskis K, Kryukov G, Hodis E, Lawrence DP, Fisher S, Getz G, Gabriel SB, Carter SL, Flaherty KT, Wargo JA, Garraway LA (2014) MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov 4 (1): 61–68.
Weisberg E, Nonami A, Chen Z, Liu F, Zhang J, Sattler M, Nelson E, Cowens K, Christie AL, Mitsiades C, Wong K-K, Liu Q, Gray N, Griffin JD (2014) Identification of Wee1 as a novel therapeutic target for mutant RAS-driven acute leukemia and other malignancies. Leukemia; e-pub ahead of print 5 May 2014; doi:10.1038/leu.2014.149.
Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS, Schadendorf D, Root DE, Garraway LA (2012) A genome-scale RNA interference screen implciates NF1 loss in resistance to RAF inhibition. Cancer Discov 3 (3): 350–362.
Wilson MA, Zhao F, Letrero R, D'Andrea K, Rimm DL, Kirkwood JM, Kluger HM, Lee SJ, Schuchter LM, Flaherty KT, Nathanson KL (2014) Correlation of somatic mutations and clinical outcome in melanoma patients treated with carboplatin, paclitaxel, and sorafenib. Clin Cancer Res 20 (12): 3328–3337.
Wu X, Marmarelis ME, Hodi FS (2013) Activity of the heat shock protein 90 inhibitor ganetespib in melanoma. PLoS ONE 8 (2): e56134.
Xia J, Jia P, Hutchinson KE, Dahlman KB, Johnson D, Sosman J, Pao W, Zhao Z (2014) A meta-analysis of somatic mutations from next generation sequencing of 241 melanomas: a road map for the study of genes with potential clinical relevance. Mol Cancer Ther 13 (7): 1918–1928.
Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PI, Waldmann H (2013) Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 497 (7451): 638–642.
Acknowledgements
Work in the Smalley lab is supported by R01 CA161107-01 and SPORE grant P50 CA168536-01A1 from the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Fedorenko, I., Gibney, G., Sondak, V. et al. Beyond BRAF: where next for melanoma therapy?. Br J Cancer 112, 217–226 (2015). https://doi.org/10.1038/bjc.2014.476
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.476
- Springer Nature Limited
Keywords
This article is cited by
-
Advances in Diagnosis and Treatment for Leptomeningeal Disease in Melanoma
Current Oncology Reports (2022)
-
Molekulargenetische Diagnostik – zielgerichtete Therapie des malignen Melanoms
Der Onkologe (2018)
-
Treatment of melanoma with selected inhibitors of signaling kinases effectively reduces proliferation and induces expression of cell cycle inhibitors
Medical Oncology (2018)
-
Melanoma cells replicate through chemotherapy by reducing levels of key homologous recombination protein RAD51 and increasing expression of translesion synthesis DNA polymerase ζ
BMC Cancer (2017)
-
MALT1 promotes melanoma progression through JNK/c-Jun signaling
Oncogenesis (2017)