
Abstract
Aim:
Pim-2 is a short-lived serine/threonine kinase, which plays a key role in metastasis of breast cancer through persistent activation of STAT3. Although the crystal structure of Pim-2 has been reported, but thus far no specific Pim-2-targeted compounds have been reported. In this study, we identified a novel Pim-2 inhibitor, HJ-PI01, by in silico analysis and experimental validation.
Methods:
The protein-protein interaction (PPI) network, chemical synthesis, molecular docking, and molecular dynamics (MD) simulations were used to design and discover the new Pim-2 inhibitor HJ-PI01. The anti-tumor effects of HJ-PI01 were evaluated in human breast MDA-MB-231, MDA-MB-468, MDA-MB-436, MCF-7 cells in vitro and in MDA-MB-231 xenograft mice, which were treated with HJ-PI01 (40 mg·kg−1·d−1, ig) with or without lienal polypeptide (50 mg·kg−1·d−1, ip) for 10 d. The apoptosis/autophage-inducing mechanisms of HJ-PI01 were elucidated using Western blots, immunoblots, flow cytometry, transmission electron microscopy and fluorescence microscopy.
Results:
Based on the PrePPI network, the potential partners interacting with Pim-2 in regulating apoptosis (160 protein pairs) and autophagy (47 protein pairs) were identified. Based on the structural characteristics of Pim-2, a total of 15 compounds (HJ-PI01 to HJ-P015) were synthesized, which showed moderate or remarkable anti-proliferative potency in the human breast cancer cell lines tested. The most effective compound HJ-PI01 exerted a robust inhibition on MDA-MB-231 cells compared with chlorpromazine and the pan-Pim inhibitor PI003. Molecular dynamics (MD) simulation revealed that HJ-PI01 had a good binding score with Pim-2. Moreover, HJ-PI01 (300 nmol/L) induced death receptor-dependent and mitochondrial apoptosis as well as autophagic death in MDA-MB-231 cells. In MDA-MB-231 xenograft mice, administration of HJ-PI01 remarkably inhibited the tumor growth and induced tumor cell apoptosis in vivo. Co-administration of HJ-PI01 with lienal polypeptide could improve the anti-tumor activity of HJ-PI01 and reduce its toxicity.
Conclusion:
The newly synthesized compound, HJ-PI01, can induce death receptor/mitochondrial apoptosis and autophagic cell death by targeting Pim-2 in human breast cancer cells in vitro and in vivo.
Similar content being viewed by others


Introduction
The proviral insertion sites in Moloney murine leukemia virus (Pim) proteins are well-known to be a family of short-lived serine/threonine kinases that are highly evolutionarily conserved in multicellular organisms1. They belong to the Ca2+/calmodulin-dependent protein kinase group. Pim has three members, Pim-1, Pim-2 and Pim-3, which show high homology with one another. Pim-1 and Pim-3 are 71% identical at the amino acid level, while Pim-1 and Pim-2 share 61% homology2. Elevated expression of the Pim kinases has been detected in hematologic malignancies and certain solid tumors3.
Regarding cancer biology, Pim family members are weak oncogenes that can contribute to tumorigenesis by selectively enhancing the cells' tumorigenic capabilities4,5. Pim kinases are overexpressed in solid tumors, such as colon cancer6, prostate cancer7 and hematologic malignancies, including lymphomas8,9, chronic lymphocytic leukemia (CLL) and acute leukemia10,11. Pim kinases are an active target for drug discovery research. Due to the known implications of Pim-1 in tumorigenesis, most compounds being examined focused on this kinase, but a few are focused on Pim-2 and Pim-3. Pim-2 is a key mechanism of metastasis in breast cancer through the persistent activation of STAT3, which may lead to the epithelial–mesenchymal transition (EMT)12. Nevertheless, once Pim-1 was inhibited, Pim-2 expression was increased. Moreover, the crystal structure of Pim-2 has been reported, but no specific Pim-2-targeted compounds have been reported to date. Pim-2 has been proposed to mediate the anti-apoptotic properties of oncogenes, such as BCR/ABL, FLT3 and Jak2 mutants13,14,15,16,17,18. To compensate for the loss of Pim-1, Pim-2 expression appears to be a later event in MMLV-induced lymphomas19,20. Thus, the discovery of drugs that target both Pim-2 and Pim-1 is very important and necessary.
Previous reports showed that the phenothiazine derivative chlorpromazine, a widely used neuroleptic drug for the control of psychosis, inhibits the proliferation of various tumor cells21. The anti-proliferative effect of chlorpromazine has been linked to mitotic arrest through the inhibition of the motor activity of the mitotic kinesin KSP/Eg5 in HCT116 and A549 cells and the induction of apoptosis-independent autophagic cell death via the Beclin-1 dependent pathway in PTEN-null U-87MG glioma cells22,23. Chlorpromazine also inhibited intracranial xenograft tumor growth in vivo. A previous study reported that a newly synthesized pan-Pim inhibitor, PI003, was obtained by modifying the chemical structure of chlorpromazine24. PI003 induced apoptosis via the death receptor and mitochondrial pathways by targeting Pim-1, Pim-2 and Pim-3 in HeLa cells. Similarly to most pan-Pim inhibitors, the efficacy of PI003 against Pim-2 was relatively poor.
In this study, we identified a newly synthesized Pim-2 inhibitor, HJ-PI01, by in silico analysis and experimental validation. We first constructed the human protein-protein interaction (PPI) network and modified it as an autophagy- and apoptosis-related PPI of Pim-2 to reveal its mechanisms. Subsequently, we synthesized and screened a candidate compound, HJ-PI01, based on the structural characteristics of Pim-2. The apoptosis- and autophagy-inducing efficacies of HJ-PI01 were verified through in vitro experiments. Then, iTRAQ and MS/MS analysis were performed to profile the differentially expressed proteins in HJ-PI01-treated MDA-MB-231 cells to explore the mechanisms of the anti-tumor efficacy of HJ-PI01. Altogether, we discovered and identified HJ-PI01 as a new compound that targets Pim-2, which sheds new light on breast cancer therapy with regard to Pim-2.
Materials and methods
Reagents
The chemicals, solvents, dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), HEPES, Triton X-100, sodium orthovanadate, sodium fluoride, edetic acid, PMSF, leupeptin and Tween-20 were purchased from Sigma–Aldrich (St Louis, MO, USA). The Dulbecco's modified Eagle's medium (DMEM), primary fibroblast cell culture medium, fetal bovine serum (FBS), trypsin/EDTA (TE) and TRIzol, as well as other cell culture plates, were obtained from Fisher Scientific (Pittsburgh, PA, USA). The DC protein assay was purchased from Bio-Rad (Bio-Rad Laboratories, Hercules, CA, USA). The TUNEL assay and Annexin V-FLUOS Staining Kit were supplied by Roche (Mannheim, Germany). The Avidin-biotin-HRP complex was purchased from Thermo (Fremont, CA, USA). All of the antibodies used in the experiments were purchased from Cell Signaling Technology (Danvers, MA, USA) and Abcam (Cambridge, UK). The lienal polypeptides were purchased from the Fengsheng Pharmaceutical Company (Jilin, China).
Network construction
To construct the Pim-2 network, we modified the global human PPI network from PrePPI, which is a database of predicted and experimentally determined human protein-protein interactions (PPIs). The predicted interactions are assigned a likelihood using a Bayesian framework that combines structural, functional, evolutionary and expression information. The unified conceptual framework of the PPI network was integrated with Cytoscape. Subsequently, we built the autophagy- and apoptosis-related PPI network of Pim-2 using the Gene Ontology (GO) consortium with the condition that many proteins that interacted with Pim-2 are involved in autophagy or apoptosis and are differentially expressed in the iTRAQ analysis.
Molecular docking
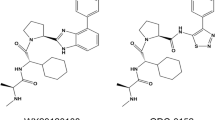
The initial three-dimensional geometric coordinates of the X-ray crystal structure of Pim-2 was retrieved from the RCSB Brookhaven Protein Data Bank (PDB) (http://www.rcsb.org/pdb/home/home.do) (PDB entry: 2IWI). Then, we used the Accelrys Discovery Studio version 3.5 with CHARMm force-field parameters to dock pre-generated conformations of drugs into Pim-2 to virtually screen modified inhibitors. We performed flexible-ligand docking to a rigid receptor with LibDock, in which the drugs were allowed to be flexible and structurally rearranged in response to Pim-2. The results were re-ranked by CDOCKER. HJ-PI01 was selected based on its scores and structure. Using the Discovery Studio program, we virtually modified chlorpromazine into HJ-PI01, which exhibits an obvious increase in every score.
Chemical synthesis
All reactions requiring anhydrous conditions were performed under an Ar or N2 atmosphere. The chemicals and solvents were either AR grade or purified by standard techniques. Thin layer chromatography (TLC) used silica gel plates GF254; the compounds were visualized by irradiation with UV light and/or by treatment with a solution of phosphomolybdic acid (20% wt in ethanol), followed by heating. Column chromatography was performed using silica gel with the eluent indicated in parentheses. 1H-NMR and 13C-NMR were performed on a Bruker Avance III 400 MHz spectrometer (Bruker, Billerica, MA, USA) using CDCl3 or DMSO-d6 as a solvent at room temperature. The chemical shifts are expressed relative to TMS (=0 ppm), and the coupling constants J are expressed in Hz. The purity of the compound screened in the biological assays was determined to be ≥97% by HPLC (Agilent 1100 HPLC system) with a photodiode array detector. An Atlantis C18 (150 mm×4.6 mm, id 5 μm) (Waters, Milford, MA, USA) was used with a gradient elution of methanol and HPLC-grade water as the mobile phase at a flow rate of 1 mL/min. The HRMS data were obtained using a Bruker micro-TOF-Q instrument or a TOF-MS instrument (Bruker, Billerica, MA, USA).
Chemical data of the synthetic compounds
HJ-PI01
1H-NMR (400 MHz, DMSO-d6) δ 7.59 (s, 2H), 7.23–7.12 (m, 4H), 7.09 (s, 2H), 2.08 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 170.35, 150.17, 126.73, 126.19, 124.47, 124.29, 117.25, 26.13.
HJ-PI02
1H-NMR (400 MHz, DMSO-d6) δ 7.18–7.10 (m, 1H), 7.01 (dd, J=6.3, 3.8 Hz, 1H), 6.98 (t, J=2.2 Hz, 2H), 6.94 (d, J=5.1 Hz, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 141.93 (s), 133.76, 123.37, 122.09, 114.95, 114.49.
HJ-PI03
1H-NMR (400 MHz, DMSO-d6) δ 7.67–7.54 (m, 2H), 7.20–7.11 (m, 4H), 7.09 (s, 2H), 1.42 (s, 9H). 13C-NMR (100 MHz, DMSO-d6) δ 150.24, 146.34, 129.44, 126.57, 125.14, 121.59, 113.67, 81.20, 28.33.
HJ-PI04
1H-NMR (400 MHz, DMSO-d6) δ 7.19–7.06 (m, 2H), 7.06–6.96 (m, 2H), 6.96–6.88 (m, 2H), 6.85 (d, J=1.6 Hz, 2H), 2.28 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 142.61, 141.94, 135.52, 133.76, 131.62, 123.89, 123.37, 122.09, 116.43, 114.95, 114.49, 114.02, 21.23.
HJ-PI05
1H-NMR (400 MHz, DMSO-d6) δ 7.72–7.48 (m, 2H), 7.26–7.08 (m, 2H), 7.15–6.97 (m, 4H), 2.28 (s, 3H), 2.08 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 170.34, 150.16, 142.34, 138.91, 126.73, 126.20, 125.48, 124.47, 124.29, 124.20, 123.62, 117.25, 117.10, 26.14, 21.23.
HJ-PI06
1H-NMR (400 MHz, DMSO-d6) δ 7.70–7.48 (m, 2H), 7.23–7.05 (m, 3H), 7.04–6.94 (m, 2H), 2.28 (s, 3H), 1.42 (s, 9H). 13C-NMR (100 MHz, DMSO-d6) δ 150.24, 146.34, 143.40, 140.80, 129.44, 128.44, 126.78, 126.57, 125.22, 125.14, 121.59, 113.67, 113.10, 81.20, 28.33, 21.23.
HJ-PI07
1H-NMR (400 MHz, DMSO-d6) δ 7.29–7.09 (m, 2H), 7.09–6.99 (m, 2H), 6.95 (ddd, J=25.7, 11.6, 8.2 Hz, 3H), 6.87–6.72 (m, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 162.00, 159.48, 141.93, 138.22, 138.19, 133.76, 128.77, 128.69, 123.37, 122.09, 115.86, 115.79, 114.95, 114.49, 108.61, 108.41, 101.03, 100.83.
HJ-PI08
1H-NMR (400 MHz, DMSO-d6) δ 7.86–7.45 (m, 2H), 7.32–7.07 (m, 3H), 7.07–6.85 (m, 2H), 2.08 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 170.35, 160.13, 157.61, 150.17, 142.92, 126.73, 126.19, 124.38, 122.04, 119.09, 117.25, 115.37, 110.02, 26.13.
HJ-PI09
1H-NMR (400 MHz, DMSO-d6) δ 7.95–7.46 (m, 2H), 7.29–6.80 (m, 5H), 1.42 (s, 9H). 13C-NMR (100 MHz, DMSO-d6) δ 158.51, 155.99, 150.23, 146.34, 129.44, 126.58, 125.13, 124.63, 121.59 (s), 113.68, 111.39, 81.20, 28.33.
HJ-PI10
1H-NMR (400 MHz, DMSO-d6) δ 7.10 (dd, J=28.0, 24.0 Hz, 2H), 6.95 (d, J=20.0 Hz, 1H), 6.79 (d, J=15.6 Hz, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 163.72, 161.20, 141.93, 141.26, 133.76 (s), 127.74, 123.37, 122.09, 116.71, 114.95, 114.49, 109.13, 108.92, 101.44.
HJ-PI11
1H-NMR (400 MHz, DMSO-d6) δ 7.60 (ddd, J=15.1, 10.3, 7.3 Hz, 2H), 7.42–7.10 (m, 3H), 7.15–6.68 (m, 3H), 2.08 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 170.35, 167.23, 164.70, 150.17, 127.09, 126.19 (s), 124.38, 117.25, 112.43, 26.13.
HJ-PI12
1H-NMR (400 MHz, DMSO-d6) δ 7.77–7.35 (m, 2H), 7.32–7.05 (m, 4H), 7.07–6.76 (m, 1H), 1.42 (s, 9H). 13C-NMR (100 MHz, DMSO-d6) δ 166.19, 163.67, 150.23, 146.34, 142.86, 129.44 (s), 127.76, 126.58, 125.13, 122.77, 121.59, 114.41, 113.67, 102.80, 81.20, 28.33.
HJ-PI13
1H-NMR (400 MHz, DMSO-d6) δ 7.24 (dd, J=16.0, 2.9 Hz, 1H), 7.16–6.91 (m, 2H), 6.97–6.54 (m, 5H), 2.28 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 162.00, 159.48, 142.61, 138.20, 135.52 (s), 131.63, 128.73, 123.89, 116.43, 115.83, 114.02, 108.51, 100.93, 21.23.
HJ-PI14
1H-NMR (400 MHz, DMSO-d6) δ 7.71 (dd, J=16.0, 2.8 Hz, 1H), 7.56 (dt, J=2.7, 1.7 Hz, 1H), 7.24–6.84 (m, 4H), 2.28 (s, 3H), 2.08 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 170.35, 160.13, 157.61, 142.92, 142.35, 138.91, 125.48, 124.20, 123.62, 122.04, 119.09, 117.10, 115.37, 110.02, 26.13, 21.23.
HJ-PI15
1H-NMR (400 MHz, DMSO-d6) δ 7.71 (dd, J=8.0, 1.4 Hz, 1H), 7.56 (d, J=7.4 Hz, 1H), 7.26–6.76 (m, 4H), 2.28 (s, 3H), 1.42 (s, 9H). 13C-NMR (100 MHz, DMSO-d6) δ 158.51, 155.99, 150.23, 143.40, 142.66, 140.80, 128.45, 126.78, 125.22, 124.63, 113.66, 113.10, 112.64, 112.44, 111.39, 81.20 (s), 28.33, 21.23.
Cell culture and the MTT assay
MDA-MB-231 cells, MDA-MB-468 cells, MDA-MB-436 cells and MCF-7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM), and HUM-CELL-0056 cells were cultured in primary fibroblast cell culture medium with 10% (v/v) heat-inactivated fetal bovine serum and incubated in a humidified incubator with 5% CO2. The cells were transferred into 96-well plates at a density of approximately 5.0×104 cells/mL. The cytotoxicity of the test compounds was measured using the MTT assay as follows: MDA-MB-231 cells, MDA-MB-468 cells, MDA-MB-436 cells and MCF-7 cells were cultured in DMEM, and HUM-CELL-0056 cells were cultured in primary fibroblast cell culture medium in the presence of a test compound for 24 h, and then 10 μL of MTT (5 mg/mL) were added to the cells in each well. After 4 h of culture, the medium was removed, and the blue formazan crystals that had formed were dissolved in dimethyl sulfoxide. The absorbance of the formazan product generated from the MTT was measured at 570 nm (Bio-Rad Model 680, Bio-Rad, Hercules, CA, USA).
Molecular dynamics (MD) simulations
MD simulations were performed with the GROMACS (version 4.5.5) software package to monitor the binding interactions between Pim-2 and HJ-PI0125. The topological parameters of the ligands were constructed with the Dundee PRODRG server26. The topology of Pim-2 was edited with Amber force field 99SB, and the small molecule was edited with Amber general force field. The complex was immersed in a cubic box of simple point charge (SPC) water molecules. Eight and eleven sodium counter-ions were added by replacing the water molecules to ensure the overall charge neutrality of the simulated receptor system. In this MD process, 200 ps simulations with a time step of 1.2 ns were performed, and the resulting trajectory files were viewed and analyzed using the VMD software.
Autophagy assay
An electron microscopy analysis (Hitachi 7000, Tokyo, Japan) was performed to observe the autophagic vacuoles. The cells were transfected with the GFP-LC3 plasmid using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. The fluorescence of GFP-LC3 was observed under a fluorescence microscope. 3-Methyl adenine (3-MA) is an established, selective inhibitor of the Class III PI3 kinase Vps34 and is a reversible inhibitor of autophagy that blocks the recruitment of lipids to the autophagosome. We investigated the effect of 3-MA to determine whether HJ-PI01-induced cell death involves an autophagic mechanism.
Apoptosis assay
The morphology of the cells undergoing apoptosis was observed under a phase-contrast microscope. For the Annexin V/PI staining assay, the cells were stained with the Annexin V-FLUOS Staining Kit according to the manufacturer's protocol (Roche, Mannheim, Germany) and observed under a fluorescence microscope. To measure the ratio of apoptotic cells, the cells were collected and fixed with 500 μL of PBS and 10 mL of 70% ethanol overnight at 4 °C; then, after washing twice with PBS, the cells were incubated with 1 mL of PI staining solution (50 mg/L PI and 1 g/L RNase A) for 30 min at 4 °C and measured by flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA).
iTRAQ and LC-MS analysis
The ER/microsomal vesicle-enriched fractions of MDA-MB-231 cells treated with or without HJ-PI01 (300 nmol/L HJ-PI01 for 24 h) were prepared using a method similar to that in a previous report27. Briefly, the proteins were digested with trypsin (10:1) overnight at 37 °C. The resulting peptides were subsequently labeled using an iTRAQ 4-plex kit (AB SCIEX, Dorchester, England) according to the manufacturer's instructions. To ensure reproducibility and exclude labeling bias, each pair of protein samples (control and HJ-PI01-treated) were labeled with two different labeling strategy. The digested samples were labeled with different iTRAQ tags as follows: control, iTRAQ 114 and HJ-PI01-treated, iTRAQ 115. After labeling, the samples were pooled and dried. For the sample fraction, the iTRAQ-labeled samples were reconstituted using a strong cation exchange (SCX) buffer A (10 mmol/L monobasic potassium phosphate, pH 2.75, and 25% acetonitrile), and the pH values of the samples were adjusted to 2.5–3 with phosphoric acid. For the LC-MS analysis, the samples were reconstituted in 0.1% formic acid. The MS analysis was performed on the TripleTOF 5600 system in the information-dependent acquisition mode. The mass spectrometry data files were processed by Protein Pilot 4.0 (AB SCIEX, Dorchester, UK) using the Paragon algorithm. The mass spectrometry data were searched against all Homo sapiens protein sequences in the Uniprot_sprot_201105 database. In the present study, only proteins whose four peak area ratios between the HJ-PI01-treated cells and control cells were >1.2 or <0.8 were accepted as differentially expressed proteins.
Western blot analysis
The MDA-MB-231 cells were treated with 300 nmol/L HJ-PI01 for 0, 12, 24, 36 and 48 h. Both the adherent and floating cells were collected. The cell pellets were resuspended in a lysis buffer, consisting of 50 mmol/L HEPES, pH 7.4, 1% Triton X-100, 2 mmol/L sodium orthovanadate, 100 mmol/L sodium fluoride, 1 mmol/L edetic acid, 1 mmol/L PMSF, 10 mg/L aprotinin (Sigma) and 10 mg/L leupeptin (Sigma), and lysed for 1 h at 4 °C. After centrifugation at 12 000 rounds per minute for 15 min, the protein content of the supernatant was determined using the Bio-Rad DC protein assay. Equal amounts of total protein were separated on 12% SDS-PAGE gels and transferred to nitrocellulose membranes; the membranes were soaked in blocking buffer (5% skimmed milk). The proteins were detected using polyclonal antibodies and visualized using anti-rabbit or anti-mouse IgG conjugated with peroxidase (HRP), and 3,3′-diaminobenzidine tetrahydrochloride (DAB) was used as the HRP substrate.
Animal model
The experimental protocol was approved by the Animal Experimental Ethics Committee of Shenyang Pharmaceutical University (Permit Number: SYPU-IACUC-C2015-0721-001). Healthy female nude mice (BALB/c, 6–8 weeks of age, non-fertile, 18–20 g of weight) were subcutaneously injected with MDA-MB-231 cells (1×107 cells/mouse). When the tumors reached 100 mm3 in volume (calculated as V=L×W×W/2), the mice were treated with hydroxypropyl-cyclodextrin (vehicle control), HJ-PI01 (40 mg·kg−1·d−1, po), or HJ-PI01 (40 mg·kg−1·d−1, po) + the lienal polypeptide (LP) (50 mg·kg−1·d−1, ip). The animals were euthanized after 10 d. The tumor tissues were isolated and frozen in liquid nitrogen or immediately fixed in formalin.
Immunohistochemical analysis of KI67 and the TUNEL assay
The tumor tissues obtained from the in vivo studies were rinsed with PBS and fixed in 4% paraformaldehyde. The samples were dehydrated in a gradient ethanol series, embedded in paraffin, and sectioned (5 μm). The deparaffinized sections were stained with KI67 antibodies. The samples were incubated overnight with biotinylated secondary antibodies. The antibodies were detected with an avidin-biotin-HRP complex (Thermo Scientific, Fremont, CA, USA) and DAB as the chromogen. The nuclei were counterstained with hematoxylin. For the TUNEL assay, the sections were permeabilized with 0.1% Triton X-100 plus 0.1% sodium citrate and then incubated with 50 μL of the TUNEL reaction mixture (Roche) for 60 min at 37 °C. After rinsing with PBS three times, 50 μL of converter-POD were added, and the tissues were incubated in a humidified chamber for 30 min at 37 °C. DAB substrate was then added, followed by counterstaining with hematoxylin. The positive cells were counted in six fields per tumor sample. The results are expressed as the average±SD of the tumors in each group.
Statistical analysis
All of the presented data and results were confirmed in at least three independent experiments. The data are expressed as the mean±SD. Statistical comparisons were made by one-way ANOVA or Student's t-test. P<0.05 was considered statistically significant.
Results
Network construction
Based on the PrePPI, we selected 602 proteins that interacted with Pim-2 (Supplementary Table S1). Subsequently, we identified the autophagy-related proteins (47 protein pairs) and apoptosis-related proteins (160 protein pairs) by functional annotation, which were used to construct the pathways of Pim-2 (Figure 1A). This result showed the potential mechanisms of Pim-2 in autophagy and apoptosis. A variety of sequence alignments were utilized to further characterize the sequence information in one cluster that included Pim-1, Pim-2 and Pim-3 (Figure 1B). The amino acid sequence of Pim-2 displayed important differences in the residues in the ATP binding pocket, LYS40, THR45, VAL62, LEU120 and ALA122, which may influence the effect of the inhibitor on Pim-2 in the subsequent docking studies.

The PPI network of Pim-2 and sequence alignment of the kinase domains of the Pim family. (A) All of the proteins that interacted with Pim-2 are listed. The red, filled circles represent apoptosis-related proteins, and the green, filled circles represent autophagy-related proteins. Moreover, the green circles with the red border represent apoptosis- and autophagy-related proteins. (B) Sequence alignment of the kinase domains of Pim-1, Pim-2 and Pim-3. The conserved catalytic residues within the binding site were highlighted in the rectangular boxes, and the regions showing the most frequent variation were colored in red.
Chemical synthesis and anti-proliferative effect of HJ-PI01
3-Iodophenol could react with 1-fluoro-2-nitrobenzene in the presence of K2CO3 to produce the Williamson reaction product 1-iodo-2-(2-nitrophenoxy) benzene, and then the product was reduced by Fe-HCl; therefore, the nitro group was converted into an amino moiety. The amino moiety was protected with an acetyl group after treatment with acetic anhydride. The condensation of the intermediate in the presence of DMEDA and K2CO3 in refluxing dioxane gave the adduct, which was finally purified by silica-gel column chromatography using hexane and ethyl acetate as an eluent to obtain the final products HJ-PI01 to HJ-PI15 (Figure 2). The potential anti-proliferative activity of all compounds was tested in MDA-MB-231 cells, MDA-MB-468 cells, MDA-MB-436 cells and MCF-7 cells (Table 1). These compounds generally exhibited moderate or remarkable anti-proliferative potency, and the most effective compound, HJ-PI01, displayed excellent inhibitory activity of up to 76.5% at a 1 μmol/L concentration in MDA-MB-231 cells. Thus, we used MDA-MB-231 cells for the subsequent experiments. According to the molecular docking results with Pim-2, we selected HJ-PI01 for the in vitro cellular examination. To examine the effect of HJ-PI01 on MDA-MB-231 cell proliferation in comparison with chlorpromazine and the pan-Pim inhibitor PI003, the MTT assay was performed to measure the inhibition of MDA-MB-231 cell proliferation following treatment with different concentrations of HJ-PI01, chlorpromazine and PI003. HJ-PI01 remarkably inhibited MDA-MB-231 cell growth in a dose-dependent manner, and treatment with 300 nmol/L of HJ-PI01 for 24 h resulted in almost 50% inhibition of MDA-MB-231 cell growth. In contrast, 750 nmol/L chlorpromazine and 460 nmol/L PI003 were required to inhibit cell growth (Figure 3A). The results showed that HJ-PI01 exhibited a substantial improvement in inhibiting MDA-MB-231 cell growth compared with chlorpromazine and PI003. We also determined the concentration-dependent effects of HJ-PI01 on normal, non-cancer HUM-CELL-0056 cells (Human Cardiac Fibroblast Cells) (Figure 3B). The 300 nmol/L HJ-PI01 treatment resulted in less than 10% inhibition of cell growth at 24 h, indicating HJ-PI01 has very weak toxicity in normal non-cancer cells.

Chemical synthesis of HJ-PI01. 3-iodophenol could react with 1-fluoro-2-nitrobenzene in the presence of K2CO3 to produce the Williamson reaction product 1-iodo-2-(2 nitrophenoxy) benzene, and then the product was reduced by Fe-HCl. The amino group was protected with an acetyl moiety after treatment with acetic anhydride. The condensation of the intermediate in the presence of DMEDA and K2CO3 in refluxing dioxane yielded the adducts HJ-PI01 to HJ-PI15.

Cell proliferation rate of MDA-MB-231 cells and the effect of HJ-PI01 on Human Cardiac Fibroblast Cells, as measured by the MTT assay. (A) The cells were treated with different concentrations of HJ-PI01 or chlorpromazine for the indicated times, and the data were representative of three independent experiments. HJ-PI01 exhibited a 1.56-fold greater inhibitory effect on the MDA-MB-231 cells than chlorpromazine. (B) The HUM-CELL-0056 cells were treated with various concentrations of HJ-PI01, 100, 200, 300, and 400 nmol/L. HJ-PI01 had a slight effect on HUM-CELL-0056 cells.
Molecular docking and molecular dynamics (MD) simulations of the HJ-PI01-Pim-2 complex
In the molecular docking assay, we found that a hydrogen bond formed between HJ-PI01 and the ILE164 residue of Pim-2, whereas the other part of the compound formed a hydrophobic interaction with the TYR203 residue of Pim-2. This binding model showed that HJ-PI01 could influence the ATP binding pocket to inhibit Pim-2. Thus, HJ-PI01 had a good binding score with Pim-2 (Figure 4A).

Molecular modeling study of the HJ-PI01 and Pim-2. (A) Cartoon showing HJ-PI01 docked into the ATP-binding site of Pim-2. (B) Time-dependence RMSDs of Pim-2 in 1.2 ns MD simulations.
The MD simulations of the HJ-PI01-Pim-2 complex were performed for 1.2 ns. The RMSD values of the Pim-2 backbone atoms in the initial, minimized structure, which are plotted in Figure 4B, were calculated through the phase of the simulation to evaluate the relative stability of the MD trajectories and the difference in the stabilities in the MD simulations. The HJ-PI01-Pim-2 complex reached equilibrium after 1 ns of the simulation phase. As shown in Figure 4B, the RMSD value for the HJ-PI01-Pim-2 complex was 0.4 Å, with less than 0.35 Å deviation. The study demonstrated that the trajectories of the MD simulations of the complex were stable after 1 ns. Based on the results of the MD simulations, the compound HJ-PI01 directly targeted the ATP-binding site of Pim-2 and stably interacted with Pim-2. Considering that Pim-2 contributes to tumorigenesis, it is reasonable to speculate that the Pim-2-targeting compound HJ-PI01 is a promising clinical agent for anti-tumor drug discovery.
HJ-PI01 induces autophagic death in MDA-MB-231 cells
As previous evidence has highlighted the important roles of autophagy in the Pim-2-regulated network in breast cancer, we explored whether autophagy is induced and required for the HJ-PI01-mediated inhibition of cell proliferation. The formation of autophagic vacuoles was assessed by transmission electron microscopy. In contrast to the normal morphology of the control cells, the HJ-PI01-treated cells showed typical characteristics of autophagy, such as the presence of autophagic vacuoles that contained cellular organelles (Figure 5A). The HJ-PI01 treatment resulted in extensive GFP-LC3 localization, suggesting that HJ-PI01 induces MDA-MB-231 cell autophagy (Figure 5B). Additionally, HJ-PI01 induced a time-dependent increase in the accumulation of the LC3-II form in MDA-MB-231 cells (Figure 5C). Moreover, the results of the Western blots also displayed a time-dependent increase in the Beclin-1 levels, which indicated that autophagy was activated. In addition, to examine whether HJ-PI01 could induce autophagic degradation, the steady-state levels of p62, which is associated with LC3 turnover and is degraded through the autophagic pathway, were analyzed by Western blot analysis. As expected, the HJ-PI01 treatment also induced p62 degradation in a time-dependent manner. To further confirm the role of HJ-PI01-induced autophagy, an autophagy inhibitor, 3-MA, was applied in combination with HJ-PI01. We found that treatment with 3-MA increased cell viability compared to the cells treated with HJ-PI01 alone (Figure 5D). These results suggested that HP-PI01 induces autophagic cell death in MDA-MB-231 cells.

HJ-PI01 induces autophagy in MDA-MB-231 cells. (A) The cellular ultrastructure was examined by transmission electron microscopy. (B) The MDA-MB-231 cells were transfected with GFP-LC3 and then treated with 300 nmol/L HJ-PI01 for 24 h; the increase in the number of GFP-LC3 puncta was observed under a fluorescence microscope. (C) The cells were treated with 300 nmol/L HJ-PI01 for the indicated time periods, followed by Western blot analysis to detect the LC3, Beclin-1 and p62 levels. β-Actin was used as an equal loading control. (D) The cells were treated with HJ-PI01 (300 nmol/L) for 24 h with or without pre-treatment with the autophagy inhibitor 3-MA (1 mmol/L). The cell viability ratios were measured using the MTT assay.
HJ-PI01 induces death receptor-dependent and mitochondrial apoptosis
Next, we examined the morphological changes in the HJ-PI01-treated MDA-MB-231 cells by phase-contrast microscopy. As shown in the Figure 6A, the control cells displayed a normal cell phenotype. In contrast, the HJ-PI01-treated MDA-MB-231 cells showed typical features of apoptosis, including the obvious appearance of apoptotic bodies. Annexin V, a Ca2+-dependent phospholipid binding protein, has a strong binding affinity for PS. Thus, we used an Annexin V-FITC/PI staining kit to assess HJ-PI01-induced cell apoptosis. When the cells were treated with HJ-PI01, a change in the subcellular localization of phosphatidylserine was observed under a fluorescence microscope, which is a typical feature of apoptosis (Figure 6B). Moreover, apoptosis was further evaluated by measuring the number of cells in the Sub-G1 phase by flow cytometry. As shown in Figure 6C, HJ-PI01 markedly induced an increase in the proportion of Sub-G1 cells, which were stained with PI, suggesting that HJ-PI01 induced DNA damage. Together, these results indicate that HJ-PI01 induces apoptotic cell death in MDA-MB-231 cells.

HJ-PI01 induces apoptosis in MDA-MB-231 cells. (A) The changes in cellular morphology were examined by phase-contrast microscopy. (B) The MDA-MB-231 cells were treated with 300 nmol/L HJ-PI01 for 24 h, and the changes in the subcellular localization of phosphatidylserine were observed under a fluorescence microscope by Annexin V/PI staining. (C) The MDA-MB-231 cells were treated with 300 nmol/L HJ-PI01 for the indicated time periods, and the number of HJ-PI01-treated cells in the Sub-G1 phase was analyzed by flow cytometry. The increased percentage of cells in the Sub-G1 phase showed that HJ-PI01 induced apoptosis.
As shown in Figure 7A, we first examined the expression levels of Fas, FADD and caspase-8 by Western blot analysis to identify the pathway by which HJ-PI01 induced apoptosis. The increased levels of these proteins suggested that the death receptor pathway was activated in HJ-PI01-induced apoptosis. Because Bcl-2 family members play vital roles in regulating the mitochondrial apoptotic pathway, the levels of Bax and Bcl-2 were detected by Western blot analysis. After treatment with HJ-PI01, the level of Bax was increased, whereas the level of Bcl-2 was decreased. Simultaneously, caspase-9 and caspase-3 were also activated by the HJ-PI01 treatment. These results clearly indicate that HJ-PI01-induced apoptosis in MDA-MB-231 cells is mediated by the mitochondrial pathway (Figure 7B). In brief, HJ-PI01 induces apoptosis through the death receptor and mitochondrial pathways. In addition, we also observed that HP-PI01 decreased the levels of Pim-2 and Pim-2 phosphorylation, but had no effect on Pim-1 and Pim-3 activity. These results suggested that HJ-PI01 directly inhibits Pim-2 (Figure 7C). To further confirm the efficacy and specificity of HJ-PI01 in Pim-2 inhibition, PI003, a pan-Pim inhibitor, was used as a positive control. Accordingly, HP-PI01 displayed more potent inhibition of p-Pim-2 than PI003, whereas PI003 significantly inhibited Pim-1 phosphorylation and Pim-3 expression, but not Pim-2 (Figure 7D). These results demonstrated that HP-PI01 exhibits good efficacy and specificity for Pim-2 compared with other pan-Pim inhibitors.

Immunoblot analysis of the anti- and pro-apoptotic proteins and cell death in the HJ-PI01-treated MDA-MB-231 cells. The MDA-MB-231 cells were treated with 300 nmol/L HJ-PI01 for the indicated time periods and then harvested and lysed. (A) The levels of the Fas, FADD, and caspase-8 proteins were analyzed by immunoblotting. (B) The levels of the Bax, Bcl-2, caspase-9, caspase-3 and PARP proteins were analyzed by immunoblotting. (C) The levels of the Pim-2, p-Pim-2, Pim-1 and Pim-3 proteins were analyzed by immunoblotting. (D) The MDA-MB-231 cells were treated with 300 nmol/L HJ-PI01 or 460 nmol/L PI003 for the indicated time periods and then harvested and lysed. The levels of the Pim-1, Pim-2, Pim-3, p-Pim-1, and p-Pim-2 proteins were analyzed by immunoblotting. β-Actin was used as an equal loading control.
Proteomics analyses of HJ-PI01-induced autophagy and apoptosis
To explore the molecular mechanisms underlying HJ-PI01-induced autophagy and apoptosis, iTRAQ and MS/MS analyses were employed to profile the differentially expressed proteins in HJ-PI01-treated MDA-MB-231 cells. A total of 1777 differentially expressed proteins were defined based on a high fold-change, of which 14 proteins were identified in the Pim-2-related PPI network. A16L1 and CISD2 are autophagy-related proteins, while PAK4, MP2K2, AKT2 and CSK22 are apoptosis-related proteins (Table 2). This result explored the mechanisms of the HJ-PI01-induced autophagy and apoptosis and the anti-cancer efficacy of Pim-2 inhibition.
Anti-tumor activity of HJ-PI01 in vivo
In this experiment, we established a xenograft mouse model using MDA-MB-231 cells to evaluate the efficacy of HJ-PI01 in vivo. As shown in Figure 8A, HJ-PI01 significantly inhibited tumor growth in nude mice compared with the control group (P<0.01), which was accompanied by obvious decreases in the body, liver, spleen and kidney weights of the mice, indicating the toxicity of HJ-PI01. Interestingly, when a lienal polypeptide was used in combination with HJ-PI01, it not only improved the anti-tumor activity of HJ-PI01 but also reduced the toxicity of HJ-PI01 (Figure 8A and 8B). Immunoreactivity for KI67, a marker of proliferation, was localized to the cell nuclei. The HJ-PI01 treatment significantly reduced the number of KI67-positive cells compared to the control treatment (Figure 8C), suggesting that HJ-PI01 inhibited tumor cell proliferation. Moreover, HJ-PI01 administration resulted in a statistically significant increase in the number of apoptotic bodies in the tumor, as visualized by TUNEL assay (Figure 8C).

Anti-tumor effects of HJ-PI01 in vivo. (A, a) Body weights of mice in each group during the treatment of the tumor xenografts. The mice were treated with hydroxypropyl-cyclodextrin (vehicle control), HJ-PI01 (40 mg·kg−1·d−1, po), or HJ-PI01 (40 mg·kg−1·d−1, po)+lienal polypeptide (LP) (50 mg·kg−1·d−1, ip). (b–d) Liver, spleen and kidney weights of the mice in each group at the end of treatment. (e) Tumor weights of the mice in each group at the end of treatment. *P<0.05, **P<0.01 vs control. (B) The tumor volume curves of each group during the treatment of the tumor xenografts. (C) Immunohistochemistry of the proliferation marker KI67 and TUNEL analysis.
Discussion
As a new class of cancer therapeutics, Pim kinase inhibitors have been frequently reported recently. Upon Pim-1 inhibition, the prevalence of increased Pim-2 expression across different cancer types suggests that Pim-2 inhibitors may be a treatment modality for a variety of cancers. In this study, we constructed the Pim-2-related PPI network for the first time, and all of the possible PPIs were shown. This result assessed the role of Pim-2 in the autophagy and apoptosis pathways. We further focused on the differences among the Pim kinase family and presented a new binding model for Pim-2 inhibitors. After selective modification of candidate pan-Pim inhibitors, we obtained a novel candidate compound, HJ-PI01. HJ-PI01 inhibited Pim-2 and induced apoptosis and autophagy in MDA-MB-231 cells. Moreover, the in vivo experiment showed that HJ-PI01 had remarkable anti-tumor efficacy.
Our results suggest that HJ-PI01 may be a new therapeutic agent for the treatment of breast cancer. Interestingly, a previous study has found that Pim-2 kinase can confer primary hematopoietic cells with resistance to rapamycin treatment28. Therefore, Pim-2 inhibitors may generate better efficacy. More importantly, we also found that HJ-PI01 induced apoptosis via the death receptor and mitochondrial pathways and induced autophagic cell death as well. Some Pim-targeted compounds induce apoptosis; here, we reported the double effect of the HJ-PI01 compound for the first time. In some circumstances, apoptosis and autophagy seem to be positively or negatively interconnected, due to the molecular switches shared between them29,30. Undoubtedly, there are multiple connections between apoptotic and autophagic processes that can jointly seal the fate of tumor cells31. In this study, the pro-death autophagy-inducing effect enhanced apoptosis. This study may explain the mechanism of candidate novel compounds. To the best of our knowledge, although some Pim-1 and pan-Pim inhibitors have been reported, no Pim-2-specific inhibitors have been described32,33,34,35,36. Compared with other compounds, HJ-PI01 has better druggability because it is based on chlorpromazine, which is an FDA-approved drug. Therefore, the above-mentioned findings may shed new light on the discovery of additional, small-molecule Pim-2 inhibitors for future breast cancer therapy.
In this study, we also used proteomics analyses to explore the molecular mechanisms underlying HJ-PI01-induced autophagy and apoptosis. Of note, we identified 14 proteins included in the PPI network, such as the autophagy-related proteins Atg16L (A16L1) and CDGSH Iron-Sulfur Domain Containing Protein 2 (CISD2). Atg16L plays an essential role in autophagy by interacting with Atg12-Atg5 to mediate the conjugation of phosphatidylethanolamine (PE) to LC337. CISD2 is a negative regulator of autophagy that contributes to antagonize BECN1-mediated cellular autophagy in the endoplasmic reticulum38. As shown in the results of our proteomics analysis, Atg16L was upregulated after HP-PI01 treatment, which indicates that autophagy was activated. Moreover, CISD2 expression was significantly decreased, indicating that HP-PI01 may induce BECN1-mediated autophagy, which has been validated by our in vitro experiment. Based on these results, explorations of the potential mechanisms of HJ-PI01-induced autophagy would further confirm the in-depth mechanisms of HP-PI01 in breast cancer therapy.
However, there are still some limitations for HJ-PI01, such as its low efficacy and increased toxicity. In our study, we found that treatment with a lienal polypeptide in combination with HJ-PI01 could improve the anti-tumor efficacy and reduce the toxicity induced by HJ-PI01. This incredible result is likely due to two possible reasons. On the one hand, the lienal polypeptide may have a certain anti-tumor activity. On the other hand, previous studies have suggested that the lienal polypeptide has immune modulating activity39; thus, some of the HJ-PI01-regulated pathways or key proteins may be affected by the lienal polypeptide. However, the detailed mechanisms remain to be further studied. Additionally, these results would also shed new light on combination drug therapies in our future studies.
Author contribution
Jian HUANG and Ling-juan ZHU conceived and designed the experiments; Yu-qian ZHAO performed the experiments; Yi-qiong YIN, Jie LIU, and Gui-hua WANG analyzed the data; Jin-hui WANG contributed reagents/materials/analysis tools; and Yu-qian ZHAO and Yi-qiong YIN wrote the paper.
Accession codes
References
Narlik-Grassow M, Blanco-Aparicio C, Carnero A . The PIM family of serine/threonine kinases in cancer. Med Res Rev 2014; 34: 136–59.
Nawijn MC, Alendar A, Berns A . For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat Rev Cancer 2011; 11: 23–34.
Blanco-Aparicio C, Carnero A . Pim kinases in cancer: diagnostic, prognostic and treatment opportunities. Biochem Pharmacol 2013; 85: 629–43.
Liu JJ, Lin M, Yu JY, Liu B, Bao JK . Targeting apoptotic and autophagic pathways for cancer therapeutics. Cancer Lett 2011; 300: 105–14.
Allen JD, Verhoeven E, Domen J, van der Valk M, Berns A . Pim-2 transgene induces lymphoid tumors, exhibiting potent synergy with c-myc. Oncogene 1997; 15: 1133–41.
Popivanova BK, Li YY, Zheng H, Omura K, Fujii C, Tsuneyama K, Mukaida N . Proto-oncogene, Pim-3 with serine/threonine kinase activity, is aberrantly expressed in human colon cancer cells and can prevent Bad-mediated apoptosis. Cancer Sci 2007; 98: 321–8.
Cibull TL, Jones TD, Li L, Eble JN, Ann Baldridge L, Malott SR, et al. Overexpression of Pim-1 during progression of prostatic adenocarcinoma. J Clin Pathol 2006; 59: 285–8.
Cohen AM, Grinblat B, Bessler H, Kristt D, Kremer A, Schwartz A, et al. Increased expression of the hPim-2 gene in human chronic lymphocytic leukemia and non-Hodgkin lymphoma. Leuk Lymphoma 2004; 45: 951–5.
Wang Z, Bhattacharya N, Weaver M, Petersen K, Meyer M, Gapter L, et al. Pim-1: a serine/threonine kinase with a role in cell survival, proliferation, differentiation and tumorigenesis. J Vet Sci 2001; 2: 167–79.
Chen LS, Redkar S, Bearss D, Wierda WG, Gandhi V . Pim kinase inhibitor, SGI-1776, induces apoptosis in chronic lymphocytic leukemia cells. Blood 2009; 114: 4150–7.
Amson R, Sigaux F, Przedborski S, Flandrin G, Givol D, Telerman A . The human protooncogene product p33pim is expressed during fetal hematopoiesis and in diverse leukemias. Proc Natl Acad Sci U S A 1989; 86: 8857–61.
Uddin N, Kim RK, Yoo KC, Kim YH, Cui YH, Kim IG, et al. Persistent activation of STAT3 by PIM2-driven positive feedback loop for epithelial-mesenchymal transition in breast cancer. Cancer Sci 2015; 106: 718–25.
Fu LL, Yang Y, Xu HL, Cheng Y, Wen X, Ouyang L, et al. Identification of novel caspase/autophagy-related gene switch to cell fate decisions in breast cancers. Cell Prolif 2013; 46: 67–75.
Agrawal S, Koschmieder S, Bäumer N, Reddy NG, Berdel WE, Müller-Tidow C, et al. Pim2 complements Flt3 wild-type receptor in hematopoietic progenitor cell transformation. Leukemia 2008; 22: 78–86.
Wernig G, Gonneville JR, Crowley BJ, Rodrigues MS, Reddy MM, Hudon HE, et al. The Jak2V617F oncogene associated with myeloproliferative diseases requires a functional FERM domain for transformation and for expression of the Myc and Pim proto-oncogenes. Blood 2008; 111: 3751–9.
Mizuki M, Ueda S, Matsumura I, Ishiko J, Schwäble J, Serve H, et al. Oncogenic receptor tyrosine kinase in leukemia. Cell Mol Biol (Noisyle-grand) 2003; 49: 907–22.
Adam M, Pogacic V, Bendit M, Chappuis R, Nawijn MC, Duyster J, et al. Targeting PIM kinases impairs survival of hematopoietic cells transformed by kinase inhibitor-sensitive and kinase inhibitor-resistant forms of Fms-like tyrosine kinase 3 and BCR/ABL. Cancer Res 2006; 66: 3828–35.
Chen Y, Fu LL, Wen X, Liu B, Huang J, Wang JH, et al. Oncogenic and tumor suppressive roles of microRNAs in apoptosis and autophagy. Apoptosis 2014; 19: 1177–89.
Breuer ML, Cuypers HT, Berns A . Evidence for the involvement of pim-2, a new common proviral insertion site, in progression of lymphomas. EMBO J 1989; 8: 743–8.
Bullock AN, Russo S, Amos A, Pagano N, Bregman H, Debreczeni JE, et al. Crystal structure of the PIM2 kinase in complex with an organoruthenium inhibitor. PLoS One 2009; 4: e7112.
Hanusova V, Skalova L, Kralova V, Matouskova P . Potential anti-cancer drugs commonly used for other indications. Curr Cancer Drug Targets 2015; 15: 35–52.
Lee MS, Johansen L, Zhang Y, Wilson A, Keegan M, Avery W, et al. The novel combination of chlorpromazine and pentamidine exerts synergistic antiproliferative effects through dual mitotic action. Cancer Res 2007; 67: 11359–67.
Shin SY, Lee KS, Choi YK, Lim HJ, Lee HG, Lim Y, et al. The antipsychotic agent chlorpromazine induces autophagic cell death by inhibiting the Akt/mTOR pathway in human U-87MG glioma cells. Carcinogenesis 2013; 34: 2080–9.
Liu Z, He W, Gao J, Luo J, Huang X, Gao C . Computational prediction and experimental validation of a novel synthesized pan-PIM inhibitor PI003 and its apoptosis-inducing mechanisms in cervical cancer. Oncotarget 2015; 6: 8019–35.
Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013; 29: 845–54.
Schuttelkopf AW, van Aalten DM . PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr 2004; 60: 1355–63.
Pathmanathan N, Balleine RL . Ki67 and proliferation in breast cancer. J Clin Pathol 2013; 66: 512–6.
Hammerman PS, Fox CJ, Birnbaum MJ, Thompson CB . Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Blood 2005; 105: 4477–83.
Xue L, Fletcher GC, Tolkovsky AM . Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol Cell Neurosci 1999; 14: 180–98.
Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A 2006; 103: 4952–7.
Liu B, Wen X, Cheng Y . Survival or death: disequilibrating the oncogenic and tumor suppressive autophagy in cancer. Cell Death Dis 2013; 4: e892.
Pierre F, Stefan E, Nédellec AS, Chevrel MC, Regan CF, Siddiqui-Jain A, et al. 7-(4H-1,2,4-Triazol-3-yl)benzo[c][2,6]naphthyridines: a novel class of Pim kinase inhibitors with potent cell antiproliferative activity. Bioorg Med Chem Lett 2011; 21: 6687–92.
Dwyer MP, Keertikar K, Paruch K, Alvarez C, Labroli M, Poker C, et al. Discovery of pyrazolo[1,5-a]pyrimidine-based Pim inhibitors: a template-based approach. Bioorg Med Chem Lett 2013; 23: 6178–82.
Dakin LA, Block MH, Chen H, Code E, Dowling JE, Feng X, et al. Discovery of novel benzylidene-1,3-thiazolidine-2,4-diones as potent and selective inhibitors of the PIM-1, PIM-2, and PIM-3 protein kinases. Bioorg Med Chem Lett 2012; 22: 4599–604.
Haddach M, Michaux J, Schwaebe MK, Pierre F, O'Brien SE, Borsan C, et al. Discovery of CX-6258. A potent, selective, and orally efficacious pan-Pim kinases inhibitor. ACS Med Chem Lett 2012; 3: 135–9.
Garcia PD, Langowski JL, Wang Y, Chen M, Castillo J, Fanton C, et al. Pan-PIM kinase inhibition provides a novel therapy for treating hematologic cancers. Clin Cancer Res 2014; 20: 1834–45.
Zavodszky E, Vicinanza M, Rubinsztein DC . Biology and trafficking of ATG9 and ATG16L1, two proteins that regulate autophagosome formation. FEBS Lett 2013; 587: 1988–96.
Chang NC, Nguyen M, Shore GC . BCL2-CISD2: An ER complex at the nexus of autophagy and calcium homeostasis? Autophagy 2012; 8: 856–7.
Jurin M, Zarković N, Ilić Z, Borović S, Hartleb M . Porcine splenic peptides (Polyerga) decrease the number of experimental lung metastases in mice. Clin Exp Metastasis 1996; 14: 55–60.
Acknowledgements
We are grateful to Prof Can-hua HUANG (Sichuan University) for his critical language editing. This study was supported by grants from the National Natural Science Fundation of China (No 81303270, U1303124, 81202403, and 81573290), Liaoning Science and Technology Project (No 2013226027-4), Jilin Science and Technology Progress Project (No 20130204058YY), Applied Basic Research Project of Sichuan Science and Technology Department (No 2013JY0154) and Shenyang Pharmaceutical University Scientific Research Fund (No ZCJJ2013407).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information is available on the website of Acta Pharmacologica Sinica.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhao, Yq., Yin, Yq., Liu, J. et al. Characterization of HJ-PI01 as a novel Pim-2 inhibitor that induces apoptosis and autophagic cell death in triple-negative human breast cancer. Acta Pharmacol Sin 37, 1237–1250 (2016). https://doi.org/10.1038/aps.2016.60
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2016.60
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Synthesis and biological evaluation of chromone derivatives against triple-negative breast cancer cells
Medicinal Chemistry Research (2023)
-
Potential of the enzyme laccase for the synthesis and derivatization of antimicrobial compounds
World Journal of Microbiology and Biotechnology (2023)
-
PIM1/STAT3 axis: a potential co-targeted therapeutic approach in triple-negative breast cancer
Medical Oncology (2022)
-
Negative regulation of AMPKα1 by PIM2 promotes aerobic glycolysis and tumorigenesis in endometrial cancer
Oncogene (2019)