
Abstract
Aim:
β, β-Dimethylacrylshikonin (DMAS) is an anticancer compound extracted from the roots of Lithospermum erythrorhizon. In the present study, we investigated the effects of DMAS on human lung adenocarcinoma cells in vitro and explored the mechanisms of its anti-cancer action.
Methods:
Human lung adenocarcinoma A549 cells were tested. Cell viability was assessed using an MTT assay, and cell apoptosis was evaluated with flow cytometry and DAPI staining. The expression of the related proteins was detected using Western blotting. The mitochondrial membrane potential was measured using a JC-1 kit, and subcellular distribution of cytochrome c was analyzed using immunofluorescence staining.
Results:
Treatment of A549 cells with DMAS suppressed the cell viability in dose- and time-dependent manners (the IC50 value was 14.22 and 10.61 μmol/L, respectively, at 24 and 48 h). DMAS (7.5, 10, and 15 μmol/L) dose-dependently induced apoptosis, down-regulated cIAP-2 and XIAP expression, and up-regulated Bax and Bak expression in the cells. Furthermore, DMAS resulted in loss of mitochondrial membrane potential and release of cytochrome c in the cells, and activated caspase-9, caspase-8, and caspase-3, and subsequently cleaved PARP, which was abolished by pretreatment with Z-VAD-FMK, a pan-caspase inhibitor. DMAS induced sustained p38 phosphorylation in the cells, while pretreatment with SB203580, a specific p38 inhibitor, blocked DMAS-induced p38 activation and apoptosis.
Conclusion:
DMAS inhibits the growth of human lung adenocarcinoma A549 cells in vitro via activation of p38 signaling pathway.
Similar content being viewed by others

Introduction
Lung adenocarcinoma is the most common histological type of lung cancer, accounting for approximately 30% to 40% of all cases of primary lung tumors1. Despite developments in surgery, radiotherapy and chemotherapy, the undesirable adverse effects of these treatments are unavoidable. Thus, more effective anticancer agents with fewer side effects for the treatment of lung adenocarcinoma are needed.
β,β-Dimethylacrylshikonin (DMAS), a shikonin derivative, is a typical component of naphthoquinone pigments isolated from the roots of Lithospermum erythrorhizon (Figure 1A)2. Previous studies have demonstrated that shikonin and its derivatives possess several biological activities, such as anti-microbial, anti-inflammatory, anti-HIV, and anti-platelet properties3,4,5,6. In addition, extensive reports have shown that shikonin and its derivatives exhibit antitumor activity against a variety of human cancers, and DMAS is one of the most effective agents. For example, DMAS exerted antitumor activity against human gastric cancer cells via Notch-1 down-regulation7. Moreover, DMAS showed a significant toxicological effect on hepatocellular carcinoma cells through increased caspase-3 activity8. DMAS also induced cell apoptosis through an increase in the Bax/Bcl-2 ratio in human colorectal cancer cells9. Recently, we showed that DMAS induced mitochondria-dependent apoptosis through the activation of ERK1/2 signaling in human gastric cancer SGC-7901 cells10.

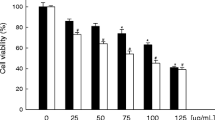
DMAS inhibits A549 cell growth. (A) The chemical structure of DMAS (MW370.4). (B) Effects of DMAS on the inhibition of A549 cell viability. The cells were treated with DMAS at different concentrations for 24 and 48 h. The cell viability was determined using an MTT assay. The viability of the control group (0.1% DMSO) was set to 100%. The data represent the mean±SD obtained from three independent experiments. bP<0.05 compared with the control group.
However, the effects of DMAS on human lung adenocarcinoma cells remain elusive. Thus, the aim of the present study was to characterize the mechanisms underlying the biological effects of DMAS on lung adenocarcinoma cells.
Materials and methods
Materials
A549 cells were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). DMAS, which was purchased from Tokyo Chemical Industry Co, Ltd (Tokyo, Japan), was dissolved in dimethyl sulfoxide (DMSO), and the DMSO content in all treatment groups was 0.1%. MTT, DAPI, SP600125, and SB203580 were purchased from Calbiochem (San Diego, CA, USA). The pan-caspase inhibitor (Z-VAD-FMK) was purchased from the Beyotime Institute of Biotechnology (Haimen, China). The cytochrome c antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibodies against ERK, phospho-ERK, JNK, phospho-JNK, P38, phospho-P38, Bcl-xL, XIAP, cIAP-2, survivin, Bax, Bak, cleaved caspase-9, cleaved caspase-8, cleaved caspase-3, cleaved PARP, and β-actin were purchased from Cell Signaling (Boston, MA, USA). The FITC-Annexin V Apoptosis Detection Kit was purchased from Becton Dickinson (San Diego, CA, USA).
Cell culture and cell proliferation assay
A549 cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum(FBS) (Hyclone, UT) and maintained at 37 °C in a humidified atmosphere of 5% CO2. For experiments, the cells were seeded onto 96-well plates at a final concentration of 5×103 cells/well and incubated in RPMI-1640 medium containing 10% FBS for 24 h. Subsequently, the cells were treated with DMAS at the indicated concentration for 24 and 48 h. The medium was removed and fresh medium containing 20 μL of MTT solution (5 mg/mL) was added to each well. After incubating for 4 h, 150 μL of DMSO was added to each well. The absorbance was detected at 570 nm using a Varioskan Flash Multimode Reader (Thermo Scientific, USA). Four duplicate wells were used for each treatment, and the experiments were repeated three times.
DAPI (4′, 6-diamidino-2-phenylindole) staining
A549 cells were plated onto six-well plates. After 24 h, the cells were treated with DMAS for the indicated time periods and subsequently fixed in cold acetone for 30 min, followed by incubation with DAPI (1 mg/mL) for 30 min. The apoptotic nuclear staining was detected using fluorescent microscopy (model IX71; Olympus, Tokyo, Japan).
Annexin V/PI assays for apoptosis
For the Annexin V/PI assays, the cells were stained with Annexin V-FITC and PI and subsequently evaluated for apoptosis using flow cytometry according to the manufacturer's instructions (BD PharMingen, San Diego, CA, USA). Briefly, 1×105 cells were washed twice with PBS and stained with 5 μL of Annexin V-FITC and 5 μL of PI in 500 μL of binding buffer for 15 min at room temperature in the dark. The apoptotic cells were determined using BD FACS Diva software (BD Biosciences, Franklin Lakes, NJ, USA). Annexin V staining indicates phosphatidylserine externalization.
Measurement of mitochondrial membrane potential
The cells (5×103 cells/well) were treated with or without DMAS (15 μmol/L) for 24 h and subsequently stained with JC-1 (BD MitoScreen JC-1 Kit, Becton Dickinson, USA) for 15 min at 37 °C. The mitochondrial membrane potential was detected using flow cytometry (FACS Canto II, Becton Dickinson, USA).
Western blot analysis
The cells were treated with the indicated DMAS concentrations for the indicated times in RPMI-1640 medium containing 10% FBS. The cells were collected in ice-cold PBS, and the cell extracts were prepared in RIPA buffer containing proteinase inhibitor cocktail (Calbiochem, San Diego, CA, USA). The protein concentrations of the cell lysates were determined, followed by boiling in gel-loading buffer for 10 min at 100 °C. Samples containing 30 μg of total protein were electrophoresed on 10% SDS-polyacrylamide gels and subsequently transferred to PVDF membranes (Millipore, Temecula, CA, USA). Following transfer, the membranes were blocked in TBST (TBS containing 0.1% Tween 20) containing 5% skimmed milk for 2 h, followed by incubation with appropriate primary antibodies overnight at 4 °C. After washing three times in TBST, the membranes were incubated with 1:1000 horseradish peroxidase-conjugated appropriate secondary antibodies for 2 h at 37 °C. The membranes were visualized using an enhanced chemiluminescence detection system (Immun-Star WesternC Kit, Bio-Rad, USA).
Immunofluorescence staining
Immunofluorescence staining was used to analyze the subcellular distribution of cytochrome c in A549 cells treated with DMAS. Cells cultured on sterile glass coverslips were fixed in cold acetone for 10 min. After permeabilization with 0.3% Triton X-100 for 20 min at room temperature, the cells were blocked in 3% bovine serum for 30 min and incubated overnight at 4 °C with anti-cytochrome c antibody (1:30) overnight at 4 °C. After washing three times in PBS, the cells were labeled with Alexa Fluor 488-conjugated secondary antibodies. DAPI was subsequently added for nuclear staining. Microscopic analysis was performed using a fluorescent microscopy (model IX71; Olympus, Tokyo, Japan).
Statistical analysis
The data are reported as the means±standard deviation (SD) of three independent experiments. The data were evaluated using one-way analysis of variance (ANOVA). Significant differences were established at P<0.05.
Results
DMAS inhibits the growth of A549 cells
The cytotoxic effects of DMAS on lung adenocarcinoma cells were determined using an MTT assay. A549 cells were treated with different concentrations of DMAS (2.5, 5, 7.5, 10, and 15 μmol/L) for 24 or 48 h, and the cell viability was subsequently examined using an MTT assay. As shown in Figure 1B, after 24 h incubation with DMAS at 2.5, 5, 7.5, 10, and 15 μmol/L, the viability of A549 cells was reduced to 98.26%±0.45%, 95.68%±1.51%, 87.40%±0.84%, 70.39%±1.31%, and 46.40% ±0.96%, respectively, compared with vehicle-treated cells. A549 cells treated with 2.5, 5, 7.5, 10, and 15 μmol/L DMAS for 48 h showed a 96.26%±1.33%, 92.69%±1.33%, 75.90%±1.57%, 56.89%±0.35%, and 17.38%±0.31% reduction in cell viability, respectively (Figure 1B). The median inhibitory concentration (IC50) of DMAS for A549 cells was 14.22 and 10.61 μmol/L at 24 and 48 h, respectively.
DMAS induces the apoptosis of A549 cells
Cell and nuclear shrinkage, apoptotic body formation, nuclear condensation, and DNA fragmentation are the hallmarks of apoptosis11. To confirm whether DMAS induces apoptosis, DMAS-treated A549 cells were subjected to DAPI staining. Nuclear condensation and apoptotic body formation were detected in the cells treated with 15 μmol/L DMAS (Figure 2A).

DMAS induced apoptosis in A549 cells. (A) DNA condensation (white arrow) was measured through DAPI staining and analyzed under a fluorescent microscope at 200× magnification. (B) The apoptotic status was evaluated using an Annexin V-FITC binding assay. Early stage apoptotic cells are shown on the lower right (Annexin V-FITC+/PI−), and late stage apoptotic cells are shown on the top right. The part (Annexin V-FITC−/PI−) was considered as Viable cells are shown on the lower left, and necrotic cells are shown on the upper left (Annexin V-FITC−/PI+).
To further verify the induction of apoptosis through DMAS in A549 cells, the apoptotic effect was evaluated using flow cytometry with Annexin V and propidium iodide (PI) double staining. As shown in Figure 2B, after 24 h incubation, the early apoptotic (Annexin V+/PI−) rates of A549 cells were 0.8%, 5.0%, 12.0%, and 23.7% in response to treatment with vehicle, 7.5, 10, and 15 μmol/L DMAS, respectively.
DMAS induces mitochondrial events associated with apoptosis in A549 cells
The Bcl-2 family proteins play pivotal roles in the regulation of apoptosis and comprise both pro- and anti-apoptotic proteins. These proteins activate or inhibit the release of several effectors, leading to the activation of caspases in apoptosis12. To investigate the expression of apoptosis-related proteins, pro-apoptotic proteins (Bax, Bak) and anti-apoptotic proteins (cIAP-2, XIAP, Bcl-xL, survivin) were detected through Western blot analysis using A549 cells treated with varying concentrations of DMAS (0, 7.5, 10, and 15 μmol/L) for 24 h. The results demonstrated that DMAS treatment decreased the expression of cIAP-2 and XIAP (Figure 3A). However, the down-regulation of XIAP was less obvious than the dose-dependent down-regulation of cIAP-2. There was no significant change in the expression of Bcl-xL and survivin in A549 cells. We also examined whether DMAS affected the expression of pro-apoptotic proteins (Bax and Bak), and observed an increase in the levels of Bax and Bak with the increasing doses of DMAS (Figure 3B).

DMAS induced mitochondrial events associated with apoptosis in A549 cells. Western blot analysis for anti-apoptotic proteins: Bcl-xL, cIAP-2, XIAP, and survivin (A) and pro-apoptotic proteins: Bax and Bak (B) in whole cell extracts of A549 cells treated with DMAS for 24 h. Actin protein levels were also measured as controls. (C) The detection of mitochondrial membrane potential through flow cytometry. A549 cells were treated with or without DMAS (15 μmol/L) for 24 h, followed by staining with JC-1 for 15 min at 37 °C and flow cytometry. (D) A549 cells were fixed and labeled for cytochrome c (green) and DNA (blue). (E) Western blot analysis for cleaved PARP, cleaved caspase-9, cleaved caspase-8, cleaved caspase-3 in whole cell extracts of A549 cells treated with DMAS for 24 h. Actin protein levels were also measured as controls. (F) A549 cells were treated with DMAS in the presence or absence of Z-VAD-FMK (10 μmol/L) for 24 h. The protein extracts were prepared and subjected to Western blot analysis using antibodies against cleaved caspase-3 and cleaved PARP. Actin protein levels were also measured as controls. (G) A549 cells were treated with DMAS in the presence or absence of Z-VAD-FMK (10 μmol/L) for 24 h. The apoptotic status was determined using an Annexin V-FITC binding assay.
The loss of mitochondrial membrane potential has been associated with the initiation and activation of some apoptotic signaling cascades13. This event can be triggered through the translocation of Bax from the cytoplasm to mitochondria, resulting in the release of cytochrome c from mitochondria into the cytoplasm14,15. Therefore, first we determined whether DMAS induced the loss or disruption of mitochondrial membrane potential in A549 cells. A549 cells were treated with 15 μmol/L DMAS for 24 h, harvested, stained with JC-1 dye and analyzed through flow cytometry. As shown in Figure 3C, the membrane potential decreased from 81.9% to 33.2% after treatment with 15 μmol/L DMAS. Next, we examined the distribution and subcellular localization of cytochrome c to confirm whether the disruption of mitochondrial membrane potential through DMAS resulted in the release of cytochrome c from the mitochondria into the cytoplasm. A549 cells were treated with or without DMAS (15 μmol/L) for 24 h, and the distribution of cytochrome c was visualized using a confocal laser microscope. The DMAS-treated cells showed blurred morphology in contrast to the obviously clear appearance in non-DMAS treated cells (Figure 3D), suggesting the translocation of cytochrome c from mitochondria to the cytoplasm in DMAS-treated cells.
Caspases are enzymes responsible for the execution of apoptosis. To understand the molecular mechanism of DMAS-induced apoptosis in A549 cells, the cleavage of caspase-3, caspase-8, caspase-9, and PARP was examined using Western blot analysis. A549 cells treated with DMAS (0, 7.5, 10, and 15 μmol/L) for 24 h showed the dose-dependent elevation of cleaved caspase-3, caspase-8, and caspase-9 (Figure 3E). Cleaved PRAP, another specific feature of apoptosis, was also increased with the increasing doses of DMAS (Figure 3E).
To further verify the role of caspase activation in DMAS-induced apoptosis, we pre-treated A549 cells with the pan-caspase inhibitor Z-VAD-FMK (10 μmol/L) prior to DMAS treatment. The DMAS-induced cleavage of caspase-3 and PARP and apoptotic cell death were almost completely blocked with Z-VAD-FMK pretreatment (Figure 3F and 3G). These results strongly suggested that the mitochondrial-mediated caspase cascade pathway plays an essential role in DMAS-induced apoptosis.
DMAS induces sustained JNK and p38 activation in A549 cells
The mitogen-activated protein kinase (MAPK) signaling pathway has been implicated in several events of cellular stress-induced apoptotic cell death16,17. Hence, we observed the activation of ERK, JNK, and p38 after DMAS treatment. As shown in Figure 4, the phosphorylation of ERK, JNK, and p38 was significantly increased in response to the DMAS treatment for 2 h in a dose-dependent manner. In addition, the phosphorylation of JNK and p38 lasted for 24 h. These data suggested that the sustained activation of JNK and p38 might be involved in DMAS-induced apoptosis in A549 cells.

Effects of DMAS on the MAPK signaling pathway. A549 cells were treated with DMAS at the indicated concentrations for the indicated times, and total cellular extracts were prepared and subjected to Western blot analysis to measure the levels of phosphorylated ERK (A), JNK (B), and p38 (C). The membranes were reprobed with antibodies against total ERK, JNK, and p38 for normalization.
The p38 signaling pathway is involved in DMAS-induced apoptosis in A549 cells
To determine whether the DMAS-induced sustained activation of JNK and p38 signaling pathways plays an important role in apoptosis, we pretreated A549 cells with specific inhibitors for JNK (SP600125) and p38 (SB203580) for 1 h, followed by DMAS treatment for 24 h. As shown in Figure 5A and 5B, pretreatment with SB203580 markedly inhibited DMAS-mediated p38 activation and blocked caspase-3 and PARP cleavage. However, the DMAS-induced cleavage of caspase-3 and PARP was not inhibited after pretreatment with SP600125 (Figure 5C). In addition, SB203580 reduced DMAS-induced apoptosis in A549 cells (Figure 5D), and the pan-caspase inhibitor Z-VAD-FMK had no effect on the DMAS-induced activation of p38 (Figure 5E). These results confirmed that DMAS-induced apoptosis in A549 cells is mediated through the sustained activation of the p38 signaling pathway.

DMAS-induced A549 cell apoptosis is mediated through p38 activation. (A) A549 cells were pretreated with or without SB203580 (10 μmol/L), followed by treatment with DMSO (vehicle) or DMAS (15 μmol/L) for 24 h. Protein extracts were prepared and subjected to Western blot analysis to measure the levels of phosphorylated p38. Total p38 protein levels were also measured as controls. (B) A549 cells were pretreated or not with SB203580 (10 μmol/L), followed by treatment with DMSO (vehicle) or DMAS (15 μmol/L) for 24 h. Protein extracts were prepared and subjected to Western blot analysis using antibodies against cleaved caspase-3 and cleaved PARP. Actin protein levels were also measured as controls. (C) A549 cells were pretreated or not with SP600125 (25 μmol/L), followed by treatment with DMSO (vehicle) or DMAS (15 μmol/L) for 24 h. Protein extracts were prepared and subjected to Western blot analysis using antibodies against cleaved caspase-3 and cleaved PARP. Actin protein levels were also measured as controls. (D) A549 cells were treated with DMAS in the presence or absence of SB203580 (10 μmol/L) for 24 h. The apoptotic status was determined using an Annexin V-FITC binding assay. (E) A549 cells were pretreated with or without Z-VAD-FMK (10 μmol/L), followed by treatment with DMAS (15 μmol/L) for 24 h. Protein extracts were prepared and subjected to Western blot analysis to measure the levels of phosphorylated p38. Total p38 protein levels were also measure as controls.
Discussion
Shikonin and its derivatives are the active components of Lithospermum erythrorhizon root extract, a traditional Chinese medicine18. These compounds might be promising anticancer agents, as multiple antitumor effects in vivo and in vitro have been reported19. Among the shikonin derivatives, DMAS exhibits strong antitumor effects2. In the present study, we investigated the effects of DMAS on human lung adenocarcinoma A549 cells. The results demonstrated that DMAS exhibited significant cytotoxic effects against A549 cells, in a dose- and time-dependent manner, and DMAS-induced apoptotic cell death in A549 cells was evidenced through an increase in early apoptotic cells (Annexin V+/PI−).
The balance between pro- and anti-apoptotic members of the Bcl-2 family is crucial for the induction of apoptosis20. Mitochondria play an essential role in the signal transduction of apoptosis21. Mitochondrial dysfunction is central to the apoptotic pathway22. Apoptotic stimuli often disturb mitochondrial function through a decrease in mitochondrial membrane potential. The translocation of apoptotic proteins from the cytosol to mitochondria results in the release of cytochrome c and second mitochondria-derived activator of caspases (SMAC) through the loss of mitochondrial membrane potential23. Consequently, multiple caspases are activated, leading to subsequent cell death via apoptosis. Among caspase family proteins, caspase-3 is predominantly responsible for apoptosis execution24. Caspase-3 can be cleaved and activated through extrinsic or death receptor-mediated and intrinsic or mitochondria-dependent pathways25,26. In the present study, we showed that DMAS decreased the expression of cIAP-2 and XIAP, increased the expression of Bax and Bak, and induced the loss of mitochondrial membrane potential and subsequent release of cytochrome c in A549 cells, consistent with the activation of mitochondria-dependent apoptosis. The DMAS-mediated activation and cleavage of caspase-9, caspase-3, and PARP and the reduction in DMAS-induced apoptosis in A549 cells through the pan-caspase inhibitor Z-VAD-FMK suggested that the mitochondrial-mediated caspase cascade pathway plays a critical role in DMAS-induced apoptosis. Taken together, these results suggested that DMAS regulates the expression of apoptosis-related proteins, induces the release of cytochrome c, and triggers caspase-dependent cell apoptosis.
MAPK has also been implicated in the regulation of diverse cellular processes, including embryogenesis, proliferation, differentiation and apoptosis27. The MAPK family comprises three protein kinases: ERK1/2, JNK, p38, and ERK528. Emerging evidence suggests that the activation of p38 contributes to apoptosis. Chun et al showed that platycodin D induced growth inhibition and apoptosis in AGS human gastric cancer cells through the sustained activation of p3829. Wang et al demonstrated that curcumin induced apoptosis through the p38-dependent up-regulation of FasL in Huh7 cells30. Recently, Hsieh et al reported that arctigenin induced the apoptosis of breast cancer MDA-MB-231 cells through the ROS/p38 pathway31. Consistently, we observed that the sustained activation of p38 was involved in DMAS-induced growth inhibition and apoptosis in A549 cells. SB203580, a specific inhibitor of p38, effectively blocked DMAS-induced p38 activation and attenuated DMAS-induced apoptosis. These results suggested that the pro-apoptotic effect of DMAS in A549 cells is mediated through the activation of the p38 signaling pathway.
In conclusion, we presented experimental evidence that strongly supports the antitumor effects of DMAS on lung adenocarcinoma cells. Thus, we propose that DMAS could be further developed as a potential anticancer agent for the activation of p38 signaling pathway through the inhibition of lung adenocarcinoma cell growth and induction of apoptosis, suggesting a new therapeutic strategy for the treatment of lung adenocarcinoma. However, further studies are warranted to fully uncover the molecular mechanism underlying the role of DMAS in lung adenocarcinoma models in vitro and in vivo.
Author contribution
Hai-bing WANG designed the study; Hai-bing WANG and Xiao-qiong MA performed the experiments; Hai-bing WANG analyzed the data and wrote the manuscript.
References
Hanagiri T, Baba T, So T, Yasuda M, Sugaya M, Ono K, et al. Time trends of surgical outcome of patients with non-small cell lung cancer. J Thorac Oncol 2010; 5: 825–9.
Xuan Y, Hu X . Naturally-occurring shikonin analogues — a class of necroptotic inducers that circumvent cancer drug resistance. Cancer Lett 2009; 274: 233–42.
Shen CC, Syu WJ, Li SY, Lin CH, Lee GH, Sun CM . Antimicrobial activities of naphthazarins from Arnebia euchroma. J Nat Prod 2002; 65: 1857–62.
Tanaka S, Tajima M, Tsukada M, Tabata M . A comparative study on anti-inflammatory activities of the enantiomers, shikonin and alkannin. J Nat Prod 1986; 49: 466–9.
Chen X, Yang L, Zhang N, Turpin JA, Buckheit RW, Osterling C, et al. Shikonin, a component of Chinese herbal medicine, inhibits chemokine receptor function and suppresses human immunodeficiency virus type 1. Antimicrob Agents Chemother 2003; 47: 2810–6.
Ko FN, Lee YS, Kuo SC, Chang YS, Teng CM . Inhibition on platelet activation by shikonin derivatives isolated from Arnebia euchroma. Biochim Biophys Acta 1995; 1268: 329–34.
Zhen-Jun S, Yuan-Yuan Z, Ying-Ying F, Shao-Ju J, Jiao Y, Xiao-Wei Z, et al. β,β-dimethylacrylshikonin exerts antitumor activity via Notch-1 signaling pathway in vitro and in vivo. Biochem Pharmacol 2012; 84: 507–12.
Wu YY, Wan LH, Zheng XW, Shao ZJ, Chen J, Chen XJ, et al. Inhibitory effects of β,β-dimethylacrylshikonin on hepatocellular carcinoma in vitro and in vivo. Phytother Res 2012; 26: 764–71.
Fan Y, Jin S, He J, Shao Z, Yan J, Feng T, et al. Effect of β,β-dimethylacrylshikonin on Inhibition of human colorectal cancer cell growth in vitro and in vivo. Int J Mol Sci 2012; 13: 9184–93.
Shen XJ, Wang HB, Ma XQ, Chen JH . β,β-Dimethylacrylshikonin induces mitochondria dependent apoptosis through ERK pathway in human gastric cancer SGC-7901 cells. PLoS One 2012; 7: e41773.
Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S . Identification of a factor that links apoptotic cells to phagocytes. Nature 2002; 417: 182–7.
Ola MS, Nawaz M, Ahsan H . Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem 2011; 351: 41–58.
Mantena SK, Sharma SD, Katiyar SK . Berberine inhibits growth, induces G1 arrest and apoptosis in human epidermoid carcinoma A431 cells by regulating Cdki-Cdk-cyclin cascade, disruption of mitochondrial membrane potential and cleavage of caspase-3 and PARP. Carcinogenesis 2006; 27: 2018–27.
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, et al. Molecular characterizaion of mitochondrial apoptosis-inducing factor. Nature 1999; 397: 441–6.
Hengartner MO . The biochemistry of apoptosis. Nature 2000; 407: 770–6.
Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000; 288: 870–4.
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME . Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995; 270: 1326–31.
Wang R, Yin R, Zhou W, Xu D, Li S . Shikonin and its derivatives: a patent review. Expert Opin Ther Pat 2012; 22: 977–97.
Andújar I, Recio MC, Giner RM, Ríos JL . Traditional chinese medicine remedy to jury: the pharmacological basis for the use of shikonin as an anticancer therapy. Curr Med Chem 2013; 20: 2892–8.
Cory S, Adams JM . The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002; 2: 647–56.
Tait SW, Green DR . Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010; 11: 621–32.
Ly JD, Grubb DR, Lawen A . The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis 2003; 8: 115–28.
Shimizu S, Narita M, Tsujimoto Y . Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999; 399: 483–7.
HONG HJ, LIU JC, CHENG TH, CHAN P . Tanshinone IIA attenuates angiotensin II-induced apoptosis via Akt pathway in neonatal rat cardiomyocytes. Acta Pharmacol Sin 2010; 31: 1569–75.
Igney FH, Krammer PH . Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2002; 2: 277–88.
Hu W, Kavanagh JJ . Anticancer therapy targeting the apoptotic pathway. Lancet Oncol 2003; 4: 721–9.
Raman M, Chen W, Cobb MH . Differential regulation and properties of MAPKs. Oncogene 2007; 26: 3100–12.
Klein AM, Zaganjor E, Cobb MH . Chromatin-tethered MAPKs. Curr Opin Cell Biol 2013; 25: 272–7.
Chun J, Joo EJ, Kang M, Kim YS . Platycodin D induces anoikis and caspase-mediated apoptosis via p38 MAPK in AGS human gastric cancer cells. J Cell Biochem 2013; 114: 456–70.
Wang WZ, Li L, Liu MY, Jin XB, Mao JW, Pu QH, et al. Curcumin induces FasL-related apoptosis through p38 activation in human hepatocellular carcinoma Huh7 cells. Life Sci 2013; 92: 352–8.
Hsieh CJ, Kuo PL, Hsu YC, Huang YF, Tsai EM, Hsu YL . Arctigenin, a dietary phytoestrogen, induces apoptosis of estrogen receptor-negative breast cancer cells through the ROS/p38 MAPK pathway and epigenetic regulation. Free Radic Biol Med 2014; 67: 159–70.
Acknowledgements
This work was financially supported through grants from the National Natural Science Foundation of China (No 81403143) and the Zhejiang Provincial Natural Science Foundation of China (No LY12H28005).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, Hb., Ma, Xq. β, β-Dimethylacrylshikonin induces mitochondria-dependent apoptosis of human lung adenocarcinoma cells in vitro via p38 pathway activation. Acta Pharmacol Sin 36, 131–138 (2015). https://doi.org/10.1038/aps.2014.108
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2014.108
- Springer Nature Singapore Pte Ltd.