
Abstract
Although nickel-based polyanionic compounds are expected to exhibit a high operating voltage for batteries based on the Ni2+/3+ redox couple activity, some rare experimental studies on the electrochemical performance of these materials are reported, resulting from the poor kinetics of the bulk materials in both Li and Na nonaqueous systems. Herein, the electrochemical activity of the Ni2+/3+ redox couple in the mixed-polyanionic framework Na4Ni3(PO4)2(P2O7) is reported for the first time. This novel material exhibits a remarkably high operating voltage when cycled in sodium cells in both carbonate- and ionic liquid-based electrolytes. The application of a carbon coating and the use of an ionic liquid-based electrolyte enable the reversible sodium ion (de-)insertion in the host structure accompanied by the redox activity of Ni2+/3+ at operating voltages as high as 4.8 V vs Na/Na+. These results present the realization of Ni-based mixed polyanionic compounds with improved electrochemical activity and pave the way for the discovery of new Na-based high potential cathode materials.
Similar content being viewed by others

Introduction
Lithium-ion batteries have been largely recognized as the most efficient electrochemical energy storage devices for both portable electronics and electric vehicle applications.1, 2 However, the growth and diversification of the energy storage market trigger interest in low-cost and environmentally friendly alternative systems. In addition, recent concerns over the cost and future availability of lithium highlight the urgent need to exploit alternative energy storage systems.3 In these terms, sodium (Na)-ion batteries are attractive candidates because of the electrochemistry that is similar to the well-established lithium-ion technology.4 To date, the LiFePO4 olivine with (PO4)3− polyanionic framework, owing to its superior thermal stability and low cost, is considered to be one of the best electrode materials for lithium-ion batteries, mostly in view of its stable operating voltage and satisfactory specific capacity. However, the intrinsic tunability of the operating voltage of polyanionic compounds because of the presence of different transition metals such as Mn, Co and Ni5, 6, 7 has triggered interest in alternative frameworks exhibiting higher cell voltages. According to theoretical prediction, lithium nickel phosphate has a remarkably high working potential (~5.1 V vs Li+/Li) because of the Ni2+/3+ redox activity.8 Unfortunately, several studies have demonstrated that the implementation of this material is restricted by several drawbacks such as the intrinsic sluggish kinetics attributable to the low electronic conductivity, the poor lithium transport in commonly used electrolyte systems and the structural instability of the delithiated phases upon cycling that require further studies to obtain high-performance Ni-based phosphates as high voltage cathodes for battery application.8, 9, 10, 11, 12
With regard to Na-based systems, considerable research has also been devoted to NASICON (sodium (Na) Super Ionic CONductor)-type polyanionic materials, such as NaMPO4,13, 14 Na2MP2O7 (where M is a transition metal)15, 16 and Na3V2(PO4)3,17, 18 as cathode materials for room temperature Na-ion batteries. However, the low energy densities resulting from the low theoretical capacity and, generally, cell voltage, as well as the poor rate capability compared with their lithium counterparts, have inhibited the widespread development of Na-ion batteries.19 Unlike LiMPO4, the NaMPO4 thermodynamically stable phase obtained under the conventional synthesis conditions is the maricite phase that is known to be electrochemically inactive with respect to the olivine phase.20, 21 Among the polyanionic frameworks, pyrophosphate compounds offer a stable three-dimensional (P2O7)4− framework with multiple sites for Na ions. Fe-based pyrophosphates have been reported to exhibit enhanced rate capability performance most likely because of the presence of open diffusion pathways for Na ions. Indeed, first principle calculations confirmed a relatively low migration barrier for diffusion of Na ions.15
In this context, a new polyanionic compound of the general formula Na4M3(PO4)2(P2O7) with the Fe2+/3+ redox couple has been proposed by Kang and colleagues22, 23 as a promising cathode in terms of its superior Na mobility and thermal stability. The material has a theoretical capacity of 129 mAh g−1 and an operating potential of ∼3.2 V vs Na+/Na, higher than the potential exhibited by NaFePO4 and Na2FeP2O7.13, 24 According to first principle calculations, the P2O7 dimers can buffer the volume change during cycling (<4%) and offer Na diffusion pathways in the three-dimensional open framework that may ensure long-term cycling performance. Cobalt-based polyanionic compounds show electrochemical properties similar to the Ni-based analogs in Li systems with low conductivity and poor thermal stability.25, 26 However, the Na4Co3(PO4)2(P2O7) synthesized by a sol–gel method shows an average working potential of ~4.4 V vs Na+/Na and a high rate capability for Na ions.27 A combination of different polyanionic groups could contribute to outstanding ion diffusion and voltage profile in Na-ion batteries as well as structure stability during the Na (de-)insertion process. Interestingly, a theoretical prediction reported by Islam and colleagues28 has pointed out that doping the Na4Fe3(PO4)2(P2O7) with Ni can lead to a significant increase in the cell voltage.
In light of such results, we report the investigation of a Ni-based polyanionic material as a potential high voltage cathode for rechargeable Na batteries. In this work, the pristine and carbon-coated materials, that is, Na4Ni3(PO4)2(P2O7) and Na4Ni3(PO4)2(P2O7)/C, were stoichiometrically prepared by a conventional solid-state method. This study represents an intriguing investigation in terms of both structural and electrochemical characterization of the Ni redox activity in mixed polyanionic compounds. The electrochemical activity of the Ni2+/3+ redox couple in Na cells, predicted by previous theoretical calculations, is experimentally reported in this paper for the first time. The attractive possibility of using high operating voltage Na-based cathode materials highlights the need of electrolyte media with a wide electrochemical stability window, opening a new unexplored research field for Na-ion batteries.
Materials and methods
Synthesis of materials
All chemicals were used as received without further purification. Syntheses were carried out by simple and time-efficient solid-state reactions. For the precursor preparation, sodium pyrophosphate decahydrate (Na4P2O7·10H2O, ⩾99%, Alfa Aesar, Karlsruhe, Germany), nickel (II) acetate tetrahydrate (Ni(OCOCH3)2·4H2O, ⩾98%, Alfa Aesar) and ammonium dihydrogen phosphate (NH4H2PO4, ⩾99%, Merck Millipore, Darmstadt, Germany) were mixed by ball-milling in acetone according to stoichiometric proportions. Citric acid monohydrate (C6H8O7·H2O, ⩾99%, Alfa Aesar) was used as a carbon source for the carbon-coated sample. After intensive milling, the mixture was dried at room temperature and ground. The precursor powder obtained was preheated at 300 °C for 6 h and then annealed at 500 °C (heating rate: 2 °C min−1) for 10 h in an aluminum oxide crucible using a tubular furnace under steady argon flow. Cooling to room temperature and grinding followed. The carbon-coated material was synthesized under the same conditions using citric acid monohydrate as the organic carbon source in the precursor preparation. A schematic representation of the synthesis procedure is reported in Supplementary Figure S1.
Material characterization
X-ray diffraction (XRD) analysis was performed in Bragg–Brentano geometry using a Bruker D8 Advance diffractometer (Bruker, Karlsruhe, Germany). All diffractograms were recorded with CuKα radiation (λ=0.154 nm) in the 2θ range of 10°–90° with a step size of 0.01°. The structures were refined using the Rietveld method with the DIFFRAC PLUS TOPAS 4.2 software (Bruker). Morphological and structural characterizations were conducted via ZEISS 1550VP Field Emission scanning electron microscopy (Carl Zeiss, Jena, Germany) and Cs-corrected FEI Titan 80–300 kV high-resolution transmission electron microscopy operated at an acceleration voltage of 80 and 300 kV (FEI, Eindhoven, The Netherlands). The elemental distribution was investigated using an energy-dispersive X-ray spectrometer (Oxford Instruments, Munich, Germany). Thermal gravimetric analysis was performed using a thermogravimetric analyzer (TGA-209F, Netzsch, Selb, Germany) under O2 flow of 20 ml min−1 within the temperature range of 30–600 °C (final materials) and 30–800 °C (precursors) at a 5 °C min−1 scan rate. Tap density measurements were performed on a tap density analyzer (Autotap, Quantachrome, Boynton Beach, FL, USA), with 1000 taps per sample.
Electrochemical characterization
The electrochemical tests were performed using three electrode Swagelok-type cells assembled in an argon-filled glove box. Sodium half-cells were assembled by using glass fiber filters (Whatman, Maidstone, UK) as separators soaked in various electrolyte solutions and sodium metal as the counter and reference electrodes. The 1 M solution of NaPF6 (99%, Alfa Aesar) in ethylene carbonate/diethyl carbonate (1:1 w/w) (BASF, Ludwigshafen, Germany, Battery Grade) and the 10 mol% solution of NaTFSI (sodium bis(trifluoromethanesulfonyl)imide, 99.5%, SOLVIONIC, Varilhes, France) in Py14FSI (N-butyl-N-methyl-pyrrolidinium bis(fluorosulfonyl)imide) were used as electrolytes. Sodium metal was cut from sodium pieces (99.8%, Acros Organics, Morris Plains, NJ, USA), roll-pressed and punched on the current collector. The electrodes were prepared by doctor-blade casting a slurry prepared by dispersing 80 wt% of active material, 10 wt% of carbon black as conductive agent (Super C65, IMERYS, Bodio, Switzerland) and 10 wt% of polyvinylidenefluoride (6020 Solef, Arkema Group, Paris, France) binder in N-methyl-2-pyrrolidone (anhydrous, 99.5%, Sigma-Aldrich, St Louis, MO, USA) on Al foil as the current collector. After drying, electrodes with a 12 mm diameter were punched, pressed and dried again. The mass loading was ~2.8 mg cm−2. The cells were assembled in an argon-filled glove box with H2O and O2 contents <1 p.p.m. A Maccor 4000 Battery system (Maccor, Tulsa, OK, USA) was used for the galvanostatic charge/discharge test between 3.0 and 4.9 V vs Na+/Na in the organic electrolyte and between 3.0 and 5.1 V vs Na+/Na in the ionic liquid electrolyte at a current density of 10 mA g−1. Cyclic voltammetry was performed on a multichannel potentiostat/galvanostat (VMP3, Biologic Science Instruments, Claix, France) using a scan rate of 0.01, 0.05, 0.1 and 0.5 mV s−1 in a voltage range of 3.0–5.0 V vs Na+/Na (organic electrolyte) and 3.0–5.2 V vs Na+/Na (ionic liquid electrolyte). All the potential values in this study are in reference to the Na+/Na reference electrode. All measurements were performed in climatic chambers at a temperature of 20±1 °C.
Results and discussion
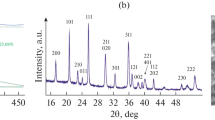
The structural properties of the Na4Ni3(PO4)2(P2O7) have been determined by XRD analysis. Figure 1 shows the Rietveld refinement and the schematic representation of the crystal structure deduced for Na4Ni3(PO4)2(P2O7). Rietveld refinement of the powder XRD pattern confirms that the Na4Ni3(PO4)2(P2O7) is isostructural to Na4Fe3(PO4)2(P2O7) and crystallizes in the orthorhombic Pn21a space group with a=18.0006(3) Å, b=6.4933(1) Å and c=10.4115(2) Å. The refinement has been performed with satisfactory agreement factors (Rwp=4.79%, Rp=3.45%, goodness of fit=2.13) (ICSD (Inorganic Crystal Structure Database) 236316).27 The diffractogram reported in Figure 1a presents no evidence of impurity phases, suggesting that the synthesis conditions adopted are suitable for obtaining pure Na4Ni3(PO4)2(P2O7). The refined cell parameters and atomic occupancy values are summarized and presented, respectively, in Supplementary Tables S1 and S2. Rietveld refinement of the XRD pattern of the carbon-coated analog (Na4Ni3(PO4)2(P2O7)/C) (Supplementary Figure S2) shows that the carbon coating process has no effect on the structural characteristics of the material that crystallizes in the same space group with comparable cell parameters. The crystal structures of Na4Ni3(PO4)2(P2O7) viewed along the [001] and [010] directions are displayed in Figure 1b. As the isostructural Na4Co3(PO4)2(P2O7) and Na4Fe3(PO4)2(P2O7), the crystal structure of Na4Ni3(PO4)2(P2O7) is composed of three-dimensional network layers of [Ni3P2O13]∞ jointly composed by NiO6 octahedra and (PO4)3− tetrahedra in the bc plane that are bridged by the (P2O7)4− dimers generating large channels along the b-axis with a low activation barrier energy for Na+ ion diffusion.23, 29 The Na ions occupy four distinguishable sites, among which two (Na1 and Na4) are located in the large channels parallel to the b-axis. The Ni-based polyanionic materials have so far been considered as an electrochemically inactive cathode material in both Li and Na systems because of the low electronic conductivity resulting in the poor alkali ion diffusion activity.12 However, in this study, the structure deriving from the mixed polyanionic Na4Ni3(PO4)2(P2O7) may stabilize the charged Ni analog and open pathways for the improved and reversible Na+ diffusion with respect to the NaNiPO4 and Na2NiP2O7 polyanionic materials that can contribute to the improved electrochemical reactivity.

(a) Rietveld refinement and powder X-ray diffraction (XRD) for Na4Ni3(PO4)2(P2O7). (b) Schematic representation of the crystal structure of Na4Ni3(PO4)2(P2O7) viewed along the [001] and [010] directions.
Scanning electron microscopy and high-resolution transmission electron microscopy analyses were conducted to assess the morphological and structural aspects of the synthesized powders. Figures 2a and b present the scanning electron microscopy images of Na4Ni3(PO4)2(P2O7) and Na4Ni3(PO4)2(P2O7)/C. As shown in Figure 2a, the pristine Na4Ni3(PO4)2(P2O7) aggregates in shapeless micrometric particles (that is, polymorphic morphology). After the carbon coating treatment, the particle size of the powder is favorably reduced with well-defined edges. The tap density of carbon-coated Na4Ni3(PO4)2(P2O7) material is ∼1.01 g cm−3, that is, smaller than the tap density of the pristine Na4Ni3(PO4)2(P2O7) material at 1.24 g cm−3, that can be attributed to the reduced particle size and the carbon coating. The elemental mapping by energy-dispersive X-ray spectroscopy of Na, Ni, O, P and C has been performed to check the metal element distribution in Na4Ni3(PO4)2(P2O7) (Supplementary Figure S3). The analysis reveals a homogeneous distribution of the elements investigated, confirming the elemental composition. The transition electron microscopy analysis of Na4Ni3(PO4)2(P2O7)/C, reported in Figure 2c, confirms the shapeless character of the particles, showing a sub-micrometric dimension with an average diameter of ∼500 nm. In addition, the high-resolution transmission electron microscopy image in Figure 2d clearly shows a lattice fringe of d=0.680 nm, corresponding to the (201) plane diffraction of Na4Ni3(PO4)2(P2O7)/C. This material shows structural instability under an intense electron beam demonstrated by the disappearance of the lattice fringe after a prolonged electron beam irradiation (Supplementary Figure S4), opening the question about the crystal stability of the mixed polyanionic structure under nonconventional conditions.30 Thermal gravimetric analysis (Supplementary Figure S5) was performed to investigate the thermal stability and carbon content of the prepared materials. The thermogram of pristine Na4Ni3(PO4)2(P2O7) shows a slight weight loss before 100 °C, attributable most likely to water desorption. The weight loss detected from 300 °C is related to the oxidation of residual carbon belonging to the organic salt precursor. Meanwhile, no weight loss is observed after 500 °C, even when exposed in O2, reflecting a considerably high thermal stability. The content of absorbed water in the carbon-coated material is higher than in the pristine material, suggesting a higher sensitivity to moisture, most likely because of a higher surface area. The carbon content is calculated as ∼6.3% that corresponds to the weight detected within the 100–450 °C temperature range.

Scanning electron microscopy (SEM) images of (a) Na4Ni3(PO4)2(P2O7) and (b) Na4Ni3(PO4)2(P2O7)/C powders. (c, d) Transmission electron microscopy (TEM) images of Na4Ni3(PO4)2(P2O7)/C performed on an FEI Titan 80–300 kV electron microscope with image Cs-corrector at an accelerating voltage of 80 kV.
The investigation of Ni redox activity in the polyanionic framework has been performed by galvanostatic cycling tests and cyclic voltammetry analysis. In an attempt to study the effect of the polyanionic framework in the Ni redox couple, three different Ni-based compounds were synthesized, that is, NaNiPO4 (the phosphate), Na2NiP2O7 (the pyrophosphate) and Na4Ni3(PO4)2(P2O7) (the mixed polyanionic compound). Na2NiP2O7 and NaNiPO4 polyanionic compounds have been prepared using the same solid-state method. As the Fe-based compound, the stoichiometric Na2NiP2O7 crystallizes in the triclinic P-1 space group. The XRD pattern obtained is shown in Supplementary Figure S6a. NaNiPO4 has also been prepared according to the synthesis conditions reported in the Supplementary Information. However, orthorhombic Na4Ni3(PO4)2(P2O7) was obtained instead of maricite NaNiPO4 (the XRD pattern obtained is presented in Supplementary Figure S6b), reflecting the thermodynamic stability of the Na4Ni3(PO4)2(P2O7) phase under our experimental conditions.
To investigate the electrochemical processes occurring during sodium (de-)insertion, the carbon-coated materials have been selected as the material of choice, owing to its enhanced conductivity with respect to the pristine material.31 Moreover, in an attempt to enlarge the electrochemical stability window of the electrolyte and enable the investigation of the Ni redox activity at higher potential values, an ionic liquid-based electrolyte, that is, NaTFSI in Py13FSI (1:9 in mole ratio), was employed.32, 33 A comparative electrochemical investigation between the carbon-coated Na4Ni3(PO4)2(P2O7) and Na2NiP2O7 is shown in Figure 3, showing the differential capacity vs voltage (dQ/dV) curves of Na4Ni3(PO4)2(P2O7)/C and Na2Ni(P2O7)/C. The dQ/dV curve of Na4Ni3(PO4)2(P2O7)/C shows three anodic peaks at 4.61, 4.67 and 5.00 V with two reversible cathodic peaks at 4.58 and 4.92 V, elucidating the redox activity of the Ni redox couple in a sodium cell accompanied by reversible Na ion (de-)insertion. For the dQ/dV curve of the Na2NiP2O7/C electrode, no electrochemical activity is observed, suggesting that Ni is not active in the pyrophosphate framework under our experimental conditions. From the comparison, it is incidentally determined that the mixed phosphate frameworks enable the Ni2+/3+ redox activity in polyanionic structures compared with those of pyrophosphates.

Initial differential capacities vs potential (dQ/dV) curves of the Na4Ni3(PO4)2(P2O7)/C and Na2NiP2O7/C in the NaTFSI: Py13FSI (1:9 mole ratio) electrolyte at a current density of 10 mA g−1.
To further understand the sodium (de-)insertion process associated with the Ni activity in such a framework, cyclic voltammetry and charging/discharging measurements were performed with NaTFSI in Py13FSI (1:9 mole ratio) electrolyte, and the corresponding results are shown in Figures 4a and b. The cyclic voltammetry profile shown in Figure 4a exhibits three anodic peaks at 4.61, 4.67, and 5.00 V and only two cathodic peaks at 4.58 and 4.92 V, in accordance with the dQ/dV profile reported in Figure 3. The potential profile reported in Figure 4b reveals the de-insertion of 2.7 Na equivalents followed by the insertion of 1.3 Na equivalents, corresponding to a discharge capacity of 63 mAh g−1, representing a promising degree of reversibility for this unexplored material. However, based on the Ni2+/3+ redox reaction, a theoretical capacity of 127.2 mAh g−1 is expected, referring to 3 equivalents of Na+. The investigation of a wide potential window shows clearly that there is no further redox process at potentials higher than 5.0 V. In addition, electrolyte decomposition, aluminum current collector corrosion and possible oxidation of the carbon deriving from the carbon coating process, which may also affect the cycling performance, cannot be excluded at such high potential values vs Na+/Na.34, 35 Cyclic voltammetry tests have also been performed in standard carbonate-based electrolyte (1 M NaPF6 in ethylene carbonate/diethyl carbonate, 1:1 w/w) (see Supplementary Figure S7). The cyclic voltammetry curves obtained at various scan rates (Supplementary Figure S7a) confirm the reversibility of the sodium (de-)insertion also at higher scan rates, despite the increased cell polarization. This observation is in good agreement with the charge/discharge voltage profile (Supplementary Figure S7b), suggesting a reversible redox process occurring at such a high working potential. The material reveals a satisfactory cycling stability. However, the redox peaks decrease upon cycling, most likely because of poor kinetics. More detailed description and discussion can be found in the Supplementary Information section. Regardless of its high working potential, the material exhibits relatively low coulombic efficiency and poor specific capacity compared with other reported Fe- or Co-based polyanionic compounds in both organic and ionic liquid electrolytes.36, 37 The inferior electrochemical behavior of the Ni-based compound with respect to Fe- and Co-based analogs may be attributed to the intrinsic lower ionic conductivity of the former material. Sanz et al.12 have reported that the Ni-based compound exhibits the lowest ionic conductivity of ∼2.1 × 10−7 S cm−1 at 300 °C, with respect to Fe, Mn and Co.

Cyclic voltammetry curve in the potential range of 3.0–5.2 V at a scan rate of 0.01 mV s−1 (a) and first cycle galvanostatic discharge/charge profile between 3.0 and 5.1 V at a current density of 10 mA g−1 (b) of Na4Ni3(PO4)2(P2O7)/C in NaTFSI:Py13FSI (1:9 mole ratio) electrolyte.
Potential profiles and cycling performance of Na4Ni3(PO4)2(P2O7)/C in both carbonate- and ionic liquid-based electrolytes are presented in Supplementary Figure S8. The profiles reveal first discharge capacities of ∼40 and 63 mAh g−1, respectively, in carbonate-based (3.0 to 4.9 V) and ionic liquid-based (3.0 to 5.1 V) electrolytes. However, the wider potential window inevitably leads to lower coulombic efficiencies because of oxidative decomposition reactions. As an example, at the highest potential (that is, 5.1 V vs Na+/Na, corresponding to 5.4 V vs Li+/Li), the decomposition of Py13FSI ionic liquid is known to occur.33 The conductive agent, Super C65, is playing a role at such potential values. The use of carbon nanotubes, for example, results in the 25% discharge capacity increase in carbonate-based electrolyte (see Supplementary Figure S9). However, extensive electrolyte decomposition occurs, resulting in an overall lower coulombic efficiency. A different electrode current collector (aluminum) and binder may also be used. For example, higher anodic stability (~5.5 V vs Na+/Na) was reported using polytetrafluoroethylene as a binder, carbon paper as the current collector and 0.5 M NaTFSI in Py13TFSI as the electrolyte. However, oxidative decomposition would always occur.38
To better understand the factors affecting the electrochemical performance exhibited, further structural investigation has been performed. It has been proposed that structural reorganization upon cycling could affect the reversibility of the sodium (de-)insertion. Indeed, the structural changes during Na (de-)insertion may derive from the instability of the desodiated electrode, most likely because of the distortion of P2O7 dimers and changes in the unit cell volume.8, 23
The investigation of the Na (de-)insertion mechanism in the Na4Ni3(PO4)2(P2O7)/C electrode was carried out by ex situ XRD measurements at different states of charge. Figure 5a displays the typical charge/discharge profile of a Na/Na4Ni3(PO4)2(P2O7)/C cell with points and letters indicating the potential values at which XRD analysis has been performed. When charging the electrode to 5.0 V in the ethylene carbonate/diethyl carbonate-based electrolyte, the initial charge capacity is higher than the theoretical capacity that, at such a high working potential, may be attributed to the cathode–electrolyte interface film formation of the Ni-based electrode materials and oxidative electrolyte decomposition.39, 40 The patterns collected in the 10–60° 2θ range are displayed in Figure 5a. The main reflections of the mixed polyanionic phase are shown to be maintained upon charge and discharge, whereas moderate peak shifting and intensity changes occur, suggesting that neither phase transition nor irreversible structural reorganization occurs upon sodium de-insertion. The amplified XRD patterns in the 2θ range of 10–25° and 25–35° in Figures 5b and c clearly show the peak shifts during the electrochemical reaction. Interestingly, although the (200) peak at ∼10° is moving to a higher 2θ value, other major peaks such as (011), (111), (220) and (022) show a slight shift to lower 2θ when the Na is extracted from the host material. During the discharge process, the peaks gradually move back to the original positions, suggesting the reversibility of the structural modifications associated with the volume change during the Na (de-)insertion process. To investigate the occupancy of the four different Na atoms upon sodium (de-)insertion, Rietveld refinement of the ex situ XRD results has been performed. The evolution of the refined lattice parameters (a, b, c) is shown in Figures 6a–c. The a lattice parameter value instantaneously decreases when the Na ions are electrochemically removed from the crystal structure, whereas the b and c axis parameters increase. The results herein presented differ slightly from those already reported for the Fe-based analog system, suggesting different structural changes in the potential range investigated.26, 27 The Na occupancies, reported in Table S3, indicate that the sodium cations located in the large channels constructed by the P2O7 dimers, that is, the Na1 and Na4 sites, can be electrochemically removed first at relatively low potentials, resulting in the decrease of the a lattice parameter and leading to a volume change of 1.6%. When charging up to 5.0 V, the other two sodium cations, that is, from Na2 and Na3 sites, are extracted subsequently, leading to the rapid increase in the b parameter. When discharged back to 3.0 V, the observed lattice parameters are slightly different from those of the pristine sample, suggesting a not fully reversible electrochemical process, indicated as well by the electrochemical characterization above. The low reversibility may be ascribed to the collapse of [Ni3P2O13]∞ layers along the a axis to a certain extent blocking the Na diffusion channels that confirms the instability of the desodiated electrode and reflects the low columbic efficiency of the charge–discharge process. However, the superior thermal stability and small volume changes during cycling of the material demonstrate the advantages of using mixed polyanionic structures to construct a large diffusion channel for Na ions, proving this strategy to be an efficient way to develop Ni-based polyanionic materials as conceivable high voltage cathodes.

(a) Typical charge/discharge profile and ex situ X-ray diffraction (XRD) pattern of the Na4Ni3(PO4)2(P2O7)/C electrode at various charge and discharge states. (b, c) Amplified XRD patterns in the 2θ range of 10–25° and 25–35°, respectively.

Lattice parameter (a, b, c) change during electrochemical desodiation/sodiation process.
The integration of the two framework types in the same lattice, namely the [PO4] and [P2O7] groups, enables the electrochemical activity of the Ni2+/3+ redox reaction at high potential. This study represents a step forward in the design of high voltage cathode materials for Na-ion batteries. However, this research field faces several challenges. Electrochemically stable electrolytes and/or electrolyte additives, forming a cathode–electrolyte interphase with protective (electronic insulating, but ion-conducting) properties, thus mitigating or avoiding oxidative decomposition reactions, should be identified to enable such high voltage cathode materials in Na-ion batteries.
Conclusions
In summary, the Ni2+/3+ redox activity in the mixed polyanionic framework has been experimentally demonstrated for the first time in sodium cells. Contrary to the pyrophosphate structure (Na2NiP2O7), the combination of phosphate and pyrophosphate groups enables the reversible Na ion (de-)insertion accompanied by the Ni2+/3+ redox activity. The Na4Ni3(PO4)2(P2O7) has been synthesized via a conventional solid-state method. The carbon coating process and the implementation of an ionic liquid-based electrolyte enables the reversible (de-)insertion of 1.3 Na equivalents at an operating potential as high as 4.8 V vs Na+/Na. Certainly, we are aware of the improvable electrochemical performance of the material; however, this is not the main objective of the present work. Meanwhile, searching for high voltage cathode materials opens a new and exciting perspective in the search for optimized and improved electrolyte solutions with a wider electrochemical stability window. Nevertheless, we believe that this study holds promise for obtaining cathode materials of a high working potential. The mixed polyanionic structure, combining phosphate and pyrophosphate frameworks, allows, thanks to the P2O7 dimers, the construction of large Na diffusion channels and, in consequence, small volume changes during cycling, representing a step towards overcoming the drawbacks of Ni analogs with low ionic/electronic conductivity and poor structural stability. The work presented herein shows, for the first time, the realization of Ni-based mixed polyanionic compounds and paves the way for the discovery of new Na-based high potential cathode materials.
References
Tarascon, J. M. & Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–367 (2001).
Scrosati, B. & Garche, J. Lithium batteries: status, prospects and future. J. Power Sources 195, 2419–2430 (2010).
Tarascon, J. M. Is lithium the new gold? Nat. Chem. 2, 510–510 (2010).
Slater, M. D., Kim, D., Lee, E. & Johnson, C. S. Sodium-Ion Batteries. Adv. Funct. Mater. 23, 947–958 (2013).
Fisher, C. A. J., Hart Prieto, V. M. & Islam, M. S. Lithium battery materials LiMPO4 (M=Mn, Fe, Co, and Ni): insights into defect association, transport mechanisms, and doping behavior. Chem. Mater. 20, 5907–5915 (2008).
Padhi, A. K., Nanjundaswamy, K. S. & Goodenough, J. B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 144, 1188–1194 (1997).
Wang, H., Yang, Y., Liang, Y., Cui, L.-F., Sanchez Casalongue, H., Li, Y., Hong, G., Cui, Y. & Dai, H. LiMn1−xFexPO4 nanorods grown on graphene sheets for ultrahigh-rate-performance lithium ion batteries. Angew. Chem. 123, 7502–7506 (2011).
Ong, S. P., Chevrier, V. L., Hautier, G., Jain, A., Moore, C., Kim, S., Ma, X. & Ceder, G. Voltage, stability and diffusion barrier differences between sodium-ion and lithium-ion intercalation materials. Energy Environ. Sci. 4, 3680–3688 (2011).
Feng, Y., Zhang, H., Fang, L., Ouyang, Y. & Wang, Y. Designed synthesis of a unique single-crystal Fe-doped LiNiPO4 nanomesh as an enhanced cathode for lithium ion batteries. J. Mater. Chem. A 3, 15969–15976 (2015).
Bramnik, N. N., Nikolowski, K., Trots, D. M. & Ehrenberg, H. Thermal stability of LiCoPO4 cathodes. Electrochem. Solid State Lett. 11, A89–A93 (2008).
Kim, S.-W., Kim, J., Gwon, H. & Kang, K. Phase stability study of Li1−xMnPO4 (0 ⩽ x ⩽ 1) cathode for Li rechargeable battery. J. Electrochem. Soc. 156, A635–A638 (2009).
Sanz, F., Parada, C., Rojo, J. M. & Ruíz-Valero, C. Synthesis, structural characterization, magnetic properties, and ionic conductivity of Na4MII3(PO4)2(P2O7 (MII=Mn, Co, Ni). Chem. Mater. 13, 1334–1340 (2001).
Oh, S.-M., Myung, S.-T., Hassoun, J., Scrosati, B. & Sun, Y.-K. Reversible NaFePO4 electrode for sodium secondary batteries. Electrochem. Commun. 22, 149–152 (2012).
Tripathi, R., Wood, S. M., Islam, M. S. & Nazar, L. F. Na-ion mobility in layered Na2FePO4F and olivine Na[Fe,Mn]PO4 . Energy Environ. Sci. 6, 2257–2264 (2013).
Kim, H., Shakoor, R. A., Park, C., Lim, S. Y., Kim, J.-S., Jo, Y. N., Cho, W., Miyasaka, K., Kahraman, R., Jung, Y. & Choi, J. W. Na2FeP2O7 as a promising iron-based pyrophosphate cathode for sodium rechargeable batteries: a combined experimental and theoretical study. Adv. Funct. Mater. 23, 1147–1155 (2013).
Barpanda, P., Ye, T., Avdeev, M., Chung, S.-C. & Yamada, A. A new polymorph of Na2MnP2O7 as a 3.6 V cathode material for sodium-ion batteries. J. Mater. Chem. A 1, 4194–4197 (2013).
Saravanan, K., Mason, C. W., Rudola, A., Wong, K. H. & Balaya, P. The first report on excellent cycling stability and superior rate capability of Na3V2(PO4)3 for sodium ion batteries. Adv. Energy Mater. 3, 444–450 (2013).
Sharma, N., Serras, P., Palomares, V., Brand, H. E. A., Alonso, J., Kubiak, P., Fdez-Gubieda, M. L. & Rojo, T. Sodium distribution and reaction mechanisms of a Na3V2O2(PO4)2F electrode during use in a sodium-ion battery. Chem. Mater. 26, 3391–3402 (2014).
Yabuuchi, N., Kubota, K., Dahbi, M. & Komaba, S. Research development on sodium-ion batteries. Chem. Rev. 114, 11636–11682 (2014).
Lee, K. T., Ramesh, T. N., Nan, F., Botton, G. & Nazar, L. F. Topochemical synthesis of sodium metal phosphate olivines for sodium-ion batteries. Chem. Mater. 23, 3593–3600 (2011).
Avdeev, M., Mohamed, Z., Ling, C. D., Lu, J., Tamaru, M., Yamada, A. & Barpanda, P. Magnetic structures of NaFePO4 maricite and triphylite polymorphs for sodium-ion batteries. Inorg. Chem. 52, 8685–8693 (2013).
Kim, H., Park, I., Seo, D.-H., Lee, S., Kim, S.-W., Kwon, W. J., Park, Y.-U., Kim, C. S., Jeon, S. & Kang, K. Understanding the electrochemical mechanism of the new iron-based mixed-phosphate Na4Fe3(PO4)2(P2O7 in a Na rechargeable battery. Chem. Mater. 25, 3614–3622 (2013).
Kim, H., Park, I., Seo, D.-H., Lee, S., Kim, S.-W., Kwon, W. J., Park, Y.-U., Kim, C. S., Jeon, S. & Kang, K. New iron-based mixed-polyanion cathodes for lithium and sodium rechargeable batteries: combined first principles calculations and experimental study. J. Am. Chem. Soc. 134, 10369–10372 (2012).
Barpanda, P., Liu, G., Ling, C. D., Tamaru, M., Avdeev, M., Chung, S.-C., Yamada, Y. & Yamada, A. Na2FeP2O7: a safe cathode for rechargeable sodium-ion batteries. Chem. Mater. 25, 3480–3487 (2013).
Hautier, G., Jain, A., Ong, S. P., Kang, B., Moore, C., Doe, R. & Ceder, G. Phosphates as lithium-ion battery cathodes: an evaluation based on high-throughput ab initio calculations. Chem. Mater. 23, 3495–3508 (2011).
Herle, P. S., Ellis, B., Coombs, N. & Nazar, L. F. Nano-network electronic conduction in iron and nickel olivine phosphates. Nat. Mater. 3, 147–152 (2004).
Nose, M., Nakayama, H., Nobuhara, K., Yamaguchi, H., Nakanishi, S. & Iba, H. Na4Co3(PO4)2P2O7: a novel storage material for sodium-ion batteries. J. Power Sources 234, 175–179 (2013).
Wood, S. M., Eames, C., Kendrick, E. & Islam, M. S. Sodium ion diffusion and voltage trends in phosphates Na4M3(PO4)2P2O7 (M=Fe, Mn, Co, Ni) for possible high-rate cathodes. J. Phys. Chem. C 119, 15935–15941 (2015).
Sanz, F., Parada, C., Amador, U., Monge, M. A. & Valero, C. R. Na4Co3(PO4)2P2O7, a new sodium cobalt phosphate containing a three-dimensional system of large intersecting tunnels. J. Solid State Chem. 123, 129–139 (1996).
Recham, N., Chotard, J. N., Dupont, L., Delacourt, C., Walker, W., Armand, M. & Tarascon, J. M. A 3.6 V lithium-based fluorosulphate insertion positive electrode for lithium-ion batteries. Nat. Mater. 9, 68–74 (2010).
Gong, Z. & Yang, Y. Recent advances in the research of polyanion-type cathode materials for Li-ion batteries. Energy Environ. Sci. 4, 3223–3242 (2011).
Shin, J.-H., Henderson, W. A. & Passerini, S. Ionic liquids to the rescue? Overcoming the ionic conductivity limitations of polymer electrolytes. Electrochem. Commun. 5, 1016–1020 (2003).
Zhou, Q., Henderson, W. A., Appetecchi, G. B., Montanino, M. & Passerini, S. Physical and electrochemical properties of N-Alkyl-N-methylpyrrolidinium Bis(fluorosulfonyl)imide ionic liquids: PY13FSI and PY14FSI. J. Phys. Chem. B 112, 13577–13580 (2008).
Yamamoto, O., Ogumi, Z., Morita, M., Krause, L. J., Lamanna, W., Summerfield, J., Engle, M., Korba, G., Loch, R. & Atanasoski, R. Corrosion of aluminum at high voltages in non-aqueous electrolytes containing perfluoroalkylsulfonyl imides; new lithium salts for lithium-ion cells. J. Power Sources 68, 320–325 (1997).
La Mantia, F., Huggins, R. A. & Cui, Y. Oxidation processes on conducting carbon additives for lithium-ion batteries. J. Appl. Electrochem. 43, 1–7 (2013).
Guerfi, A., Dontigny, M., Charest, P., Petitclerc, M., Lagacé, M., Vijh, A. & Zaghib, K. Improved electrolytes for Li-ion batteries: mixtures of ionic liquid and organic electrolyte with enhanced safety and electrochemical performance. J. Power Sources 195, 845–852 (2010).
Guerfi, A., Duchesne, S., Kobayashi, Y., Vijh, A. & Zaghib, K. LiFePO4 and graphite electrodes with ionic liquids based on bis(fluorosulfonyl)imide (FSI)− for Li-ion batteries. J. Power Sources 175, 866–873 (2008).
Kundu, D., Tripathi, R., Popov, G., Makahnouk, W. R. M. & Nazar, L. F. Synthesis, structure, and Na-ion migration in Na4NiP2O7F2: a prospective high voltage positive electrode material for the Na-ion battery. Chem. Mater. 27, 885–891 (2015).
Jarry, A., Gottis, S., Yu, Y.-S., Roque-Rosell, J., Kim, C., Cabana, J., Kerr, J. & Kostecki, R. The formation mechanism of fluorescent metal complexes at the LixNi0.5Mn1.5O4−δ/carbonate ester electrolyte interface. J. Am. Chem. Soc. 137, 3533–3539 (2015).
Kim, J.-H., Pieczonka, N. P. W., Lu, P., Liu, Z., Qiao, R., Yang, W., Tessema, M. M., Sun, Y.-K. & Powell, B. R. In situ formation of a cathode–electrolyte interface with enhanced stability by titanium substitution for high voltage spinel lithium-ion batteries. Adv. Mater. Interfaces 2, 1500109 (2015).
Acknowledgements
HZ and BQ gratefully acknowledge the financial support from the Chinese State Scholarship Fund.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the NPG Asia Materials website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, H., Hasa, I., Buchholz, D. et al. Exploring the Ni redox activity in polyanionic compounds as conceivable high potential cathodes for Na rechargeable batteries. NPG Asia Mater 9, e370 (2017). https://doi.org/10.1038/am.2017.41
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/am.2017.41
- Springer Japan KK
This article is cited by
-
A comprehensive understanding of the anionic redox chemistry in layered oxide cathodes for sodium-ion batteries
Science China Chemistry (2021)
-
Electrochemical investigations of high-voltage Na4Ni3(PO4)2P2O7 cathode for sodium-ion batteries
Journal of Solid State Electrochemistry (2020)
-
Unlocking the potential of weberite-type metal fluorides in electrochemical energy storage
npj Computational Materials (2019)
-
Ni-based cathode materials for Na-ion batteries
Nano Research (2019)