
Abstract
The precise method for synthesizing well-defined polymer microspheres via π-allylnickel-catalyzed living coordination polymerization of allene derivatives under dispersion polymerization conditions is described. The π-allylnickel-catalyzed living coordination polymerization of allene derivatives such as phenoxyallene was carried out in protic solvents such as methanol in the presence of stabilizers such as polyvinylpyrrolidone to produce polymer microspheres with a narrowly dispersed diameter distribution (Dw/Dn) at a high yield. For example, polymer microspheres with a number-average diameter (Dn) of 1.10 μm and Dw/Dn of 1.01 were obtained at a yield of 97%; these microspheres are composed of a polymer with a controlled molecular weight (Mn=11 700) and narrow molecular weight distribution (Mw/Mn=1.06). The size of the resulting polymer microspheres proved to be affected by polymerization conditions such as the initial monomer concentrations, monomer structures, polymerization media and stabilizers. Unlike conventional dispersion polymerization systems, the use of a bifunctional monomer, 1,4-diallenoxybenzene, for this living dispersion polymerization successfully produced well-defined crosslinked living polymer microspheres that exhibited high solvent tolerance. The postpolymerization of functional allene monomers in the living dispersion polymerization was also successful, resulting in functionalized polymer microspheres that could be applied in solid-phase organic synthesis and solid-supported transition metal catalysis.
Similar content being viewed by others


Introduction
Polymer microspheres are frequently applied in scientific and industrial fields such as coatings, electronics, biomedical engineering and organic synthesis.1, 2, 3, 4, 5, 6 Precision synthesis of polymer microspheres with a regulated size, a narrow size distribution and surface functionality is very important for the further development of polymer microspheres for advanced applications. The crosslinking of polymer microspheres is also likely to be very important for achieving sufficient reagent resistance, thermal stability and good mechanical properties.7
Among the heterogeneous polymerization techniques, dispersion polymerization is an effective method for obtaining polymer microspheres with narrowly dispersed diameter distributions.8, 9 Dispersion polymerization is defined as a heterogeneous polymerization process that starts from a homogeneous solution of all the starting materials (i.e., the monomer, initiator and stabilizer), whereas the polymer microspheres precipitate out during the progress of the polymerization.10 Accordingly, the polymerization should be carried out in good solvents for the monomers but poor solvents for the polymers being formed. To establish precision dispersion polymerization, the living anionic polymerization of styrene was studied in hydrocarbon solvents.11, 12, 13 Although this technique successfully provides narrowly dispersed polymer microspheres that are composed of well-defined polymer chains, the monomers and solvents are strictly limited so as not to interrupt the living nature of the polymerization. In general, dispersion polymerization of less polar conventional monomers such as styrene is performed in polar protic media such as alcohols. Thus, the free radical mechanism is most suitable to prevent the deactivation of the growing species.8, 14 Although the precision synthesis of polymer microspheres by the controlled radical dispersion polymerization technique has been reported, this method seems to be insufficient for the preparation of well-defined polymer microspheres consisting of regulated polymer chains, presumably because the concentration of the growing species in the polymer microspheres is too high to suppress side reactions.15, 16, 17, 18, 19, 20, 21, 22 As mentioned above, the formation of a networked structure within the polymer microspheres is often required for the practical applications of the resulting polymer microspheres. However, free radical dispersion polymerization is often accompanied by the coagulation of the microspheres or yields microspheres with broader size distributions, with only a few exceptions where successful results were independently obtained based on the free radical two-stage monomer feeding technique and the reversible addition fragmentation chain transfer polymerization technique by Winnik and co-workers23, 24 and Tan et al., respectively.25
Several approaches have successfully been reported for the synthesis of surface-functionalized polymer microspheres. Seeded polymerization is one such approach where the functional polymers produced in the postpolymerization are adsorbed physically onto the seed polymer particles.26, 27, 28 In this case, however, the combination of the seed polymer particles and the functional polymers produced in the postpolymerization appears to be strictly limited to avoid the nucleation of new polymer particles from the functional polymers in the postpolymerization process. Another method is to use functional macromonomers that can be copolymerized during the dispersion polymerization of conventional monomers, such as styrene, in which the functional polymer segments originated from the macromonomers segregate onto the surface of the polymer microspheres.29 In this case, the structure of the functional macromonomers also appears to be limited because they should have lower surface energies than the polymers obtained from conventional monomers. The postpolymerization of functional monomers from the initiators chemically bound onto the surface of the polymer microspheres is also effective for obtaining surface-functionalized polymer microspheres.30 This method is more general in terms of the structure of the comonomers, whereas the multistep synthesis of microspheres with initiation groups and postpolymerization processes that preferably do not involve transfer reactions are required to carry out the effective functionalization of the surface.31, 32
If the dispersion polymerization can be performed on the basis of the ideal living polymerization technique, which is totally free from side reactions such as deactivation and transfer reactions, it is expected that well-defined polymer microspheres with crosslinked structures and surface functionality can be produced simply by the use of multifunctional and functional comonomers, respectively. The π-allylnickel-catalyzed living coordination polymerization of allene derivatives that we have been studying is an attractive candidate for performing precision dispersion polymerization because this method is based on the highly chemoselective insertion of allene moieties into the π-allylnickel growing species. In fact, this living polymerization can be applied to monomers bearing diverse functional groups such as alkyl, aryl, alkoxy, carboalkoxy, hydroxy and amide to produce well-defined polymers bearing the corresponding functional groups.33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47 Additionally, this living polymerization can be carried out in protic polar media such as alcohols without any deactivations of the growing species.47
In this paper, we describe the results of the π-allylnickel-catalyzed living dispersion polymerization of allene derivatives that enables the precision synthesis of well-defined polymer microspheres. The effect of the polymerization conditions on the morphologies of the polymer microspheres was studied in detail. Precision synthesis of crosslinked polymer microspheres and surface-functionalized polymer microspheres was performed by sequential copolymerization. On the basis of the present synthetic method of well-defined polymer microspheres, two preliminary examples that demonstrate the excellent ability to design functional polymer microspheres are also described.
Experimental procedure
Materials
Allyl trifluoroacetate was obtained from Tokyo Chemical Industry (Tokyo, Japan) and was distilled under nitrogen. Triphenylphosphine (PPh3) was obtained from Wako Pure Chemical Industry (Osaka, Japan) and was purified by recrystallization from diethyl ether. Toluene was dried over sodium and distilled under nitrogen. Ethanol (EtOH) and methanol (MeOH) were dried over magnesium ethoxide and magnesium methoxide, respectively, and were distilled under nitrogen. A toluene solution of [(π-allyl)-NiOCOCF3]2/PPh3 (1) was prepared as described previously.33 Monomers (2a–g) were prepared as reported previously.33, 34, 48 Polyvinylpyrrolidone (PVP) (Mw=10 000 and 360 000) and poly(ethylene glycol) (Mw=8500) were obtained from Tokyo Chemical Industry and were used as received. Propyl cellulose and 2-hydroxypropyl cellulose were obtained from Wako Pure Chemical Industry and were used as received. PVP (Mw=29 000 (2.9% H2O), Mw=40 000 (4.6% H2O) and Mw=1 300 000 (3.8% H2O)), poly(propylene glycol) (Mw=8300 (0.007% H2O)), triblock copolymers of poly(ethylene glycol) and poly(propylene glycol) (ethylene glycol (EG):propylene glycol (PG):EG=40:20:40 (Mw=8400, EG:PG=5.49:1 (0.13% H2O)) and EG:PG:EG=20:70:20 (Mw=5800, EG:PG=0.602:1 (0.02% H2O))), methyl cellulose (30.5% methoxy content (1.5% H2O)), ethyl cellulose (48.8% ethoxy content (0.8% H2O)) and 2-hydroxyethyl cellulose (viscosity-average molecular weight (Mv)=90 000) were obtained from Sigma-Aldrich (Tokyo, Japan) and were used as received. All the polymerizations were carried out under nitrogen. Other reagents were used as received.
Characterization
Nuclear magnetic resonance (NMR) spectra were recorded on a JEOL ECP-300 instrument (JEOL, Tokyo, Japan) using tetramethylsilane as an internal standard (300 and 75 MHz for 1H NMR and 13C NMR, respectively). Fourier transform infrared (IR) spectra were recorded on a JASCO FT/IR-5300 (Tokyo, Japan) or a Nicolet iS10 instrument (Tokyo, Japan). Gel permeation chromatography (GPC) measurements were performed on a Shimadzu LC-10AS (Kyoto, Japan) equipped with Tosoh TSK-gel GMHHR-M tandem columns using CHCl3 as an eluent at 35 °C. Polystyrene standards were used for calibration. Gas chromatography (GC) analyses were performed on a Shimadzu GG-14B (Kyoto, Japan) or GC-2014 instrument (Kyoto, Japan, SE-30, 2 m, 100–230 °C, heating rate of 10 °C min−1). The morphologies, particle sizes and size distributions of the polymer microspheres were determined with a scanning electron microscope (SEM, JEOL JSM-5600 or Shimadzu SS-550) and optical microscope (Shimadzu OLS4000). All of the SEM and optical microscope size data reflect the averages of more than 100 particles and are calculated by formulas described previously.49 The contact angle measurements of the layers of the polymer microspheres were performed on a NICK LSE-ME1 contact angle meter (Saitama, Japan) using a water droplet (1.0 μl). Three measurements were taken across each sample, with their average being used for analysis. The layers of the polymer microspheres were prepared by the drop casting of suspensions of the polymer microspheres in MeOH (0.5 ml, 0.05 g ml−1) on a glass substrate. After the slow evaporation of the solvent at ambient temperature, the formed polymer layers were further dried under vacuum at ambient temperature for 1 h.
Dispersion polymerization of allene derivatives (Typical Procedure)
To a test tube equipped with a three-way stopcock and a magnetic stirring bar and containing PVP (Mw=40 000, 0.010 g, 5 wt%) a toluene solution of 1 was added (0.10 M, 0.15 ml, 0.015 mmol). After the removal of the toluene under vacuum, EtOH (1.5 ml) and 2a (198.3 mg, 1.5 mmol, [2a]/[1]=100) were added at ambient temperature and the mixture was stirred at 350 r.p.m. at that temperature for 6 h. After confirming the complete conversion of 2a by GC, the resulting polymer microspheres were separated by centrifugation and washed three times with MeOH, and the sample was then subjected to SEM measurement. The yield of the polymer microspheres was determined by dissolving the particles in tetrahydrofuran (THF) (2 ml) and then precipitating them into MeOH/water (v v−1=50/50, 100 ml). Poly(2a): 97% yield (0.17 g, 1.45 mmol). 1H-NMR (CDCl3): δ 2.84–3.10 (–CH2–, br, 2H × y), 5.08–5.31 (CH2=, –CH–, br, 2H × x+1H × x), 6.34 (=CH–, br, 1H × y), 6.79–7.05 (–C6H5, 5H), x: y (in Scheme 1)=17: 83. 13C-NMR (CDCl3): δ 25.6, 29.7, 33.4, 79.6, 116.1, 118.5, 120.5, 122.2, 138.3, 139.2, 141.3, 157.3. IR (neat): 3063, 3040, 2909, 2350, 1669, 1593, 1489, 1456, 1237, 1167, 1109, 1032, 833, 754 cm−1.
Poly(2b): 92% yield (0.30 g, 1.9 mmol). 1H-NMR (CDCl3): δ 2.76–2.97 (–CH2–, br, 2H × y), 3.57 (–OCH3, s, 3H), 4.96–5.68 (CH2=, –CH–, br, 2H × x+1H × x), 6.22 (=CH–, br, 1H × y), 6.54–7.11 (–C6H4–, 4H), x: y=18: 82. 13C-NMR (CDCl3): δ 24.4, 28.2, 33.2, 55.4, 80.2, 114.4, 116.5, 117.4, 139.3, 140.3, 143.1, 151.5, 154.7. IR (neat): 3043, 2906, 2833, 1591, 1505, 1226, 1105, 1036, 826, 756 cm−1.
Poly(2c): 94% yield (0.34 g, 1.90 mmol). 1H-NMR (CDCl3): δ 2.98–3.26 (–CH2–, br, 2H × y), 5.24–5.46 (CH2=, –CH–, br, 2H × x+1H × x), 6.55 (=CH–, br, 1H × y), 7.14–7.42 (–C10H7, 7H), x: y=20: 80. 13C-NMR (CDCl3): δ 28.8, 33.1, 110.0, 116.2, 118.2, 119.1, 124.0, 126.2, 126.9, 127.4, 128.9, 129.6, 134.0, 138.7, 139.1, 142.5, 155.1. IR (neat): 3057, 2903, 2836, 1630, 1599, 1510, 1466, 1217, 1019, 841, 808, 745 cm−1.
Poly(2d): 91% yield (0.38 g, 1.8 mmol). 1H-NMR (CDCl3): δ 2.74–2.97 (–CH2–, br, 2H × y), 5.00–5.66 (CH2=, –CH–, br, 2H × x+1H × x), 6.18 (=CH–, br, 1H × y), 6.55–7.18 (–C6H4–, 4H), x: y=18: 82. 13C-NMR (CDCl3): δ 25.3, 28.6, 33.1, 79.9, 113.2, 115.0, 117.5, 132.5, 138.4, 139.1, 141.7, 156.1. IR (neat): 3057, 2903, 2836, 1630, 1599, 1510, 1466, 1217, 1019, 841, 808, 745 cm−1.
Poly(2e): 94% yield (0.22 g, 1.9 mmol). 1H-NMR (CDCl3): δ 2.96 (–CH2–, br, 2H), 6.34 (CH2=, br, 1H), 6.70–7.80 (–C6H5, 5H). 13C-NMR (CDCl3): δ 30.3, 34.2, 125.5, 126.3, 128.2, 128.8, 135.7, 137.2, 137.8, 151.5. IR (neat): 3024, 2981, 2911, 1942, 1597, 1460, 1377 cm−1.
Poly(2a)-b-poly(2b): 89% yield (0.44 g, 3.7 mmol). 1H-NMR (CDCl3) δ 2.63–3.08 (–CH2–, br, 2H × y), 3.65 (–OCH3, s, 3H), 5.06–5.78 (CH2=, –CH–, br, 2H × x+1H × x), 6.33 (=CH–, br, 1H × y), 6.58–7.22 (–C6H4–, 4H). 13C-NMR (CDCl3): δ 27.6, 28.4, 29.3, 30.3, 55.4, 114.4, 115.4, 116.2, 117.3, 120.3, 122.1, 128.4, 129.5, 132.0, 139.1, 140.3, 142.4, 151.1, 154.7. IR (neat): 3043, 2930, 2834, 1669, 1594, 1503, 1490, 1440, 1295, 1211, 1103, 1034, 903, 823, 750 cm−1.
Synthesis of crosslinked polymer microspheres by one-shot monomer addition (Typical Procedure)
To a test tube equipped with a three-way stopcock and a magnetic stirring bar and containing PVP (Mw=40 000, 0.010 g, 5 wt%) a toluene solution of 1 was added (0.10 M, 0.15 ml, 0.015 mmol). After the removal of the toluene under vacuum, EtOH (0.5 ml) and an EtOH (1.0 ml) solution of 2a (0.20 g, 1.50 mmol, [2a]/[1]=100) and 2f (0.055 g, 0.30 mmol, [2f]/[1]=20) at ambient temperature were added, and the mixture was stirred at 350 r.p.m. at that temperature. During the polymerization for 24 h, the consumption of both 2a and 2f reached ~50%, as monitored by GC, whereas no polymerization took place when the reaction was prolonged for an additional 24 h. The resulting polymer microspheres were collected in 52% yield (0.14 g) by centrifugation and washed three times with MeOH. The obtained polymer microspheres were suspended again in CHCl3 (5 ml) and filtered (CHCl3-insoluble:CHCl3-soluble=98:2). IR (neat): 3040, 2922, 1669, 1594, 1490, 1456, 1219, 1167, 1104, 1023, 888, 751, 690 cm−1.
Synthesis of crosslinked polymer microspheres by sequential monomer addition (Typical Procedure)
To a test tube equipped with a three-way stopcock and a magnetic stirring bar and containing PVP (Mw=40 000, 0.010 g, 5 wt%) a toluene solution of 1 was added (0.10 M, 0.15 ml, 0.015 mmol). After the removal of the toluene under vacuum, EtOH (1.5 ml) and 2a (0.20 g, 1.50 mmol, [2a]/[1]=100) were added at ambient temperature, and the mixture was stirred at 350 r.p.m. at that temperature for 6 h. After confirming the complete conversion of 2a by GC, an EtOH solution of 2f (0.60 M, 0.50 ml, 0.30 mmol, [2f]/[1]=20) was added, and the reaction was continued for an additional 24 h at that temperature. After confirming the complete conversion of 2f by GC, the resulting polymer microspheres were separated by centrifugation and washed three times with MeOH. Finally, the crosslinked polymer microspheres were collected in 87% yield (0.23 g). The obtained polymer microspheres were suspended again in CHCl3 (5 ml) and filtered (CHCl3-insoluble:CHCl3-soluble=91:9). IR (neat): 3042, 2922, 1669, 1594, 1490, 1456, 1219, 1167, 1104, 1023, 888, 751, 690 cm−1.
Synthesis of crosslinked polymer microspheres possessing a hydroxy group by the sequential addition of phenoxyallene (2a), 1,4-Bis(allenoxy)benzene (2f) and 4-(hydroxymethyl)phenoxyallene (2g)
To a test tube equipped with a three-way stopcock and a magnetic stirring bar and containing PVP (Mw=40 000, 0.010 g, 5 wt%), a toluene solution of 1 was added (0.10 M, 0.15 ml, 0.015 mmol). After the removal of the toluene under vacuum, EtOH (1.5 ml) and 2a (0.20 g, 1.50 mmol, [2a]/[1]=100) were added at ambient temperature, and the mixture was stirred at 350 r.p.m. at that temperature for 6 h. After confirming the complete conversion of 2a by GC, an EtOH solution of 2f (0.60 M, 0.50 ml, 0.30 mmol, [2f]/[1]=20) was added, and the polymerization was continued for an additional 24 h at that temperature. After confirming the complete conversion of 2f by GC, an EtOH solution of 2g (0.60 M, 0.50 ml, 0.30 mmol, [2g]/[1]=20) was added, and the polymerization was continued for an additional 24 h at that temperature. After confirming the complete conversion of 2g by GC, the resulting polymer microspheres were separated by centrifugation and then washed three times with MeOH, suspended again in CHCl3 (5 ml) and filtered. Finally, the crosslinked polymer microspheres were obtained as a CHCl3-insoluble fraction in 79% yield (0.25 g). IR (neat): 3325, 3040, 2901, 1666, 1593, 1490, 1456, 1290, 1220, 1166, 1104, 1023, 888, 824, 751, 688 cm−1.
Condensation of 4-iodobenzoic acid onto crosslinked polymer microspheres possessing a hydroxy group
To a round-bottom flask, hydroxy-functionalized crosslinked polymer microspheres were added (3a) (0.051 g, prepared from 2a, 2f and 2g in the ratio of 1:2a:2f:2g=1:100:50:20; hydroxy content: 0.80 mmol g), 4-iodobenzoic acid (0.050 g, 0.20 mmol, 5.0 equiv), dicyclohexylcarbodiimide (0.042 g, 0.20 mmol, 5.0 equiv), N,N-dimethyl-4-aminopyridine (0.0010 g, 0.040 mmol, 1.0 equiv) and THF (1.0 ml) at ambient temperature. After being stirred for 24 h, the resulting polymer microspheres (3b) were separated by centrifugation and washed three times with THF. IR (neat): 3044, 2922, 2898, 1718, 1667, 1593, 1490, 1456, 1431, 1336, 1291, 1273, 1218, 1167, 1104, 1008, 996, 891, 825, 751, 690, 667 cm−1.
Cleavage of methyl 4-iodobenzoate from polymer microspheres
To a round-bottom flask-esterified crosslinked polymer microspheres were added (3b) (obtained from hydroxy-functionalized crosslinked polymer microspheres (0.051 g) that were prepared from 2a, 2f and 2g in the ratio of 1:2a:2f:2g=1:100:50:20), NaOMe (0.00040 g, 0.0080 mmol, 0.10 equiv), THF (0.8 ml) and MeOH (0.2 ml) at ambient temperature. After being stirred at 45 °C for 24 h, the polymer microspheres were filtered and washed with MeOH. The filtrate was concentrated, and the residue was purified by silica gel column chromatography (eluent: CH2Cl2/n-hexane, v v−1=4/1) to give methyl 4-iodobenzoate (0.011 g, 0.040 mmol) as a white solid. 1H NMR (CDCl3): δ 3.91 (-OCH3, s, 3H), 7.74 (-C6H4-, d, 4H, J=8.6 Hz), 7.80 (-C6H4-, d, 2H, J=8.6 Hz). IR (neat): 2960, 2947, 1715, 1585, 1438, 1391, 1365, 1285, 1273, 1116, 1107, 1006, 846, 756, 686 cm−1.
Solid-phase Suzuki–Miyaura reaction of phenylboronic acid with a 4-iodobenzoate moiety attached on the crosslinked polymer microspheres
To a round-bottom flask-esterified crosslinked polymer microspheres were added (3b) (obtained from hydroxy-functionalized crosslinked polymer microspheres (0.062 g) that were prepared from 2a, 2f and 2g in the ratio of 1:2a:2f:2g=1:100:50:20), Pd(OAc)2 (0.0010 g, 0.0040 mmol, 0.10 equiv), PPh3 (0.0040 g, 0.016 mmol, 0.40 equiv), N,N-dimethylformamide (DMF) (1.0 ml), sodium carbonate (0.011 g, 0.10 mmol, 2.5 equiv) and phenylboronic acid (0.015 g, 0.12 mmol, 3.0 equiv) at ambient temperature. After being stirred for 24 h at 100 °C, the resulting polymer microspheres (3c) were separated by centrifugation and washed with water, MeOH and ethyl acetate.
The cross-coupling product was cleaved from the polymer microspheres in methyl ester form as follows. To a round-bottom flask reacted crosslinked polymer microspheres (3c), NaOMe (0.00040 g, 0.0080 mmol, 0.10 equiv), THF (0.80 ml) and MeOH (0.20 ml) were added at ambient temperature. After being stirred at 45 °C for 24 h, the polymer microspheres were filtered and washed with MeOH. The filtrate was concentrated, and the residue was purified by silica gel column chromatography (eluent: CH2Cl2/n-hexane, v v−1=4/1) to give methyl 4-phenylbenzoate (4) in 72% yield (0.0090 g, 0.035 mmol) as a white solid. 1H NMR (CDCl3): δ 3.94 (-OCH3, s, 3H), 7.40–7.47 (-C6H4-C6H5, 3H), 7.62–7.68 (-C6H4-C6H5, 4H), 8.11 (-C6H4-C6H5, d, 2H, J=8.8 Hz). IR (neat): 2960, 2927, 2855, 1719, 1608, 1487, 1438, 1405, 1314, 1589, 1261, 1183, 1114, 1099, 1020, 858, 802, 750, 702 cm−1.
Preparation of palladium-supported cross-linked polymer microspheres
First, the condensation of 4-(diphenylphosphino)benzoic acid onto the crosslinked polymer microspheres possessing a hydroxy group was performed as follows. To a round-bottom flask crosslinked polymer microspheres (0.10 g, prepared from 2a, 2f and 2g in the ratio of 1:2a:2f:2g=1:100:50:20, hydroxy content: 0.80 mmol g−1), 4-(diphenylphosphino)benzoic acid (0.028 g, 0.09 mmol, 3.0 equiv), dicyclohexylcarbodiimide (0.019 g, 0.09 mmol, 3.0 equiv), N,N-dimethyl-4-aminopyridine (0.004 g, 0.03 mmol, 0.3 equiv) and THF (1.0 ml) were added at ambient temperature. After being stirred at that temperature for 24 h, the resulting polymer microspheres were separated by centrifugation and washed three times with THF.
Next, palladium was loaded onto polymer microspheres by the following procedure. To a round-bottom flask phosphine-containing polymer microspheres, PdCl2 (0.0016 g, 0.090 mmol, 3.0 equiv) and DMF (1.0 ml) were added at ambient temperature. After being stirred for 24 h, the resulting polymer microspheres were separated by centrifugation and washed three times with DMF to give palladium-supported crosslinked polymer microspheres (0.090 g).
Suzuki–Miyaura coupling reaction using palladium-supported crosslinked polymer microspheres as a catalyst
To a round-bottom flask iodobenzene (0.10 g, 0.50 mmol), palladium-supported crosslinked polymer microspheres (0.0020 g, 1.0 mol% of palladium), potassium carbonate (0.346 g, 2.50 mmol, 2.5 equiv), phenylboronic acid (0.073 g, 0.60 mmol, 1.2 equiv), DMF (2.5 ml) and dodecane (0.085 g, 0.50 mmol, an internal standard) were added at ambient temperature. After being stirred at 100 °C for 24 h, the reaction mixture was subjected to GC analysis.
Results and discussion
Living dispersion polymerization
Following the conditions for the radical dispersion polymerization of styrene,9, 14 polymerization of phenoxyallene (2a) ([2a]0=1.0, [2a]/[1]=100) by [(π-allyl)-NiOCOCF3]2/PPh3 (1) was performed at ambient temperature in EtOH containing PVP (5 wt%) under magnetic stirring at 350 r.p.m. (Scheme 1). As a result, the polymerization system became heterogeneous within 1 min, and the conversion of 2a was complete within 4–5 h, resulting in a suspension of polymer microspheres (Supplementary Figure S1 and Supplementary Movie S1). SEM observations clearly indicated the production of polymer microspheres with a narrowly dispersed diameter distribution (Dw/Dn), where the number-average diameter (Dn) and Dw/Dn were determined to be 1.10 and 1.01 μm, respectively. The size distribution of the microspheres was also evaluated in the EtOH dispersion state using optical microscopy (Supplementary Figure S2). As a result, Dn and Dw/Dn were estimated to be 1.15 and 1.03, respectively, almost identical to the values obtained with the SEM measurements in the dried state. When all of the microspheres were dissolved in THF and precipitated into MeOH/water (v v−1=50/50), the polymers were isolated at a 97% yield with the number-average molecular weight and molecular weight distribution estimated by GPC to be Mn=11 700 and Mw/Mn=1.06.
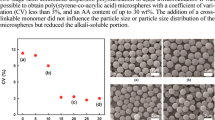
The dispersion polymerization of 2a by 1 was similarly performed under the various feed ratios of [2a]/[1], from which the polymer microspheres were consistently produced at a high yield with Mn increasing linearly as a function of [2a]/[1] ratio (Supplementary Table S1 and Figure 1). These results indicate that the π-allylnickel-catalyzed coordination dispersion polymerization of 2a occurs via a living mechanism. It is of note that the size of the polymer microspheres was not affected by the monomer feed ratio if the polymerization was started from the same initial monomer concentration ([2a]0=1.0 M, [1]0=1.0 × 10−2–5.0 × 10−2 M) (Supplementary Table S1).

Dependence of Mn and Mw/Mn of poly(2a) on [2a]/[1] ratio. Mn, molecular number; Mw, molecular weight.
The living character of this system was further demonstrated by the postpolymerization experiment. After the complete conversion of 2a (100 equiv), another 100 equiv of 2a was added, and postpolymerization was conducted for an additional 24 h. In GPC, the elution peak shifted to the higher molecular weight region (Mn=21 100, Mw/Mn=1.10) compared with that of the starting poly(2a) (Mn=11 600, Mw/Mn=1.09), whereas the narrow molecular weight distribution was preserved. SEM observations showed that the polymer microspheres had a larger diameter (Dn=1.36 μm, Dw/Dn=1.01) compared with those before the postpolymerization of 2a (Dn=1.10 μm, Dw/Dn=1.01), whereas the narrowly dispersed diameter distribution was maintained (Supplementary Figure S3). It is of note that the number-average volume (Dn3) of the microspheres obtained by the postpolymerization process was found to be Dn3=10.5 μm3, which corresponds to the duplicated volume before the postpolymerization (Dn3=5.58 μm3).
Regarding the size of the resulting polymer microspheres, the polymerization conditions, such as the initial monomer concentration, the polarity of the reaction media and the structure, molecular weight and concentration of the stabilizers were found to be influential in the present living dispersion system, in accordance with the reports on the free radical dispersion polymerization of nonpolar monomers such as styrene (see Supplementary Tables S2–S6 and Supplementary Figures S4–S8).9, 50, 51, 52, 53 Moreover, the monomer structure was found to be quite effective for the control of the size of the resulting polymer microspheres. The living dispersion polymerization of allene derivatives such as 4-methoxyphenoxyallene (2b), 2-naphthoxyallene (2c), 4-bromophenoxyallene (2d) and phenylallene (2e) was performed in EtOH containing 5 wt% of PVP at ambient temperature (Table 1 and Supplementary Figure S9). In all cases, the polymerization occurred successfully to yield well-defined polymer microspheres with narrowly dispersed diameter distributions. The resulting polymer microspheres were composed of well-defined polymers with regulated molecular weight and a narrow molecular weight distribution. It is of note that the monomer structure was found to strongly affect the size of the resulting polymer microspheres. In particular, when 2e was used as a monomer, Dn exceeded 3 μm. The polymer microspheres obtained in the present study often exhibited structural color owing to the highly regulated size distribution. For example, by casting the suspension of the microspheres obtained in entry 2 onto a glass substrate, we could observe greenish-yellow color or green color, depending on the viewing angle (Supplementary Figure S10).
Living dispersion block copolymerization
The dispersion copolymerization of 2a and 2b was performed in EtOH containing 5 wt% of PVP at ambient temperature (Scheme 2). After the complete conversion of 2a (100 equiv), 2b (100 equiv) was added, and the copolymerization was conducted for an additional 24 h. As clearly shown in Figure 2, the elution peak in GPC shifted to the higher molecular weight region ((b); Mn=24 100, Mw/Mn=1.10) compared with that of the starting poly(2a) ((a); Mn=12 000, Mw/Mn=1.09), whereas the narrow molecular weight distribution was preserved. This result indicates that the copolymerization occurred not only from the surface but also from the inside of the particles. SEM observations showed that the polymer microspheres obtained after the postpolymerization of 2b had a larger size (Dn=1.37 μm, Dw/Dn=1.01) compared with those before the postpolymerization (Dn=1.10 μm, Dw/Dn=1.01), whereas the diameter distribution remained sufficiently narrow. Peaks for both monomer units were detected in the 1H NMR spectrum of the block copolymer, where the composition of two monomers in poly(2a)-b-poly(2b) (2a:2b=100:112) agreed well with the feed ratio of the monomers (2a:2b=100:100) (Supplementary Figure S11). If the microspheres of poly(2a) having deactivated growing species and the comonomer (2b) were mixed in EtOH under the postpolymerization conditions for 1 h, the microspheres of poly(2a) partially provided secondary aggregated complex textures (Supplementary Figure S12), indicating that the comonomer (2b) can swell the microspheres during and/or in advance of the postpolymerization. Because the postpolymerization of the comonomer should proceed inside of the polymer microspheres, it is essential to have good compatibility between the comonomer and the polymer microspheres of the first monomer, possibly limiting the combination of the monomers suitable for the dispersion block copolymerization.

Gel permeation chromatography (GPC) traces and scanning electron microscope (SEM) images of poly(2a) produced by dispersion polymerization of 2a (100 equiv) (a) and a block copolymer obtained by the postpolymerization of 2b (100 equiv) (b).
Synthesis of crosslinked polymer microspheres
Although crosslinked polymer microspheres with narrowly dispersed diameter distribution are attractive for many applications, they are difficult to access using the conventional free radical dispersion polymerization technique.23, 24, 25 Because the present living dispersion polymerization can provide block copolymers by simple postpolymerization as mentioned above, the synthesis of crosslinked polymer microspheres was similarly performed by using 1,4-diallenoxybenzene (2f) as a crosslinker.
First, the one-shot living dispersion copolymerization was performed in EtOH at ambient temperature in the presence of 5 wt% of PVP by feeding mixed monomers of 2a and 2f (2a: 100 equiv; 2f: 10–30 equiv) (Scheme 3a). In all cases, the consumption of the monomers stopped before reaching complete conversion to produce the crosslinked polymer microspheres at lower yields. The consumption of the monomers did not occur even after a prolonged reaction period, presumably because the crosslinked polymer microspheres could not absorb the monomers that suppressed the chain growth inside the particles. Although the yield of the crosslinked polymer microspheres was not sufficiently high, SEM observations indicate that the produced crosslinked polymer microspheres had a narrowly dispersed diameter distribution, and the size of the resulting crosslinked microspheres was smaller than that of the non-crosslinked microspheres obtained from 2a (Table 2 and Figures 3a–c).

Scanning electron microscope (SEM) images of the obtained crosslinked polymer microspheres listed in Table 7, entries 1 (a), 2 (b) and 3 (c), and Table 8, entries 1 (d), 2 (e), 3 (f) and 4 (g).
In contrast, sequential copolymerization of 2a and 2f was found to be more effective for obtaining crosslinked polymer microspheres at higher yields. After the complete conversion of 2a (100 equiv), various amounts of 2f (10–50 equiv) were added and copolymerization was conducted for an additional 24 h (Scheme 3b). In all cases, 2f was consumed completely to produce polymer microspheres at higher yields. All crosslinked polymer microspheres proved to have a narrowly dispersed diameter distribution, as supported by SEM observations (Table 3 and Figures 3d–g). It was found that all crosslinked polymer microspheres produced after the postpolymerization of 2f were slightly larger than those of poly(2a), possibly supporting the hypothesis that the postpolymerization occurs inside of each polymer microsphere.
The solvent-resistant properties of the crosslinked polymer microspheres are presented in Tables 2 and 3 (i.e., the samples prepared by both the one-shot and sequential copolymerizations), as evaluated by submerging them for 10 min with CHCl3, which is a good solvent for non-crosslinked polymer microspheres of poly(2a). For the samples prepared by the one-shot copolymerization, the microspheres exhibited high solvent resistance irrespective of the added amount of 2f; the spherical particles recovered almost completely, and the recovered samples retained the original spherical shape (Figures 4a–c). In the case of the samples prepared by sequential copolymerization, the solvent resistance was affected by the amount of the crosslinker. That is, the sample obtained from 2a (100 equiv) and 2f (10 equiv) in Table 3, entry 1 dissolved completely upon contact with CHCl3 (Figure 4d), whereas the samples obtained by the use of a larger amount of 2f (⩾20 equiv; Table 3, entries 2–4) successfully gave entirely cross-inked polymer microspheres that hardly dissolved in CHCl3 and retained the original spherical shape (Figures 4e–g).

Scanning electron microscope (SEM) images of the CHCl3-treated samples listed in Table 7, entries 1 (a), 2 (b) and 3 (c), and Table 8, entries 1 (d), 2 (e), 3 (f) and 4 (g).
Synthesis of functionalized crosslinked polymer microspheres
Based on the crosslinking technique described above, solvent-resistant polymer microspheres bearing functional groups on their surfaces can be designed by continuing the postpolymerization of functional monomers from the living growing ends that still exist after crosslinking the polymer microspheres. For example, polymer microspheres possessing hydroxy functional groups were prepared by the postpolymerization of 4-(hydroxymethyl)phenoxyallene (2g) from the living end of the crosslinked polymer microspheres. After the preparation of the crosslinked polymer microspheres from 2a (100 equiv) and 2f (20 equiv) under the conditions shown in entry 2 (Tables 3), 2g (20 equiv) was added to the reaction mixture as a third comonomer, and postpolymerization was carried out for several hours at ambient temperature (Scheme 3c). SEM observation showed that the polymer microspheres retained the narrowly dispersed particle shape after the postpolymerization of 2g (Dn=1.11 μm, Dw/Dn=1.01), with the size and size distribution very close to those before the copolymerization of 2g (Dn=1.10 μm, Dw/Dn=1.01) (Figure 5a). A broad peak for νOH was observed at 3325 cm−1 in the IR spectrum of the sample obtained after the copolymerization of 2g (Figure 5b). Owing to the hydroxy groups attached to the polymer microspheres, it is expected that the polymer microspheres will have a hydrophilic surface. According to Chen et al.,54 the contact angle of the water droplet on the thin layers of the polymer microspheres can be used to evaluate the hydrophilicity of the surface of the polymer microspheres. Therefore, a thin layer of the polymer microspheres was prepared by drop casting the suspension of the polymer microspheres in EtOH on a glass substrate followed by the slow evaporation of EtOH, and the contact angle of the water droplet on the thin layer was evaluated. Figures 5c and d show the contact angles of the water droplet on the layer of the polymer microspheres obtained before and after the post-polymerization of 2g. As expected, the contact angle was decreased from 122° to 44° by the postpolymerization of 2g, indicating that the surface nature of the polymer microspheres changed from hydrophobic to hydrophilic by the postpolymerization of the OH-functionalized monomer (2g).

(a) Scanning electron microscope (SEM) image of OH-functionalized crosslinked polymer microspheres. (b) Infrared (IR) spectra of the crosslinked polymer microspheres obtained before and after the postpolymerization of 2g. (c and d) Profiles of water droplets on the layer of polymer microspheres obtained before (c) and after (d) postpolymerization of 2g.
Applications of functionalized crosslinked polymer microspheres
Two preliminary experiments were performed to demonstrate the potential of the OH-functionalized crosslinked polymer microspheres described in the preceding section in applications using solid-phase synthesis and solid-supported catalysis as examples.
First, the Suzuki–Miyaura cross-coupling reaction was carried out using the OH-functionalized crosslinked polymer microspheres under the conditions described in a previous report on the solid-phase organic reaction using the commercially available OH-functionalized Wang resin (Scheme 4a).55 The OH-functionalized crosslinked polymer microspheres (3a) were prepared by the block copolymerization of 2a, 2f and 2g ([1]:[2a]:[2f]:[2g]=1:100:50:20). The polymer microspheres (3a) thus obtained are expected to have 0.80 mmol g−1 of the hydroxy group, assuming that all hydroxy groups were successfully incorporated on the resin. The hydroxy content of the present resin was almost comparable to that of the commercially available Wang resin (1.2 mmol g−1 of the hydroxy-content). Unlike the Wang resin, which is commonly used as a swollen form in organic solvents, the polymer microspheres (3a) do not swell in organic solvents because of their highly crosslinked core structure. The condensation reaction of 4-iodobenzoic acid (5 equiv) with the hydroxy groups on 3a was performed in THF at ambient temperature for 24 h in the presence of N,N'-dicyclohexylcarbodiimide (5 equiv) and N,N-dimethyl-4-aminopyridine (0.1 equiv). A peak attributable to νC=O was observed at 1718 cm−1 in the IR spectrum of the resin (3b) obtained after the condensation, whereas a broad peak for νOH at 3325 cm−1 disappeared completely (Supplementary Figure S13). The Suzuki–Miyaura cross-coupling reaction of the aryl iodide moieties on the resin with phenylboronic acid (3 equiv) was carried out in DMF at 100 °C for 24 h by use of Pd(OAc)2/4 PPh3 (10 mol%). Finally, the product (4) was isolated at a 72% yield (with respect to the aryl iodide moieties loaded onto the crosslinked polymer microspheres) by cleavage from the microspheres (3c) using the transesterification reaction under basic conditions followed by purification using silica gel column chromatography.
Next, a solid-supported Pd catalyst was prepared, and its activity was demonstrated in the Suzuki–Miyaura cross-coupling reaction (Scheme 4b). That is, the OH-functionalized crosslinked polymer microspheres prepared by the block copolymerization of 2a, 2f and 2g ([1]:[2a]:[2f]:[2g]=1:100:50:20) were subjected to a condensation reaction with 4-diphenylphosphinobenzoic acid (3 equiv) by N,N'-dicyclohexylcarbodiimide (3 equiv) and N,N-dimethyl-4-aminopyridine (0.1 equiv) in THF at ambient temperature for 24 h. Formation of a palladium complex on the surface of the polymer microspheres was performed by mixing with PdCl2 (3 equiv) in DMF at ambient temperature for 1 h, and the resulting supported microspheres were washed three times with DMF. The catalytic activity of the Pd-supported polymer microspheres was evaluated in the Suzuki–Miyaura cross-coupling reaction of iodobenzene and phenylboronic acid. As a result, the reaction took place smoothly in the presence of the supported catalyst containing 1 mol% of the palladium salt to produce the target biphenyl product at a 92% GC yield. The catalytic activity was retained after repeating the recovery and reuse of the supported catalyst for at least three times to consistently produce biphenyl at high yields (Supplementary Table S7).
Conclusions
The π-allylnickel-catalyzed coordination dispersion polymerization of less polar allene derivatives such as phenoxyallene and its derivatives in polar polymerization media was shown to proceed in a living manner to produce polymer microspheres consisting of a well-defined particle size and polymer chain. It was found that the monomer structure, the initial monomer concentration, the polymerization solvents and the stabilizers affect the size of the resulting polymer microspheres, whereas the molecular weight and its distribution were predetermined by the ratio of the monomer to initiator.
The use of 1,4-diallenoxybenzene as a comonomer for the postpolymerization successfully provides crosslinked polymer microspheres that exhibit high solvent resistance. The surface of the crosslinked polymer microspheres can be functionalized by sequential copolymerization of functional allene monomers from the living growing ends in the crosslinked polymer microspheres. As applications of this technique, crosslinked hydroxy-functionalized polymer microspheres were prepared and used for solid-phase organic synthesis. Additionally, their further functionalization was found to produce a supported-palladium catalyst suitable for use in the cross-coupling reaction.
Because the π-allylnickel-catalyzed living coordination polymerization can cover a wide range of functional monomers, it is expected that the dispersion polymerization using this living system will provide well-defined functionalized polymer microspheres consisting of versatile functional groups. Accordingly, the macromolecular design of functional polymer microspheres and their applications are currently being investigated.

π-Allylnickel-catalyzed living coordination dispersion polymerization of allene derivatives.

Dispersion block copolymerization of 2a and 2b.

(a and b) One-shot copolymerization (a) and sequential block copolymerization (b) of 2a and 2f. (c) Synthesis of OH-functionalized crosslinked polymer microspheres by sequential copolymerization of 2a, 2f and 2g.

(a) Solid-phase synthesis on OH-functionalized crosslinked polymer microspheres. (b) Preparation of supported-Pd polymer microspheres.
References
Ugelstad, J., Berge, A., Ellingsen, T., Schmid, R., Nilsen, T. N., Mork, P. C., Stenstad, P., Hornes, E. & Olsvik, O. Preparation and application of new monosized polymer particles. Prog. Polym. Sci. 17, 87–161 (1992).
Xia, Y. N., Gates, B., Yin, Y. D. & Lu, Y. Monodispersed colloidal spheres: old materials with new applications. Adv. Mater. 12, 693–713 (2000).
Mathiowitz, E., Jacob, J. S., Jong, Y. S., Carino, G. P., Chickering, D. E., Chaturvedi, P., Santos, C. A., Vijayaraghavan, K., Montgomery, S., Bassett, M. & Morrell, C. Biologically erodable microsphere as potential oral drug delivery system. Nature 386, 410–414 (1997).
Fruchtel, J. S. & Jung, G. Organic chemistry on solid supports. Angew. Chem. Int. Ed. 35, 17–42 (1996).
Merrifield, R. B. Solid phase peptide synthesis. 1. Synthesis of a tetrapeptide. J. Am. Chem. Soc. 85, 2149–2154 (1963).
Jiang, L., Zhao, Y. & Zhai, J. A lotus-leaf-like superhydrophobic surface: a porous microsphere/nanofiber composite film prepared by electrohydrodynamics. Angew. Chem. Int. Ed. 43, 4338–4341 (2004).
Zosel, A. & Ley, G. Influence of cross-linking on structure, mechanical-properties, and strength of latex films. Macromolecules 26, 2222–2227 (1993).
Lok, K. P. & Ober, C. K. Particle-size control in dispersion polymerization of polystyrene. Can. J. Chem. 63, 209–216 (1985).
Lovell, P. A. in Emulsion Polymerization and Emulsion Polymers (eds Lovell, P. A. & El-Aasser, M. S. ) (Wiley: Chichester, UK, 1997).
Barrett, K. E. J. & Thomas, H. R. Kinetics of dispersion polymerization of soluble monomers. I. Methyl methacrylate. J. Polym. Sci. Part A 7, 2621–2650 (1969).
Awan, M. A., Dimonie, V. L. & El-Aasser, M. S. Anionic dispersion polymerization of styrene. 1. Investigation of parameters for preparation of uniform micron-size polystyrene particles with narrow molecular weight distribution. J. Polym. Sci. Part A 34, 2633–2649 (1996).
Awan, M. A., Dimonie, V. L. & El-Aasser, M. S. Anionic dispersion polymerization of styrene. 2. Mechanism of particle formation. J. Polym. Sci. Part A 34, 2651–2664 (1996).
Kim, J., Jeong, S. Y., Kim, K. U., Ahn, Y. H. & Quirk, R. P. Anionic dispersion polymerization.1. Control of particle size. J. Polym. Sci. Part A 34, 3277–3288 (1996).
Jayachandran, K. N. N. & Chatterji, P. R. Preparation of linear and crosslinked polymer microspheres by dispersion polymerization. J. Macromol. Sci. Polym. Rev. 41, 79–94 (2006).
Holderle, M., Baumert, M. & Mulhaupt, R. Comparison of controlled radical styrene polymerizations in bulk and nonaqueous dispersion. Macromolecules 30, 3420–3422 (1997).
Gabaston, L. I., Jackson, R. A. & Armes, S. P. Living free-radical dispersion polymerization of styrene. Macromolecules 31, 2883–2888 (1998).
Song, J. S. & Winnik, M. A. Monodisperse, micron-sized reactive low molar mass polymer microspheres by two-stage living radical dispersion polymerization of styrene. Macromolecules 39, 8318–8325 (2006).
Min, K. & Matyjaszewski, K. Atom transfer radical dispersion polymerization of styrene in ethanol. Macromolecules 40, 7217–7222 (2007).
Sugihara, S., Sugihara, K., Armes, S. P., Ahmad, H. & Lewis, A. L. Synthesis of biomimetic poly(2-(methacryloyloxy)ethyl phosphorylcholine) nanolatexes via atom transfer radical dispersion polymerization in alcohol/water mixtures. Macromolecules 43, 6321–6329 (2010).
Shim, S. E., Jung, H., Lee, H., Biswas, J. & Choe, S. Living radical dispersion photopolymerization of styrene by a reversible addition–fragmentation chain transfer (RAFT) agent. Polymer 44, 5563–5572 (2003).
Ki, B., Yu, Y. C., Jeon, H. J., Yu, W.-R., Ryu, H. W. & Youk, J. H. Dispersion polymerization of styrene using poly(4-vinylpyridine) macro-RAFT agent under UV radiation. Fibers Polym. 13, 135–138 (2012).
Warren, N. J. & Armes, S. P. Polymerization-induced self-assembly of block copolymer nano-objects via RAFT aqueous dispersion polymerization. J. Am. Chem. Soc. 136, 10174–10185 (2014).
Song, J. S., Tronc, F. & Winnik, M. A. Two-stage dispersion polymerization toward monodisperse, controlled micrometer-sized copolymer particles. J. Am. Chem. Soc. 126, 6562–6563 (2004).
Song, J. S. & Winnik, M. A. Cross-linked, monodisperse, micron-sized polystyrene particles by two-stage dispersion polymerization. Macromolecules 38, 8300–8307 (2005).
Tan, J., Rao, X., Wu, X., Deng, H., Yang, J. & Zeng, Z. Photoinitiated RAFT dispersion polymerization: a straightforward approach toward highly monodisperse functional microspheres. Macromolecules 45, 8790–8795 (2012).
Hawkett, B. S., Napper, D. H. & Gilbert, R. G. Seeded emulsion polymerization of styrene. J. Chem. Soc. Faraday Trans. 76, 1323–1343 (1980).
Wang, W., Dimonie, V. L., Sudol, E. D. & El-Aasser, M. S. Seeded dispersion polymerization. J. Appl. Polym. Sci. 84, 2710–2720 (2002).
Song, Z., Daniels, E. S., Sudol, E. D., Klein, A. & El-Aasser, M. S. Seeded dispersion polymerization of MMA using submicron PMMA particles as seed: a mechanistic study Colloid Polym. Colloid Polym. Sci. 292, 645–952 (2014).
Capek, I. Surface active properties of polyoxyethylene macromonomers and their role in radical polymerization in disperse systems. Adv. Colloid Interface Sci. 88, 295–357 (2000).
Kawaguchi, H. Functional polymer microspheres. Prog. Polym. Sci. 25, 1171–1210 (2000).
Zetterlund, P. B., Kagawa, Y. & Okubo, M. Controlled/living radical polymerization in dispersed systems. Chem. Rev. 108, 3747–3794 (2008).
Zetterlund, P. B., Thickett, S. C., Perrier, S., Bourgeat-Lami, E. & Lansalot, M. Controlled/living radical polymerization in dispersed systems: an update. Chem. Rev. 115, 9745–9800 (2015).
Tomita, I., Kondo, Y. & Takagi, K. A novel living coordination polymerization of methoxyallene by π-allylnickel catalyst. Macromolecules 27, 4413–4414 (1994).
Endo, T., Takagi, K. & Tomita, I. Design and synthesis of polymerizable cumulated double bond system. Living coordination polymerization of alkylallenes by π-allylnickel catalyst. Tetrahedron 53, 15187–15196 (1997).
Takagi, K., Tomita, I. & Endo, T. A novel living coordination polymerization of phenylallene derivatives by π-allylnickel catalyst. Macromolecules 30, 7386–7390 (1997).
Takagi, K., Tomita, I. & Endo, T. Block copolymerization of alkoxyallenes with phenylallene by the living coordination system with π-allylnickel catalyst. Polym. Bull. 39, 685–692 (1997).
Takagi, K., Tomita, I. & Endo, T. Living coordination polymerization of alkylallenes by π-allylnickel catalyst: observation of an extremely high polymerizability of 1,2-cyclononadiene. Chem. Lett. 26, 1187–1188 (1997).
Takagi, K., Tomita, I. & Endo, T. Living coordination polymerization of N-allenylamides by π-allylnickel catalysts. Macromolecules 31, 6741–6747 (1998).
Taguchi, M., Tomita, I., Yoshida, Y. & Endo, T. Block copolymerization of allene derivatives with 1,3-butadiene by living coordination polymerization with π-allylnickel catalyst. J. Polym. Sci. Part A 37, 3916–3921 (1999).
Taguchi, M., Tomita, I. & Endo, T. Living coordination polymerization of allene derivatives bearing hydroxy groups by π-allylnickel catalyst. Angew. Chem. Int. Ed. 39, 3667–3669 (2000).
Taguchi, M., Tomita, I., Yoshida, Y. & Endo, T. Living coordination polymerization of end-allenyloxy poly(ethylene glycol) macromonomer by a π-allylnickel catalyst. Macromol. Chem. Phys. 201, 1025–1030 (2000).
Taguchi, M., Tomita, I., Yoshida, Y. & Endo, T. Preparation of end-π-allylnickel macroinitiator from poly(ethylene glycol) allenyl methyl ether and its application to the living coordination polymerization of isonitriles. J. Polym. Sci. 39, 495–499 (2001).
Wang, J., Tomita, I. & Endo, T. Synthesis of well-defined glycopolymers by π-allylnickel-catalyzed living coordination polymerization. Macromolecules 34, 4294–4295 (2001).
Takagi, K., Tomita, I. & Endo, T. Allylnickel-catalyzed living coordination polymerization of allenes with ester functionality. Polym. Bull. 50, 335–342 (2003).
Kino, T. & Tomita, I. Synthesis of poly(alcohol)s by hydroboration/oxidation of poly(methylallene) prepared by π-allylnickel-catalyzed living coordination polymerization. Polym. Bull. 55, 251–258 (2005).
Mochizuki, K. & Tomita, I. π-Allylnickel-catalyzed living coordination polymerization of allene having homochiral phenylcarbamoyloxy-substituted binaphthyl function. Macromolecules 39, 6336–6340 (2006).
Kino, T., Taguchi, M., Tazawa, A. & Tomita, I. Living coordination polymerization of allene derivatives in protic solvents: remarkable acceleration of polymerization and increase of 1,2-polymerization selectivity. Macromolecules 39, 7474–7478 (2006).
Sato, E., Yokozawa, T. & Endo, T. Polyaddition of diallenes: radical polyaddition of dithiols to 1,4-bis(allenyloxy)benzene. Macromolecules 26, 5185–5186 (1993).
Bai, F., Yang, X. L. & Huang, W. Q. Synthesis of narrow or monodisperse poly(divinylbenzene) microspheres by distillation–precipitation polymerization. Macromolecules 37, 9746–9752 (2004).
Vanzo, E. Particle-size control in dispersion polymerization. J. Appl. Polym. Sci. 16, 1867–1868 (1972).
Shen, S., Sudol, E. D. & Elaasser, M. S. Control of particle-size in dispersion polymerization of methyl-methacrylate. J. Polym. Sci. Part A 31, 1393–1402 (1993).
Paine, A. J., Luymes, W. & McNulty, J. Dispersion polymerization of styrene in polar-solvents. 6. Influence of reaction parameters on particle-size and molecular-weight in poly(n-vinylpyrrolidone)-stabilized reactions. Macromolecules 23, 3104–3109 (1990).
Paine, A. J. Dispersion polymerization of styrene in polar-solvents. 7. A simple mechanistic model to predict particle-size. Macromolecules 23, 3109–3117 (1990).
Chen, W., Fadeev, A. Y., Hsieh, M. C., Oner, D., Youngblood, J. & McCarthy, T. J. Ultrahydrophobic and ultralyophobic surfaces: Some comments and examples. Langmuir 15, 3395–3399 (1999).
Frenette, R. & Friesen, R. W. Biaryl synthesis via suzuki coupling on a solid support. Tetrahedron Lett. 35, 9177–9180 (1994).
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas ‘New Polymeric Materials Based on Element-Blocks (No. 2401)’ (JSPS KAKENHI Grant Number JP24102007). We are also grateful to Shimadzu Co. Ltd for the measurement of OM image on a Shimadzu OLS4000 instrument.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the NPG Asia Materials website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yamauchi, A., Shirai, A., Kawabe, K. et al. Well-defined polymer microspheres formed by living dispersion polymerization: precisely functionalized crosslinked polymer microspheres from monomers possessing cumulated double bonds. NPG Asia Mater 8, e307 (2016). https://doi.org/10.1038/am.2016.123
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/am.2016.123
- Springer Japan KK