
Abstract
Low temperature is a major environmental factor that limits the growth, yield, and geographical distribution of Cavendish and Dajiao bananas (Musa spp.). Dajiao bananas exhibit a significantly higher cold tolerance than the Cavendish cultivar. However, the underlying mechanisms involved in cold tolerance regulation in Dajiao plants have not been determined. In this study, we investigated the mechanisms underlying the differences in cold tolerance at the cellular level between the cold-sensitive Cavendish and cold-tolerant Dajiao banana types through comparative metabolomics and transcriptomics analyses in calli exposed to cold treatment under dark conditions. A higher accumulation of lipids was observed in Dajiao calli cells compared to Cavendish cells under cold stress. After cold treatment, 4,626 and 5,516 differentially expressed genes (DEGs) were identified in Cavendish and Dajiao banana cells, respectively. By integrating the transcriptomic and metabolomic datasets, we discovered that the linoleic acid and α-linoleic acid metabolism and the Abscisic acid (ABA)-independent Mitogen-activated protein kinase (MAPK) cascade-Inducer of CBF expression 1 (ICE1) signal transduction pathway (including Mitogen-activated protein kinase kinase kinase 1 [MEKK1], MAPK5, ICE1, and Cold-regulated 47 [COR47]) played crucial roles in the cold tolerance of Dajiao bananas. Our study provides new insights into cold response regulation and novel cold tolerance mechanisms, providing valuable leads and targets for the genetic improvement of cold tolerance in bananas.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Banana (Musa spp.) is one of the most popular fruits consumed globally and is also a vital food crop in developing countries, second only to rice, wheat, and maize (Singh et al. 2016; Alemu 2017; Paul et al. 2017). However, frequent cold waves in subtropical regions during the winter and early spring have negatively impacted banana yields (Chinnusamy et al. 2007; Meng et al. 2020). The most commonly cultivated banana cultivars, Cavendish (Musa AAA group, cv. Cavendish) is particularly sensitive to low temperatures. Dajiao (Musa ABB group, cv. Dajiao), which also belongs to the Musa genus, exhibits strong cold resistance and can withstand temperatures as low as 0 to 4°C (Fig. 1a). Thus, this Dajiao banana cultivar is an ideal genetic resource for the identification of cold-tolerant associated genes, to improve banana cold tolerance and develop new cold-tolerant banana cultivars (Yang et al. 2012, 2015).

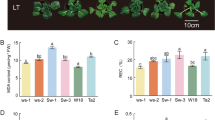
Different physiological responses of Cavendish and Dajiao to cold stress. (a) Cavendish (left) and Dajiao (right) banana growth phenotypes exposed to different cold treatment durations. (b) Cavendish and Dajiao seedlings were exposed to 8°C for 0 h and 24 h. (c) Cavendish and Dajiao embryogenic cell suspension (ECS) phenotypes subjected to cold treatment at 8°C for 0 h, 6 h, 24 h, or 48 h. (d) Hydrogen peroxide accumulation in the Cavendish and Dajiao ECSs under 8°C treatment was detected by DAB staining. (e) Cellular damage in the Cavendish and Dajiao ECSs under 8°C treatment detected via trypan blue staining. (f) Area occupied by trypan blue staining (%). (g) Peroxide Staining Gray Scale Value. *An asterisk indicates a significant difference between the two cultivars according to the student's t-test (*P < 0.05, **P < 0.01, ***P < 0.001)
Studying the molecular mechanisms underlying cold tolerance in banana has been challenging due to their complex genetic regulation and the highly heterozygous genetic background of banana cultivars (D'Hont et al. 2012; Wang et al. 2019). Numerous previous studies have been carried out to identify and functionally validate cold tolerance genes in banana seedlings. At the transcriptional level, the rapid activation and selective expression of Inducer of CBF expression 1 (ICE1) and DNA-binding repeat MYB transcription factor S3 (MYBS3) in Dajiao bananas, as well as the expression of other cold stress-specific genes, potentially underlies the increased resistance to cold stress of Dajiao bananas compared to Cavendish (Yang et al. 2015). Proteomic analysis suggested that the MAPK3-ICE1-POD P7 pathway potentially contributed to the increased cold tolerance in Dajiao by regulating cellular signaling and homeostasis under cold stress (Gao et al. 2021). Furthermore, overexpression of the Dajiao ICE1 and MYBS3 genes in Cavendish banana increased the cold tolerance of transgenic plants, while RNAi silencing of Mitogen-activated protein kinase 3 (MAPK3) in Dajiao reduced its cold tolerance (Dou et al. 2016; Gao et al. 2021). Moreover, phosphoproteomics studies have shown that the MKK2-MAPK3 module may also contribute to greater cold tolerance in Dajiao plants (Gao et al. 2017).
Cells are the basic unit of life activity and can be observed in greater detail to identify molecular changes when exposed to specific environmental conditions. Compared with traditional whole-plant studies, the plant cell suspension system is a superior test model for the in-depth investigation of plant stress responses at the cellular levels and the underlying transcriptional and metabolic regulatory pathways. This is because the cell suspension system has many benefits, including good dispersion, consistent cell shape, and cell cluster size, allowing for uniformity of stress treatments and good experimental reproducibility (Nogueira et al. 2007). However, no studies have reported on the investigation of cold tolerance mechanisms in banana cells to identify and mine cold-tolerant genes. In this study, we cultured banana embryogenic cell suspensions (ECSs) induced from male banana inflorescences under dark conditions and subjected them to low-temperature treatment. This highly controlled cell suspension system enabled us to eliminate the complexity caused by different cell types and the effects of light signaling pathway-related proteins under photooxidative stress, allowing us to precisely investigate the molecular mechanisms at the cellular level associated with the differential cold tolerance between the cold-sensitive Cavendish and cold-tolerant Dajiao banana cultivars. High-throughput "omics" methods, such as genomics, transcriptomics, proteomics, and metabolomics, have been widely utilized to explore the mechanisms underlying plant adaptation to different biotic and abiotic stresses and gain a deeper understanding of various stress responsive biological pathways (Raza et al. 2021a, b). By employing a combined metabolomic and transcriptomic analysis approach, our study aimed to identify and assess the correlation between differentially expressed genes and metabolites between Cavendish and Dajiao banana ECSs under cold stress. The comparison of the adaptive responses to cold stress of these two varieties at the cellular level will allow us to gain novel insights into the molecular mechanisms underlying cold tolerance in bananas.
Materials and methods
Plant materials and cold treatment
ECSs were induced from banana male inflorescences. Male inflorescences were sterilized, inoculated into the embryogenic tissue induction media, and transferred to liquid media for shaking liquid culture to obtain the embryogenic cell suspensions. The established suspension cell lines were maintained at 27°C and 110 rpm in the dark (Hu et al. 2010). The ECSs of two different banana cultivars, the cold-sensitive Cavendish and cold-tolerant Dajiao, were used in this study. The ECSs were cultured with shaking at 110 rpm and then transferred to an artificial climate chamber without light for 7 days to acclimate. After acclimation, the ECSs were cultured at 28°C and 60–80% relative humidity. The low-temperature treatment was initiated at 12:00 AM on day 8, with the temperature reduced to 8°C, which was reached after approximately 20 min. Three biological replicates were performed for both banana varieties at each time point. Samples of the suspended cell precipitates were collected from each biological replicate at different time points (0 h, 6 h, and 24 h at 8°C and recovered at 28°C for 24 h). The samples were then quickly frozen in liquid nitrogen and stored at -80°C until further use.
Histochemical staining
To assess the cold tolerance of the two banana genotypes, the ECSs of Cavendish and Dajiao were subjected to low-temperature treatment at 8°C for 0 h, 6 h, 24 h, and 48 h. A trypan blue staining solution was used according to the manufacturer's instructions (Beijing Solaibao Technology Co., Ltd., Beijing, China) to determine the cell membrane integrity and calculate the area occupied by the trypan blue dye using the Image J software. Furthermore, the accumulation of reactive oxygen species (ROS) was measured using a hydrogen peroxide detection kit (DAB method) (Servicebio Technology Co., Ltd., Wuhan, China), and the peroxide staining grayscale value was calculated using the ImageJ software (a darker color corresponded to a lower grayscale value). All the experiments were conducted in triplicate following the manufacturer's protocol.
Metabolite extraction and profiling
To extract the cell metabolites, the frozen samples were subjected to vacuum freeze-drying using a freeze-dryer Scientz-100F (SCIENTZ, Ningbo, China). The resulting freeze-dried samples were ground into a fine powder using a mixer mill MM 400, Retsch (Verder Scientific, Newtown, PA, United States) operated at 30 Hz for 1.5 min. Subsequently, 50 mg of each powdered sample was weighed and dissolved in 1.2 mL of 70% methanol extract. The samples were vortexed for 30 s every 30 min for a total of six times. After that, the supernatant was aspirated following centrifugation at 12,000 rpm and 4°C for 3 min. The samples were filtered through 0.22 μm microporous filters during the preparation process. The filtered samples were then subjected to ultra-performance liquid chromatography and tandem mass spectrometry (UPLC‒MS/MS) analysis. To monitor the reproducibility of the analytical process, a quality-control sample was prepared from a mixture of sample extracts and inserted into the apparatus auto-sampler after every 10 test samples during the analysis process.
The instrument systems used for data acquisition in this study were primarily UPLC (SHIMADZU Nexus X2) and MS/MS (Applied Biosystems 4500 QTRAP). The analyses were performed using a chromatographic column (Agilent SB-C18 1.8 µm, 2.1 mm × 100 mm) and a mobile phase consisting of ultrapure water with 0.1% formic acid (phase A) and acetonitrile with 0.1% formic acid (phase B). The elution gradient started with 5% phase B at 0.00 min and increased linearly to 95% within 9.00 min, then maintained at 95% for 1 min, decreased to 5%, and equilibrated at 5% for 14 min. The flow rate was 0.35 mL/min, and the column temperature was maintained at 40°C. The injection volume was 4 μL. The main parameters for the mass spectrometer were an electrospray ionization source (ESI) temperature of 550°C and an ion spray voltage of 5500 V (positive ion mode)/-4500 V (negative ion mode). The ion source gas I (GSI), gas II (GSII), and curtain gas (CUR) pressures were set to 50, 60, and 25 psi, respectively, and the collision-induced ionization parameter was set to high. Tuning and mass calibration of the instrument were performed using 10 and 100 μmol/L polypropylene glycol solution in QQQ and LIT modes, respectively. QQQ scans were conducted using the multiple reaction monitoring (MRM) mode with the collision gas (nitrogen) set to medium. The decluttering potential (DP) and collision energy (CE) of each MRM ion pair were optimized to measure the charge state of each metabolite. A specific set of MRM ion pairs were monitored for each period based on the eluted metabolites during this specific period. The in-house metabolic database of MetWare Biotechnology Co., Ltd., in Wuhan, China, was utilized, and the MRM mode with triple quadrupole mass spectrometry was used. MRM was used for the qualitative and quantitative analysis of the metabolites present in the samples. The MultiQuant software was used to integrate and correct the chromatographic peaks, with the peak area representing the relative content of the corresponding compound.
Transcriptome sequencing
Total RNA was extracted from the samples, and the RNA integrity, as well as the presence of DNA contamination, were assessed using agarose gel electrophoresis. The RNA purity was determined using the NanoPhotometer® spectrophotometer (IMPLEN, CA, United States) by measuring the OD260/280 and OD260/230 ratios, while RNA quantification was performed using Qubit® RNA Assay Kit in Qubit®2.0 Flurometer (Life Technologies, CA, United States). Finally, RNA integrity analysis was carried out using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, United States). At least 1 μg of total RNA was used to construct cDNA libraries. The libraries generated were pooled according to established requirements of effective concentration and targeted sequencing depth, and they were sequenced using Illumina sequencing technology, which generated 150 bp paired-end reads. Sequencing was carried out by MetWare Biotechnology Co., Ltd. (Wuhan, China). The raw read data were filtered with Fastp v0.19.3 (Chen et al. 2018), which removed the adapter sequences from the reads. Paired reads with an N base percentage exceeding 10% of the base number of the reads were removed, and those with a number of low-quality bases (Q ≤ 20) greater than 50% of the read length were excluded. Clean reads were used for all subsequent analyses. The banana genome and associated annotations were downloaded from https://banana-genome-hub.southgreen.fr/node/50/7720975. HISAT v2.1.0 was used to construct an index (Kim et al. 2015), which was subsequently used to align the clean reads to the banana reference genome. Gene alignments were calculated by featureCounts v1.6.2, with subsequent calculation of fragments per kilobase million (FPKM) values based on gene length to quantify gene expression levels (Liao et al. 2014). The FPKM method is currently the most widely used method for estimating gene expression levels. Differential expression analysis was performed using DESeq2 v1.22.1 to identify cold-responsive genes with varying expression patterns between the two banana genotypes. P values were corrected using the Benjamini and Hochberg method. After P value correction, a threshold of |log2-fold change| was used to determine significant differences in gene expression (Love et al. 2014; Varet et al. 2016). Enrichment analysis of differentially expressed genes (DEGs) was performed using a hypergeometric test. For Kyoto Encyclopedia of Genes and Genomes (KEGG), a hypergeometric distribution test based on each pathway was used (Kanehisa et al. 2008). Gene Ontology (GO) enrichment analysis was performed based on the GO terms (Ashburner et al. 2000).
Gene expression analysis by real-time quantitative PCR (qRT-PCR)
Total RNA isolation was performed from three biological replicates of Cavendish and Dajiao ECS samples treated at 8°C for 0 h, 6 h, 24 h, and 28°C for 24 h. First-strand cDNA was synthesized according to the Prime Script™ RT-PCR kit protocol (Takara, Japan). Primer pairs for qRT-PCR were designed using Primer Premier 5.0 (Premier Biosoft, Palo Alto, CA, United States). PCR was performed with a LightCycler 480 II and its installed software (Roche, Germany) following the protocol of the LightCycle Premix EX Taq (Perfect Real Time) kit (Takara, Japan). The total volume for each reaction mixture was 10 μL, consisting of 0.4 μL of primer F, 0.4 μL of primer R, 1 μL of cDNA, 5 μL of 2 × SYBR Green PCR Master Mix, and 3.2 μL of RNase-free water. The cycling program was as follows: initial denaturation at 95°C for 2 min, followed by denaturation at 95°C for 10 s, annealing, and elongation at 60°C for 30 s (40 cycles). The relative transcript levels in each sample were quantified by the 2–ΔΔCT method (Livak and Schmittgen 2001) using the 25S ribosomal RNA gene as a reference gene for normalization (Van Den Berg et al. 2007). The expression levels of each gene in Cavendish at 0 h was used as a control, and its value was set to 1. The relative expression levels of the corresponding genes in other samples (including the 0 h time point for Dajiao) were determined relative to the Cavendish 0 h control. The information on the genes used in qRT-PCR analysis is listed in Supplementary Table 1.
Statistical analysis
Image J software was used to calculate the area occupied by trypan blue staining and the grayscale quantification of peroxide staining intensity. The statistical analysis of the data was performed using the GraphPad Prism 8.0 software (GraphPad, L.A., California, United States). Duncan's multiple range test (P < 0.05) and Student's t-test were used to assess the statistical significance of the differences (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
The Dajiao ECS exhibits greater tolerance to cold stress compared to the Cavendish ECS
After the seedlings were subjected to low-temperature treatment, distinct differences in cold tolerance were observed between the Cavendish and Dajiao banana cultivars. When the plants were exposed to 8°C for 24 h, the leaves of Cavendish banana seedlings exhibited severe water loss and necrosis, whereas there were no apparent changes in the leaves of Dajiao (Fig. 1b). ECSs induced from the male inflorescences of the above two banana cultivars showed different degrees of cell injury after low-temperature treatment. As the duration of cold treatment increased, the color of the Cavendish ECS transitioned to yellow. In contrast, the color of the Dajiao ECS remained white (Fig. 1c). DAB staining was performed to measure hydrogen peroxide (H2O2) accumulation in the ECSs of both cultivars after cold stress (Fig. 1d). With increasing cold stress duration, the amount of brown precipitate in the ECS of both cultivars increased. Specifically, after an 8°C cold treatment for 48 h, the Cavendish ECS had significantly lower hydrogen peroxide staining grayscale values compared to Dajiao ECS, indicating that Cavendish exhibited a greater hydrogen peroxide accumulation under cold stress (Fig. 1d, g).
Furthermore, trypan blue staining after cold stress exposure at 8°C for 6 h and revealed minor cell damage in both the Cavendish and Dajiao ECSs, with a 17.44% and 4.30% trypan blue stained area, respectively (Fig. 1e‒f). However, a considerable increase in the degree of cell damage was observed in the Cavendish ECS after exposure to cold stress for 24 and 48 h, with the trypan blue stained area accounting for 33.85% and 65.43% of the total, respectively. In contrast, the Dajiao ECS sustained lower cell damage, with trypan blue stained areas of 5.32% and 25.93% after 24 and 48 h of cold stress, respectively (Fig. 1e‒f).
Metabolomic profiles of Cavendish and Dajiao ECS in response to cold stress
To investigate the changes in the ECS metabolome of the cold-sensitive Cavendish and cold-tolerant Dajiao cultivars in response to cold stress, cell samples were collected at four different time points following treatment (8°C for 0 h, 6 h, or 24 h and recovery at 28°C for 24 h) and subjected to UPLC‒MS/MS analysis. A total of 489 metabolites were identified, which were categorized into lipids (28.02%), organic acids (16.97%), amino acids (21.88%), nucleotides (14.52%), and other compounds (18.61%) (vitamins, sugars, and alcohols) (Supplementary Table 2). Heatmap analysis was performed to illustrate the metabolic profiles in response to cold stress. After 24 h of treatment, Dajiao cells exhibited greater lipid concentration increases compared to Cavendish. (Fig. 2).

Metabolite analysis of the Cavendish and Dajiao embryogenic cell suspensions (ECSs) in response to cold stress. Heatmap of metabolite levels according to metabolite classification
The differentially accumulated metabolites (DAMs) that met the following criteria: P < 0.05 and a fold change (FC) ≥ 2 or ≤ 0.5 were identified. As a result, 190 DAMs were identified out of a total of 489 metabolites (Supplementary Table 3). Among the 190 DAMs, 86 were identified in the ECS of Cavendish, and 29 metabolites were uniquely differentially accumulated in this cultivar. Moreover, 161 DAMs were identified in the ECS of Dajiao, 104 of which were unique, indicating significant differences in the metabolite profiles between the two cultivars in response to cold stress. Interestingly, 91 of the 161 DAMs identified in Dajiao belonged to the lipid family (Table 1).
Primary metabolites of the Dajiao ECS under cold stress
To further investigate the changes in the relative contents of metabolites among the different cultivar and treatment groups, the differentially accumulated metabolites were identified using designated screening criteria and were subjected to unit variance scaling (UV), followed by K-means cluster analysis. As shown in Fig. 3a-d, the four clusters formed by the DAMs exhibited similar trends. Notably, there was an increase in the abundance of DAMs within cluster 2 in Dajiao ECS subjected to 8°C for 24 h (Fig. 3b), suggesting that cluster 2 metabolites could be specific to the Dajiao ECS cold stress response. KEGG pathway enrichment analysis revealed that these metabolites were predominantly enriched in pathways related to linoleic acid metabolism, α-linoleic acid metabolism, and unsaturated fatty acid (FA) biosynthesis (Fig. 3e).

Analysis of primary metabolites in the embryogenic cell suspensions (ECSs) of Cavendish and Dajiaounder cold stress. (a)–(d) K-means cluster analysis of differentially accumulated metabolites (DAMs) with the same change trend. Note: Sub Class represents the number of metabolites with the same trend in the class compared to the total, ∗ which represents the number of metabolites in this class ∗ . Sub Class 1 contains mainly amino acids and derivatives, at 52.63%. Sub Class 2 mainly contains lipids, at 77.60%. Sub Class 3 mainly contains nucleotides and derivatives, at 39.06%. Sub Class 4 mainly contains organic acids, at 31.58%. (e) KEGG enrichment analysis of Sub Class 2 DAMs
We further analyzed the lipid metabolites at different time points and their trends in the ECSs of Cavendish and Dajiao after cold treatment (Table 2). A total of 93 lipids were detected, which were classified into six classes, namely, phosphatidylcholine (PC), lysophosphatidylethanolamine (LPE), lysophosphytytocholine (LPC), glycerol esters, free fatty acids, and sphingolipids (Supplementary Table 3). Four types of LPEs, LPCs, and five glycerol esters accumulated in Cavendish and Dajiao ECSs. However, Dajiao accumulated these lipids earlier and to a greater extent under cold stress. Moreover, the content of four free fatty acids in the ECS of Cavendish decreased, whereas they were significantly increased in the ECS of Dajiao throughout the entire stress period. Additionally, a higher accumulation of lipids was observed in the ECS of Dajiao, including 17 LPEs, 16 LPCs, 10 glycerol esters, and 30 free fatty acids, compared to Cavendish under cold stress. Interestingly, the Cavendish ECS accumulated high amounts of sphingolipids under the entire cold stress treatment period.
Functional classification of differentially accumulated primary metabolites in the Cavendish and Dajiao ECSs with a fold change > or < 2-fold.
alog2fold change C6 h: cold-treated sample at 6 h/cold-treated sample at 0 h. log2fold change C24 h: cold-treated sample at 24 h/cold-treated sample at 0 h. log2fold change R24 h: sample after cold stress recovery for 24 h/sample that was cold treated at 0 h.
b + : upregulated; -: downregulated; = : not significantly changed.
Transcriptomic profiles of Cavendish and Dajiao ECSs in response to cold stress
To investigate the response of Cavendish and Dajiao ECSs to cold stress at the transcriptional level, total RNA was extracted from their ECSs at different time points after the cold stress treatment, and the differentially expressed genes were analyzed. We identified the statistically significant differences in gene expression using a |log2fold change| of 1 or greater and a false discovery rate (FDR) of less than 0.05 as thresholds. Venn diagram analysis was performed to illustrate the number of DEGs in the Cavendish and Dajiao ECSs after cold treatment (Fig. 4a). In the ECS of Cavendish, we identified 533 DEGs (334 upregulated and 199 downregulated) after 6 h of cold treatment and 4,093 DEGs (2,368 upregulated and 1,725 downregulated) after 24 h of cold treatment. In the ECS of Dajiao, we identified, after 6 h of cold treatment,1,200 DEGs (616 upregulated and 584 downregulated) and, after 24 h of cold treatment, 4,316 DEGs (2,545 upregulated and 1,771 downregulated). The number of cold-responsive genes was markedly greater after 24 h of cold treatment than after 6 h, indicating that extended cold treatment leads to more profound changes in the ECS transcriptome (Fig. 4a, Table 3).

Transcriptomic analysis of the embryogenic cell suspensions (ECSs) of Cavendish and Dajiao bananas in response to cold stress. (a) Venn diagram of differentially expressed genes (DEGs) in the ECSs of Cavendish and Dajiao before and after cold stress (8°C for 6 h and 24 h). (b)–(c) KEGG enrichment analysis of DEGs in the ECS of Cavendish and subjected to cold treatment at 8°C for 6 h. (b) and 24 h (c), respectively. (d)–(e) KEGG enrichment analysis of DEGs in the ECS of Dajiao subjected to cold treatment at 8°C for 6 h (d) and (e), respectively. (f)–(g) KEGG enrichment analysis of DEGs between the ECSs of Cavendish and Dajiao subjected to cold treatment at 8°C for 6 h (f) and 24 h (g), respectively
Functional enrichment analysis of DEGs
A KEGG pathway enrichment analysis was performed to identify the predominant functions of the DEGs in response to various cold treatment conditions (Fig. 4b–g). After exposure to 8°C for 6 h, DEGs in the ECS of Cavendish were primarily enriched in the oxidative phosphorylation pathway. On the other hand, the DEGs in the ECS of Dajiao were involved in fatty acid elongation, oxidative phosphorylation, plant-pathogen interactions, and MAPK signaling pathways (Fig. 4b, d). After treatment at 8°C for 24 h, a significant enrichment of the plant hormone signal transduction was observed among the DEGs of the Cavendish ECS. Moreover, the enriched pathways among the DEGs in the ECS of Dajiao were associated with plant hormone signal transduction, plant-pathogen interactions, and MAPK signaling pathways (Fig. 4c, e). Moreover, to further compare the molecular mechanism of cold tolerance in the ECSs of Cavendish and Dajiao, we further analyzed the molecular and metabolic functions of the DEGs through the KEGG pathway enrichment analysis. The results showed that after exposure to 8°C for 6 h, the major pathways involved in the cold stress response in Dajiao were phenylpropanoid biosynthesis, glutathione metabolism, MAPK signaling, and linoleic acid metabolism pathways, while the ECS of Cavendish responded to cold stress (Fig. 4f). However, after exposure to 8°C for 24 h, the cold stress response in the ECS of Dajiao was mainly relayed by plant hormone signal transduction pathways, the MAPK signaling pathway, and the brassinosteroid biosynthesis pathway (Fig. 4g).
DEG validation by qRT-PCR analysis
To confirm the accuracy of the RNA-seq transcriptomic results, 18 genes were selected for validation by qRT-PCR (Fig. 5). These genes are involved in various processes, such as cold response, fatty acid elongation, MAPK signaling, and plant-pathogen interactions. The cold-responsive genes included ICE1, Dehydration-responsive element-binding protein 1C (DREB1C), Dehydration-responsive element-binding protein 1E (DREB1E), Desiccation-responsive protein 29A (RD29A), Cold-regulated 47 (COR47), E3 SUMO-protein ligase SIZ1 (SIZ1), E3 ubiquitin-protein ligase HOS1 (HOS1), and myeloblastosis domain protein 15 (MYB15). The fatty acid elongation genes were RADIALIS-like 1 protein (RADL1), 3-K-CoA synthase 1 (KCS1), and KCS11, and the MAPK signaling genes were Mitogen-activated protein kinase kinase kinase 1 (MEKK1), MAPK3, and MAPK5. The plant-pathogen interaction genes were Pathogenesis related protein 1 (PR1), WRKY domain protein 2 (WRKY2), WRKY19, and WRKY71. In the ECS of Cavendish, after 6 and 24 h of cold treatment, ICE1, DREB1C, SIZ1, HOS1, and MYB15 were initially significantly upregulated, followed by a downregulation in expression with the increase in cold stress treatment duration. An opposite response was observed in the ECS of Dajiao, where these genes were significantly downregulated initially and then showed increased expression. Notably, the basal expression levels of these genes in the ECS of Dajiao were markedly greater than those in Cavendish (Fig. 5a, b, f, g, h). Similarly, the DREB1E and RD29A were expressed in both Cavendish and Dajiao in response to cold stress (Fig. 5c, d). The expression of COR47 gradually increased in the ECS of Cavendish with increasing cold treatment duration. In contrast, in the ECS of Dajiao, its expression was downregulated initially and then significantly upregulated (Fig. 5e). KCS1 and KCS11, involved in fatty acid elongation, were significantly upregulated in the ECS of Dajiao after 24 h of cold treatment (Fig. 5j, k). In the Cavendish ECS, the expression of MEKK1, involved in MAPK signaling, was significantly upregulated after 6 h of cold treatment but decreased to a level similar to the control after 24 h of cold treatment. Although the expression of MEKK1 in the ECS of Dajiao decreased after 6 h of cold treatment, no significant differences were observed between Dajiao and Cavendish, as the initial expression of MEKK1 was greater in Dajiao than in Cavendish (Fig. 5l). A similar expression pattern was observed for MAPK3 (Fig. 5m). On the other hand, MAPK5 was significantly upregulated in Dajiao ECS after 24 h of cold treatment (Fig. 5n). WRKY2 and WRKY19, which are involved in plant-pathogen interaction pathways, could also possess significant functions in improving cold tolerance in Dajiao (Fig. 5p, q). WRKY71 expression was upregulated in response to cold stress in both Cavendish and Dajiao, but its expression was much higher in Dajiao than in Cavendish. Specifically, its expression level after 6 h of cold treatment in Dajiao was equivalent to that in Cavendish after 24 h of cold treatment. These findings suggested that WRKY71 potentially plays a critical role in the increased cold tolerance of Dajiao bananas (Fig. 5r). The similarities in the expression profiles of the selected genes determined by qRT-PCR and the RNA-Seq data indicated that the transcriptomic data were reliable.

Expression analysis of cold-responsive genes in the ECSs of Cavendish and Dajiao in response to cold stress (at 8°C for 0 h, 6 h, and 24 h and after recovery at 28°C for 24 h). (a) Transcription factor inducer of CBF expression 1 (ICE1). (b) Dehydration-responsive element-binding protein 1C (DREB1C). (c) Dehydration-responsive element-binding protein 1E (DREB1E). (d) Desiccation-responsive protein 29A (RD29A). (e) Cold-regulated 47 (COR47). (f) E3 SUMO-protein ligase SIZ1 (SIZ1). (g) E3 ubiquitin-protein ligase HOS1 (HOS1). (h) Transcription factor myeloblastosis domain protein 15 (MYB15). (i) RADIALIS-like 1 protein (RADL1). (j) 3-K-CoA synthase 1 (KCS1). (k) 3-K-CoA synthase 11 (KCS11). (l) Mitogen-activated protein kinase kinase kinase 1 (MEKK1). (m) Mitogen-activated protein kinase 3 (MAPK3). (n) MAPK5. (o) Pathogenesis related protein 1 (PR1). (p) Transcription factor WRKY domain protein 2 (WRKY2). (q) Transcription factor WRKY domain protein 19 (WRKY19). (r) Transcription factor WRKY domain protein 71 (WRKY71). The statistical significance was determined via one- or two-way analysis of variance (ANOVA) and Dunnett's multiple comparisons test. Different lowercase letters on top of the error bars indicate significant differences at P ≤ 0.05
Integrated analysis of genes and metabolites involved in key cold tolerance pathways
To obtain a more comprehensive understanding of the Dajiao banana response mechanism under cold stress, we integrated the metabolomic and transcriptomic data to construct two simplified regulatory networks (Fig. 6a, b). The networks included the ABA signaling pathway and the lipid metabolism pathway. With regards to the ABA signaling pathway, the expression of the ABA response genes was consistent with the expression patterns of ABA and β-D-Glucopyranosyl abscisate (ABA-GE) (data not shown), which showed no significant difference between the two banana varieties under cold stress (Fig. 6a). On the other hand, the expression of WRKY2/19/71, which are transcription factors associated with the ABA-independent pathway, significantly increased after 24 h of cold treatment in the Dajiao ECS (Fig. 5p–r). To identify metabolites with the greatest changes in concentration, we constructed a map of the gene expression and metabolite accumulation profiles of the DAMs and DEGs associated with lipid metabolism. Among the included DAMs, three fatty acid metabolites, lyso phospholipids (LPCs and LPE), free fatty acids with relatively high degrees of unsaturation (linoleic acid and α-linoleic acid) and glycerol esters, had the most correlated genes and exhibited strong positive correlations with glycerol-3-phosphate acyltransferase 3 (GPAT3), fatty acid desaturase 3C (FAD3C), and lipoxygenase 5/15 (LOX5/15) genes (Fig. 6b).

Integrated analysis of the metabolomic and transcriptomic data. (a) ABA-dependent signaling pathway and related gene expression profiles. (b) Regulatory network of lipid metabolism associated with cold stress responses in Dajiao and Cavendish plants (red and green indicate upregulated and downregulated genes, respectively). Note: ABA-GE and ABA hormone levels (ng/g) are shown. The peak areas in the chromatogram represent the relative content of the corresponding metabolite, and log2 fragments per kilobase million (FPKM) was used to calculate relative gene expression. (c) A comprehensive conceptual model of the Dajiao response to cold stress (based on Yang et al. 2012; Gao et al. 2017, 2021; He et al. 2018). Note: MAPK5 is synonymous with MAPK3 in our previous articles. ABA, Abscisic acid; ABA-GE, β-D-Glucopyranosyl abscisate; PYR/PYL/RCAR, pyrabactin resistance/pyr1-like/regulatory components of ABA receptor; PP2C, type 2C protein phosphates; SnRK2, SNF1-regulated protein kinase 2; AREB/ABF, ABA responsive element binding protein/ABA responsive element binding factors; G3P, glyceraldehyde 3-phosphate; LPA, lysophosphatidic acid; LPC, lysophosphytytocholine; LPE, lysophosphatidylethanolamine; PA, phosphatidic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; DAG, diacylglycerol; TAG, triacylglycerol; MGD, monogalactosyldiacylglycerol; DGD, digalactosyldiacylglycerol; FA, fatty acid; PLA, phospholipase A; PLD, phospholipase D; PAP6, phosphatidic acid phosphatase 6; DGAT3, diacylglycerol acyltransferase 3; GPAT3, glycerol-3-phosphate acyltransferase 3; HSL, hormone-sensitive lipase; KCS, 3-K-CoA synthase; FAD3C, fatty acid desaturase 3C; LOX, lipoxygenase; WS, wax ester synthase; PIP, plasma membrane intrinsic proteins; FAD, fatty acid desaturase; COLD1, chilling tolerance divergence 1; POD P7, Peroxidase P7; GGAT, glutamate glyoxylate aminotransferase; SGAT; serine glyoxylate aminotransferase; SHMT, serine hydroxymethyltransferase; SOD, superoxide dismutase; CAT, catalase; MAPK, mitogen-activated protein kinase; MEKK1, mitogen-activated protein kinase kinase kinase 1; MKK2, mitogen-activated protein kinase kinase 2; ICE1, Inducer of CBF expression 1; WRKY2/19/71, WRKY domain protein 2/19/71; COR47, cold-regulated 47
Taking into consideration all the data presented in this study and the established literature, we propose a conceptual model for the Dajiao response to cold stress (Fig. 6c). At the cellular level under dark conditions, Dajiao responds to cold stress through an ABA-independent pathway by activating the MAPK cascade signaling pathway. This pathway recruits the transcription factors WRKY2/19/71, which potentially modulate the metabolism of linoleic acid and α-linolenic acid, thereby enhancing membrane stability and, as a result, cold tolerance.
Discussion
Lipid metabolism is a crucial pathway associated with the increased cold tolerance of Dajiao bananas
Plants regulate membrane fluidity to adapt to temperature changes, which depends on the membrane's distribution ratio and unsaturation of various lipids (Li et al. 2015). Phosphatidic acid (PA) is a lipid second messenger that rapidly accumulates in cells cultured in a suspension when exposed to cold stress (Cantrel et al. 2011; Peppino Margutti et al. 2017). It has been demonstrated that excessive accumulation of PA at low temperatures can cause the generation of ROS, triggering membrane lipid peroxidation (Moellering and Benning 2011). However, to date, there has been a scarcity of studies conducted to elucidate the mechanisms by which lipid metabolism is related to cold tolerance in bananas (Su et al. 2005). One of the striking findings in this work was the significant differences in lipid metabolism response patterns in the ECSs of Cavendish and Dajiao under low temperatures in dark conditions. KEGG enrichment analysis revealed that the cold-responsive DAMs were significantly enriched in the pathways of linoleic acid metabolism, α-linoleic acid metabolism, and unsaturated fatty acid biosynthesis (Fig. 3e). Moreover, the cold-responsive DEGs in the ECS of Dajiao were significantly enriched in fatty acid elongation and linoleic acid metabolism pathways compared to those in Cavendish. Among these genes, two upregulated lipoxygenase genes, LOX5 and LOX15 are involved in the linoleic acid pathway, which is known to be associated with cold acclimation in plants (Upadhyay et al. 2019). The findings of this study suggest that lipid metabolism pathways play an essential role in cold adaptation in banana (Yang et al. 2023).
In this study, the DEGs and metabolites in the lipid metabolism pathways were comprehensively investigated to understand the differential response of the banana cultivars Cavendish and Dajiao to cold stress. Among the six different lipid compound types analyzed, except for PC, the accumulation patterns of five lipid types in response to cold stress were significantly different between the Cavendish and Dajiao ECSs (Fig. 6b). It is not uncommon for mono- and di-galactose-diglycerides (MGD and DGD) and LPC to increase in response to cold stress, while PC is decreased. Interestingly, during cold stress, the accumulation of LPCs, LPEs, glycerol esters, and free fatty acids was augmented in Dajiao, potentially contributing to its greater cold tolerance. Specifically, under dark conditions, the Dajiao ECS primarily adapts to cold stress by (1) increasing lysophospholipid levels to enhance membrane stability and (2) increasing the levels of free fatty acids with increased unsaturation and glycerol esters, thereby enhancing membrane fluidity. Under low-temperature stress, sphingolipids function as signaling molecules (Huang et al. 2017), and their metabolite ceramide has been found to be involved in programmed cell death activation (Liang et al. 2003) and mitochondrial H2O2 accumulation (Bi et al. 2014). The background levels of sphingolipids were significantly greater in Dajiao than in Cavendish, and they distinctly increased during the Cavendish ECS cold response. As a result, an increased abundance of sphingolipids that specifically accumulate in Cavendish under cold stress might also be responsible for its adaptation to low temperature and the reduction of cell damage under cold stress.
We found that the expression of DEGs associated with lipid metabolism was upregulated after 24 h of cold treatment in the ECS of Dajiao. Among the upregulated DEGs were phospholipase A1 (PLA1), phosphatidic acid phosphatase 6 (PAP6), diacylglycerol acyltransferase 3 (DGAT3), GPAT3, KCS, FAD3C, LOX5, LOX15, and wax ester synthase (WS). PLA1 promotes the conversion of LPA to PA, and PAP6 promotes the conversion of PA to glycerol ester to enhance cellular cold tolerance. KCS encodes an enzyme involved in FA synthesis and is involved in wax and sphingolipid biosynthesis. The FA synthesis process typically results in the production of C16 and C18 FAs. However, plants require longer-chain FAs for membrane lipid formation to adapt to and withstand environmental stresses. Wax, derived from C26 to C32 FAs synthesized through the WS, and sphingolipids containing C22 and C24 FAs are crucial for adaptation. Furthermore, unsaturated FAs, namely, linoleic acid and α-linoleic acid, accumulated at significantly higher levels in the ECS of Dajiao after 24 h of cold treatment. Thus, based on the upregulation of genes involved in lipid metabolism after cold stress and their corresponding metabolomics data, we concluded that lipid metabolism plays a pivotal role in Dajiao banana adaptation to cold stress.
The MAPK cascade-ICE1 signal transduction pathway plays a critical role in the cold tolerance of Dajiao
Calcium, a key secondary messenger, plays a crucial role in plant cold stress responses. When cells are exposed to cold stress, signals are transmitted through protein phosphorylation cascades, causing a rapid increase in the Ca2+ concentration within cells (Saijo et al. 2000). ICE1 is an important cold-responsive transcription factor that is also regulated by MAPK cascade phosphorylation (Gao et al. 2021). Li and Zhao reported that mitogen-activated protein kinase 3/6 (MPK3/6) could phosphorylate ICE1 and promote its degradation (Li et al. 2017; Zhao et al. 2017), while MKK2/MPK4 were involved in the maintenance of ICE1 stability by reducing the expression of MPK3/6, thereby enhancing the cold tolerance of plants. These results indicate that the MKK2/MPK4-MPK36-ICE1 regulatory pathway plays an essential role in regulating the cold tolerance of Arabidopsis thaliana. Our previous studies demonstrated that MaMKK2, MaMAPK3 (synonymous to MAPK5 in this article), and MaICE1 are crucial for the response to and tolerance to cold stress in Dajiao. Specifically, MKK2 gene expression regulates various cellular functions and contributes to cold tolerance (Gao et al. 2021). In this study, we found a significant increase in MEKK1, MAPK5, and ICE1 expression in the ECS of Dajiao after 24 h of cold exposure (Fig. 5a, l, n). These results suggest that the MAPK cascade-ICE1 signal transduction pathway (including MEKK1, MAPK5, ICE1, and COR47) may positively regulate cold tolerance in Dajiao. ICE1, a MYC-type transcription factor, is known to induce and regulate the expression of CBF genes, thereby forming the ICE1-CBF-COR cold signal regulation pathway (Chinnusamy et al. 2003, 2007). We discovered that the cold-responsive genes DREB1E and COR47 were significantly more highly expressed in the ECS of Dajiao than in the Cavendish banana after 24 h of cold exposure (Fig. 5c, e). Furthermore, we observed that the expression of WRKY genes (WRKY2, WRKY19, and WRKY71) involved in plant-pathogen interactions were upregulated to a greater extent in the ECS of Dajiao than in that of Cavendish (Fig. 5p, q, r). These genes potentially contribute to Dajiao's improved cold tolerance. Based on previous studies, CdWRKY2-mediated sucrose biosynthesis and CBF signaling contribute to cold tolerance enhancement in Cynodon dactylon (Huang et al. 2022). Moreover, TaWRKY19 can enhance cold tolerance in Triticum aestivum by activating DREB2A, RD29A, RD29B, and COR6.6 (Niu et al. 2012), while OsWRKY71 is induced in Oryza sativa L. by cold stress and plays a significant role in regulating downstream target genes (Kim et al. 2016). These findings align with our results, indicating that WRKY2, WRKY19, and WRKY71 can positively affect cold tolerance in Dajiao bananas. Furthermore, while ABA-dependent pathways appear to recruit antioxidant and osmoprotectant mechanisms, ABA-independent pathways generally regulate protective proteins to enhance stress tolerance. WRKY transcription factors provide crosstalk between these pathways, with certain members, such as TaWRKY2 and TaWRKY19, potentially being integral to the ABA-independent pathways (Budak et al. 2013), which further confirms that the ABA-independent pathway plays a major role in the enhanced cold tolerance of Dajiao bananas.
Conclusions
By assessing cold tolerance in cell cultures, this study avoided the drawbacks of whole-plant materials, such as tissue-specific expression and expression dilution, and excluded the influence of photosynthesis-related proteins. The aim of this study was to elucidate the molecular mechanisms underlying the differences in cold tolerance between cold-sensitive Cavendish and cold-tolerant Dajiao at the cellular level. We identified a core set of key cold-responsive genes and metabolites. KEGG pathway enrichment analysis revealed the lipid metabolism and the MAPK cascade-ICE1 signal transduction pathway as key pathways directly involved in Dajiao cold stress tolerance. Overall, MEKK1, MAPK5, ICE1, and COR47 play critical roles in Dajiao's cold tolerance. Based on the findings acquired from this study and combined with the results of our group's previous researches, we constructed a comprehensive conceptual model of Dajiao banana response to cold stress (based on Yang et al. 2012; Gao et al. 2017, 2021; He et al. 2018) (Fig. 6c). Previous studies by our group have shown that under light conditions, Dajiao plants respond to cold stress by enhancing their antioxidant capacity primarily through the MAPK cascade signaling pathway. The main components include (1) reduced ROS generation through the photorespiratory proteins GGAT, SGAT, and SHMT; (2) increased scavenging of excess ROS by superoxide dismutase (SOD), catalase (CAT), and peroxidase P7 (POD P7); and (3) prevention of membrane lipid peroxidation by LOX. In this study, we observed a distinct cold tolerance adaptation mechanism in Dajiao under dark conditions compared to its response under light conditions. The MAPK phosphorylation signaling pathway was predominantly induced in Dajiao banana cells via an ABA-independent pathway. This activation led to the upregulation of key transcription factors, including ICE1 and WRKY2/19/71, resulting in changes in membrane composition. The enhanced cell membrane stability was achieved by increasing the levels of unsaturated fatty acids, such as linoleic acid and α-linoleic acid. Such adaptations contributed to Dajiao's improved acclimatization to low temperature in the dark.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request. Transcriptome raw data is submitted in NCBI public repository under BioProject (PRJNA997744).
References
Alemu MM. Banana as a cash crop and its food security and socioeconomic contribution: the case of Southern Ethiopia. Arba Minch J Environ Prot Sci. 2017;8:319–29. https://doi.org/10.4236/jep.2017.83024.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. https://doi.org/10.1038/75556.
Bi FC, Liu Z, Wu JX, Liang H, Xi XL, Fang C, et al. Loss of ceramide kinase in Arabidopsis impairs defenses and promotes ceramide accumulation and mitochondrial H2O2 bursts. Plant Cell. 2014;26:3449–67. https://doi.org/10.1105/tpc.114.127050.
Budak H, Kantar M, Kurtoglu KY. Drought tolerance in modern and wild wheat. Sci World J. 2013;2013:548246. https://doi.org/10.1155/2013/548246.
Cantrel C, Vazquez T, Puyaubert J, Rezé N, Lesch M, Kaiser WM, et al. Nitric oxide participates in cold-responsive phosphosphingolipid formation and gene expression in Arabidopsis thaliana. New Phytol. 2011;189:415–27. https://doi.org/10.1111/j.1469-8137.2010.03500.x.
Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90. https://doi.org/10.1093/bioinformatics/bty560.
Chinnusamy V, Ohta M, Kanrar S, Lee BH, Hong X, Agarwal M, et al. ICE1: a regulator of cold-induced transcriptome and freezing tolerance in Arabidopsis. Genes Dev. 2003;17:1043–54. https://doi.org/10.1101/gad.1077503.
Chinnusamy V, Zhu J, Zhu JK. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007;12:444–51. https://doi.org/10.1016/j.tplants.2007.07.002.
D’Hont A, Denoeud F, Aury JM, Baurens FC, Carreel F, Garsmeur O, et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature. 2012;488:213–7. https://doi.org/10.1038/nature11241.
Dou TX, Hu CH, Sun XX, Shao XX, Wu JH, Ding LJ, et al. MpMYBS3 as a crucial transcription factor of cold signaling confers the cold tolerance of banana. Plant Cell Tiss Organ Cult. 2016;125:93–106. https://doi.org/10.1007/s11240-015-0932-y.
Gao J, Zhang S, He WD, Shao XH, Li CY, Wei YR, et al. Comparative phosphoproteomics reveals an important role of MKK2 in banana (Musa spp.) cold signal network. Sci Rep. 2017;7:40852. https://doi.org/10.1038/srep40852.
Gao J, Dou T, He W, Sheng O, Bi F, Deng G, et al. MaMAPK3-MaICE1-MaPOD P7 pathway, a positive regulator of cold tolerance in banana. BMC Plant Biol. 2021;21:97. https://doi.org/10.1186/s12870-021-02868-z.
He WD, Gao J, Dou TX, Shao XH, Bi FC, Sheng O, et al. Early cold-induced peroxidases and aquaporins are associated with high cold tolerance in Dajiao (Musa spp. ‘Dajiao’). Front Plant Sci. 2018;9:282. https://doi.org/10.3389/fpls.2018.00282.
Hu C, Wei Y, Yi G, Huang B, Huang Y. Establishment of a high efficient Agrobacterium tumefaciens-mediated transformation system for banana. Molecular Plant Breeding. 2010;8:172–8.
Huang X, Zhang Y, Zhang X, Shi Y. Long-chain base kinase1 affects freezing tolerance in Arabidopsis thaliana. Plant Sci. 2017;259:94–103. https://doi.org/10.1016/j.plantsci.2017.03.009.
Huang X, Cao L, Fan J, Ma G, Chen L. CdWRKY2-mediated sucrose biosynthesis and CBF-signalling pathways coordinately contribute to cold tolerance in bermudagrass. Plant Biotechnol J. 2022;20:660–75. https://doi.org/10.1111/pbi.13745.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–4. https://doi.org/10.1093/nar/gkm882.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60. https://doi.org/10.1038/nmeth.3317.
Kim CY, Vo KTX, Nguyen CD, Jeong DH, Lee SK, Kumar M, et al. Functional analysis of a cold-responsive rice WRKY gene, OsWRKY71. Plant Biotechnol Rep. 2016;10:13–23. https://doi.org/10.1007/s11816-015-0383-2.
Li Q, Zheng Q, Shen W, Cram D, Fowler DB, Wei Y, et al. Understanding the biochemical basis of temperature-induced lipid pathway adjustments in plants. Plant Cell. 2015;27:86–103. https://doi.org/10.1105/tpc.114.134338.
Li H, Ding Y, Shi Y, Zhang X, Zhang S, Gong Z, et al. MPK3- and MPK6-Mediated ICE1 phosphorylation negatively regulates ICE1 stability and freezing tolerance in Arabidopsis. Dev Cell. 2017;43:630–42. https://doi.org/10.1016/j.devcel.2017.09.025.
Liang H, Yao N, Song JT, Luo S, Lu H, Greenberg JT. Ceramides modulate programmed cell death in plants. Genes Dev. 2003;17:2636–41. https://doi.org/10.1101/gad.1140503.
Liao Y, Smyth GK, Shi W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. https://doi.org/10.1093/bioinformatics/btt656.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25:402–8. https://doi.org/10.1006/meth.2001.1262.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. https://doi.org/10.1186/s13059-014-0550-8.
Meng J, Hu B, Yi G, Li X, Chen H, Wang Y, et al. Genome-wide analyses of banana fasciclin-like AGP genes and their differential expression under low-temperature stress in chilling sensitive and tolerant cultivars. Plant Cell Rep. 2020;39:693–708. https://doi.org/10.1007/s00299-020-02524-0.
Moellering ER, Benning C. Galactoglycerolipid metabolism under stress: a time for remodeling. Trends Plant Sci. 2011;16:98–107. https://doi.org/10.1016/j.tplants.2010.11.004.
Niu CF, Wei W, Zhou QY, Tian AG, Hao YJ, Zhang WK, et al. Wheat WRKY genes TaWRKY2 and TaWRKY19 regulate abiotic stress tolerance in transgenic Arabidopsis plants. Plant Cell Environ. 2012;35:1156–70. https://doi.org/10.1111/j.1365-3040.2012.02480.x.
Nogueira FC, Gonçalves EF, Jereissati ES, Santos M, Costa JH, Oliveira-Neto OB, et al. Proteome analysis of embryogenic cell suspensions of cowpea (Vigna unguiculata). Plant Cell Rep. 2007;26:1333–43. https://doi.org/10.1007/s00299-007-0327-6.
Paul JY, Khanna H, Kleidon J, Hoang P, Geijskes J, Daniells J, et al. Golden bananas in the field: elevated fruit pro-vitamin A from the expression of a single banana transgene. Plant Biotechnol J. 2017;15:520–32. https://doi.org/10.1111/pbi.12650.
Peppino Margutti M, Reyna M, Meringer MV, Racagni GE, Villasuso AL. Lipid signalling mediated by PLD/PA modulates proline and H2O2 levels in barley seedlings exposed to short- and long-term chilling stress. Plant Physiol Biochem. 2017;113:149–60. https://doi.org/10.1016/j.plaphy.2017.02.008.
Raza A, Razzaq A, Mehmood SS, Hussain MA, Wei S, He H, et al. Omics: The way forward to enhance abiotic stress tolerance in Brassica napus L. GM Crops Food. 2021a;12:251–81. https://doi.org/10.1080/21645698.2020.1859898.
Raza A, Tabassum J, Kudapa H, Varshney RK. Can omics deliver temperature resilient ready-to-grow crops? Crit Rev Biotechnol. 2021b;41:1209–32. https://doi.org/10.1080/07388551.2021.1898332.
Saijo Y, Hata S, Kyozuka J, Shimamoto K, Izui K. Over-expression of a single Ca2+-dependent protein kinase confers both cold and salt/drought tolerance on rice plants. Plant J. 2000;23:319–27. https://doi.org/10.1046/j.1365-313x.2000.00787.x.
Singh B, Singh JP, Kaur A, Singh N. Bioactive compounds in banana and their associated health benefits - A review. Food Chem. 2016;206:1–11. https://doi.org/10.1016/j.foodchem.2016.03.033.
Su W, Li M, Huang H. Effect of betaine on activities of membrane protective enzymes in banana seedlings under low temperature stress. J Southern Agric. 2005;36:21–3.
Upadhyay RK, Handa AK, Mattoo AK. Transcript abundance patterns of 9- and 13-lipoxygenase subfamily gene members in response to abiotic stresses (heat, cold, drought or salt) in tomato (Solanum lycopersicum L.) highlights member-specific dynamics relevant to each stress. Genes (basel). 2019;10:683. https://doi.org/10.3390/genes10090683.
Van Den Berg N, Berger DK, Hein I, Birch PR, Wingfield MJ, Viljoen A. Tolerance in banana to Fusarium wilt is associated with early up-regulation of cell wall-strengthening genes in the roots. Mol Plant Pathol. 2007;8:333–41. https://doi.org/10.1111/j.1364-3703.2007.00389.x.
Varet H, Brillet-Guéguen L, Coppée JY, Dillies MA. SARTools: A DESeq2- and EdgeR-based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS ONE. 2016;11:e0157022. https://doi.org/10.1371/journal.pone.0157022.
Wang Z, Miao H, Liu J, Xu B, Yao X, Xu C, et al. Musa balbisiana genome reveals subgenome evolution and functional divergence. Nat Plants. 2019;5:810–21. https://doi.org/10.1038/s41477-019-0452-6.
Yang QS, Wu JH, Li CY, Wei YR, Sheng O, Hu CH, et al. Quantitative proteomic analysis reveals that antioxidation mechanisms contribute to cold tolerance in plantain (Musa paradisiaca L.; ABB Group) seedlings. Mol Cell Proteomics. 2012;11:1853–69. https://doi.org/10.1074/mcp.M112.022079.
Yang QS, Gao J, He WD, Dou TX, Ding LJ, Wu JH, et al. Comparative transcriptomics analysis reveals difference of key gene expression between banana and plantain in response to cold stress. BMC Genomics. 2015;16:446. https://doi.org/10.1186/s12864-015-1551-z.
Yang QS, Wu SF, Hu CH, Zhang S, Deng G, Sheng O, et al. Transcriptomics and metabolomics analyses reveal the differential molecular mechanisms of cold tolerance between Cavendish and Dajiao banana cells. [Preprint]. 2023 [cited 2023 May 23]: [7 p.]. Available from: https://doi.org/10.22541/au.168483266.60185260/v1.
Zhao C, Wang P, Si T, Hsu CC, Wang L, Zayed O, et al. MAP Kinase Cascades regulate the cold response by modulating ICE1 protein stability. Dev Cell. 2017;43:618–29. https://doi.org/10.1016/j.devcel.2017.09.024.
Acknowledgements
Not applicable.
Funding
This work was supported by the Research Fund of Maoming Branch, Guangdong Laboratory for Lingnan Modern Agriculture (2021TDQD003), the Laboratory of Lingnan Modern Agriculture Project (NT2021004), the National Key R&D Program of China (2018YFD1000302), the National Natural Science Foundation of China (31872939), the Modern Agricultural Innovation Team Project of Guangdong Province (2023KJ106 and 2022KJ106) and the Banana Seed Industry Innovation Park Project of the Guangdong Provincial Department of Agriculture and Rural Affairs (2022-NJS-00–001).
Author information
Authors and Affiliations
Contributions
Q-S Yang and ZY conceived and designed the experiments. SW performed the experiments, analyzed the data and wrote the main manuscript. CH, GD, OS, TD, FB, WH, TD, CL, SL and HG provided laboratory guidance and assistance. Q-S Yang and SZ revised and edited the manuscript. Q-S Yang supervised this study and obtained funding. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Gene primer information for qRT-PCR.
Additional file 2.
All metabolite sample date.
Additional file 3.
Differentially accumulated metabolites summary.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, S., Hu, C., Zhang, S. et al. Lipid metabolism and MAPK-ICE1 cascade play crucial roles in cold tolerance of banana. HORTIC. ADV. 2, 8 (2024). https://doi.org/10.1007/s44281-024-00028-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44281-024-00028-5