
Abstract
Protein–protein interactions and enzyme-catalyzed reactions are the fundamental processes in life, and the quantification and manipulation, kinetics determination, and ether activation or inhibition of these processes are critical for fully understanding physiological processes and discovering new medicine. Various methodologies and technologies have been developed to determine the parameters of these biological and medical processes. However, due to the extreme complexity of these processes, current methods and technologies can only determine one or a few parameters. The recent development of quantitative Förster resonance energy transfer (qFRET) methodology combined with technology aims to establish a high-throughput assay platform to determine protein interaction affinity, enzymatic kinetics, high-throughput screening, and pharmacological parameters using one assay platform. The FRET assay is widely used in biological and biomedical research in vitro and in vivo and provides high-sensitivity measurement in real time. Extensive efforts have been made to develop the FRET assay into a quantitative assay to determine protein–protein interaction affinity and enzymatic kinetics in the past. However, the progress has been challenging due to complicated FRET signal analysis and translational hurdles. The recent qFRET analysis utilizes cross-wavelength correlation coefficiency to dissect the sensitized FRET signal from the total fluorescence signal, which then is used for various biochemical and pharmacological parameter determination, such as KD, Kcat, KM, Ki, IC50, and product inhibition kinetics parameters. The qFRET-based biochemical and pharmacological parameter assays and qFRET-based screenings are conducted in 384-well plates in a high-throughput assay mode. Therefore, the qFRET assay platform can provide a universal high-throughput assay platform for future large-scale protein characterizations and therapeutics development.
Graphical Abstract

Highlights
• A high-throughput quantitative FRET(qFRET) technology platform has been developed for basic biochemical, pharmacological, and system biology research and development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction of FRET and FRET quantification
Some molecules from an excited state, after excitation by light or photo, would emit photons with lower energy from the lowest-lying singlet excited state, so-called fluorescence, and undergo a thermal energy loss to the environment through vibrations. Fluorescent technologies and associated tools have revolutionized our ability to probe biological processes, such as reporter gene expression, protein trafficking, and molecular dynamics, particularly at the cellular and even single-cell levels [1,2,3]. Various fluorescence imaging techniques, such as fluorescence lifetime imaging microscopy (FLIM), fluorescence recovery after photobleaching(FRAP), fluorescence localization after photobleaching (FLAP), fluorescence loss in photobleaching(FLIP) and Förster resonance energy transfer (FRET) have been developed for cellular processes [4,5,6,7]. Many fluorescent sensors have been developed, utilizing either fluorescent proteins or small-molecule dyes, to monitor proteins, DNA, RNA, small molecules, ions, and even cellular properties, such as phosphorylation, pH, and membrane potential [8,9,10,11].
At the same time, some fluorescence technologies have played significant roles in translational research and drug discovery, such as time-resolved fluorescence energy transfer (TR-FRET)/ homogeneous time-resolved fluorescence (HTRF) assays, fluorescence polarization/fluorescence anisotropy (FP/FA) assays [12,13,14]. The TR-FRET/HTRF combines fluorescence resonance energy transfer technology (FRET) with time-resolved measurement (TR), utilizing the long half-life of Lanthanide, such as Europium cryptate (Eu)or Terbium cryptate (Tb) as a fluorescence donor to avoid background fluorescence noise, which half-life normally is very short. The principle of FP derives from the changes of polarization of a fluorophore, which is inversely related to its molecular rotation, before and after binding to another molecule: the emission light intensity parallel and perpendicular to the excitation light plane normalized by the total fluorescence emission intensity [4, 12]. The FP has been used for various drug target classes, including small molecule-protein, ligand-receptor GPCRs, and substrate-enzyme interactions to determine ligand/inhibitor-target interactions and affinity [15]. However, all the above fluorescence technologies have yet to be expanded to comprehensive quantitative measurements for both basic research and HTS drug discovery.
FRET, a physical phenomenon in which dipole-dipole resonance interaction leads to the energy transfer from an excited donor to an acceptor chromophore, has been widely used to detect molecular interactions or molecules in biomedical and chemical research, such as molecular and cellular processes, signal transduction, medical diagnostics, and optical imaging. The FRET phenomena and energy transfer efficiency were first proposed by a German scientist, Theodor Förster, in 1948 [16]. The interaction of two fluorophores, either genetic or organic molecule, occurs between oscillating dipoles with overlapped emission and excitation resonance frequencies and depends on geometric properties of the donor-acceptor pair. FRET can occur when the donor fluorophore (D) and acceptor fluorophore (A) are within a distance of 1–10 nm in general, which is comparable to the size of small organic or biological molecules, and the distances of molecular interactions (Fig. 1A). This high sensitivity of FRET efficiency to the distance of two fluorophores serves as the basis of two FRET-based applications, 1) FRET is used as spectroscopic ruler to precisely measure molecular distance between the donor and acceptor if the distance is not far from their Förster radiuses, R0; 2) the “on” and “off” status of FRET can determine the status of molecular interaction, which fits well with biomolecule interaction distance.

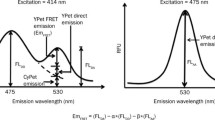
Quantitative FRET(qFRET) signal analysis and high-throughput qFRET assay: A Fluorescent emission intensity of FRET donor and acceptor mixture at acceptor emission wavelength 530 nm, when excited at 414 nm (FLDA), can be divided into three components: sensitized FRET emission, EmFRET, direct emission from free donor, a*FLDD, direct emission of the free acceptor, b*FLAA, where a and b are correlation coefficiency of donor and acceptor, respectively (See B). B The cross-wavelength correlation coefficiency of the donor, a, is defined as the fluorescent emission of the donor at the acceptor emission wavelength, 530 nm, when excited at the donor excitation wavelength, 414 nm (FLDA), divided by the fluorescence emission of the donor at donor emission wavelength, 475 nm, when excited at 475 nm (FLAA). The cross-wavelength correlation coefficiency b of the acceptor is defined as the fluorescence emission of the donor at the acceptor emission wavelength 530 nm, when excited at the donor excitation wavelength 414 nm (FLAD), divided by the fluorescence emission of the donor emission wavelength 530 nm, when excited at acceptor excitation wavelength 475 nm (FLAA). C FRET generation with Mass action of protein–protein interaction. D The qFRET assay is conducted in 384-well plate format in solution as a high-throughput assay
Accurate determination of the FRET signal is critical for measuring the distance between two fluorophores or associated molecules and the molecular interaction events associated with FRET. However, the emissions of free donors at acceptor emission wavelength and free acceptors can cause FRET signal contamination, which includes 1) Bleed-through emission of an acceptor when excited by the donor’s excitation wavelength; 2) Cross-talk of a donor emission contributes to the acceptor emission wavelength (Fig. 1A). FRET signal quantifications, such as FRET efficiency and FRET indices, requires corrections of signals for the bleed-through and the cross-talk.
A direct method to quantify the FRET signal is the ratiometric approach. When a FRET occurs between two fluorescent molecules, the acceptor fluorescence emission is increased and the donor fluorescence emission is quenched. Therefore, this makes it easy to quantify the FRET signal by the traditional ratiometric method, meaning the fluorescent signal at the acceptor emission wavelength divided by the fluorescent signal of donor emission at the donor emission wavelength [17,18,19]. However, the ratiometric method cannot correct the FRET contamination because it does not differentiate the contributions of the free donor, the free acceptor, from the total fluorescence signals. Therefore, FRET signal contamination, through the bleed-through or the cross-talk, affects the accurate FRET signal measurement and needs to be eliminated. Several more rigorous approaches, mainly for quantitative FRET imaging analysis, have been developed to elucidate the FRET signal from the contaminated signals. The first one employs “three-cube” FRET signal analysis for FRET imaging analysis [20,21,22]. To dissect the fluorescence contamination, two images are also acquired: acceptor fluorescence emission during acceptor excitation (IAA) and donor fluorescence emission during donor excitation (IDD), together with the fluorescence emission at the acceptor emission wavelength during donor excitation (IDA image). The crosstalk coefficiencies of the acceptor or donor are given as α and β, respectively, which are constant and assume only these two contaminating components [20, 23]. In this way, the sensitized FRET emission, Fc, can be calculated by the subtraction of the IDA intensity with those contributions of the free donor and free acceptor as
Based on the “three-cube FRET” analysis method, several methods were developed to extract the FRET signal and applied in biological research. A technique developed by Tsuji et al. assumes that the acceptor was not excited at the donor excitation wavelength, and thus, there was no fluorescence contamination from the donor. They applied this method to directly measure a specific messenger RNA in a single living cell [24]. This method is most appropriate for monitoring quick dynamic FRET events, as only one signal is required during detection.
Another primary method was developed to elucidate the accurate FRET signal by correcting signal bleed-through (SBT) through photobleaching or FLIM. Photobleaching is comparatively straightforward to perform on any conventional fluorescence microscope. Photobleaching mainly measures the timing and frequency of fluorescence emission, and therefore, it is not affected by the concentration-dependent emission intensities of the donor and acceptor. The rate of donor bleach is directly related to the excited state lifetime of the donor, which the FLIM method can determine. FLIM is independent of probe concentration, excitation intensity, and other factors that determine steady-state intensity-based measurements [25]. The measurement of fluorescence lifetimes can be divided into the time and frequency domains, which can be used in multi-photo FRET-FLIM microscopy. FLIM can determine FRET efficiency in a concentration-independent way and allows the determination of dynamic events at very high temporal resolution(ns). The combination of quenched and unquenched donor lifetimes obtained by FRET-FLIM allows a more precise estimate of the distance between donor and acceptor than that based on the concentration-dependent FRET signals from the donor and acceptor.
The traditional FRET-FLIM technique requires that the donor is photolabile, and the acceptor is photostable, so the FRET signal only comes from the acceptor. An opposite version of the traditional FRET-FLIM is a measurement for the photobleaching of the acceptor, which requires the acceptor to be photolabile and the donor to be photostable [26,27,28]. This approach determines the donor fluorescence's reappearance after saturating the FRET acceptor signal. The basic principle is to measure donor intensity before and after complete acceptor bleaching to provide internal control without FRET. The increase in donor intensity can be directly related to the FRET efficiency after the correction of the acceptor cross-talk in the donor emission wavelength. In principle, this method requires that acceptor bleaching be complete, which is difficult in most cases due to the low intensity of excitation light for the acceptor. This limitation was overcome by a curve-fitting method of continuous measurements of the donor and gradual acceptor photobleaching [28].
In addition to the above method, a step-by-step algorithm to remove SBT contamination in FRET images was also developed, in which the bleed-through ratios were not considered constant but were determined at each fluorescence intensity in confocal and two-photon FRET microscopy [29]. Moreover, the FRET signals have been determined using a complex matrix of α, β, γ and ζ. The α depicts contaminated FRET signal (direct excitation of acceptor by the donor excitation wavelength), the β describes spectral bleed-through of the donor, the γ characterizes the ratio of the extinction coefficient of acceptor/donor at the donor’s excitation wavelength, and the ζ represents fluorescence intensity of the acceptor’s sensitized emission signal to the fluorescence intensity that would have arisen from the quenched donor. This complex-matrix approach was used to study the cellular protein-binding affinities (KD) of dynamic protein interaction in insulin secretory granule behavior by three-dimensional FRET microscopy images from the total internal reflection fluorescence (TIRF) microscopy [30, 31].
A recent correlation coefficiency method for elucidating sensitized FRET signal(EmFRET) has been developed for spectroscopy/fluorescence plate reader, which can be adapted as high-throughput assays [32,33,34] (Fig. 1A). The fundamental principle of correlation coefficiency method is based on the facts that emissions of FRET donor or acceptor at the FRET signal emission wavelength are correlated to their emissions at another wavelength or excitation wavelength, respectively, resulting from the same chemical structure of fluorophores with or without FRET. Therefore, determining the emissions of free FRET donors or acceptors, which contribute to the bleed-through signals, can be achieved through their emissions at another wavelength or excitation wavelength times a constant for FRET donor or acceptor, respectively. This constant is the so-called correlation coefficiency constant. Specifically, the measurements of donor fluorescence intensities at its own emission wavelength(FLDD) and acceptor emission wavelength(FLDA) to establish a ratio α, and acceptor fluorescence intensities at its excitation wavelength(FLAA) and donor excitation wavelength(FLDA) to establish a ratio β in advance (Fig. 1B). The sensitized FRET signal(EmFRET) is obtained by subtracting total fluorescence intensity(FLDA) at the acceptor emission wavelength by the contribution of the donor and acceptor,
The sensitized FRET signal(EmFRET) is correlated with the interaction events of the FRET donor and acceptor (Fig. 1C). Therefore, it can be used to determine molecular interaction affinity or other biochemical or pharmacological parameters. Because the engineered FRET donor and acceptor, CyPet and YPet, have high quantum yields, all the fluorescent signals can be determined using a fluorescence plate reader in a 384-well plate, and therefore, all the assays can be performed in a high-throughput mode (Fig. 1D).
Because this method is straightforward and robust to elucidating sensitized FRET signals, which can be correlated with various biochemical reactions, it has been developed into a platform of multiple biological and pharmacological parameter determinations and high-throughput assays [35,36,37] (Graphic Abstract). The developments of qFRET-based mathematic algorithms for various reactions and experimental procedures and their application for determinations of protein binding affinity (KD), protease endopeptidase and Isopeptidase kinetics (kcat/KM), product inhibition for both enzyme-product interaction affinity(KD) and enzyme kinetics(kcat/KM), peptide transfer reaction kinetics(kcat/KM), enzyme inhibitor affinity(Ki), Ubl enzyme specificity determination, HTS for protein-protein interaction and multiple-enzyme catalyzed cascade inhibitor discovery. determinations in SUMOylation cascade. The new development of qFRET assays covers basic research and translational research and development, and it can be generally expanded to many biological and pathological processes as a generic assay platform in the future.
The qFRET method for protein–protein interaction dissociation constant K D determination in solution
Current techniques for protein interaction affinity determination
Protein–protein interaction plays pivotal roles in all the physiological processes and pathological developments of living organisms, from signal transductions, ligand–receptor interactions, transcript/translation, genome replication, cell death, and cell cycles to various diseases, such as cancers, infectious diseases, diabetes, and neurodegenerative diseases [38, 39]. Accordingly, many technologies have been developed to determine protein-protein interaction affinity, such as radioactive-labeled binding assay, ultracentrifugation, surface plasmon resonance (SPR), nuclear magnetic resonance (NMR), calorimetric methods (for example, ITC—isothermal titration calorimetry and DSC—differential scanning calorimetry), FP, and FRET (Fig. 2). These methods have significantly enable us to understand protein functions and dynamics quantitatively for basic research and translational studies. However, these methods are limited by their intrinsic capabilities, such as the requirement of tedious protein expression and purification, a large quantity of proteins, expensive instruments, or tedious procedures. Therefore, the protein interaction affinities for those “difficult-to-be expressed” proteins and large-scale networks at the proteomics level are still largely unknown so far, resulting in a poorly understanding of those proteins in physiology and pathology, and even few therapeutics that target protein–protein interactions [40,41,42,43].

Comparisons of current major technology and protein–protein interaction assay capability and affinity determination
The SPR, a quantitative spectroscopic technique, is a popular technology to determine protein-protein or protein-other molecule interactions [44,45,46]. The method detects the changes of the refractive beam caused by a protein binding event at a functional sensing surface, in which an interactive partner protein is attached in advance. The SPR sensing surface is generally a metallic gold surface interfaced with a prism. Monochromatic light is reflected from the functionalized metal surface at an angle called the SPR angle, which is generated from the plasmon of the excited metal surface electrons oscillation after the potential binding of the interactive protein or other molecules. In the event of a biomolecule interacting event, the SPR angle of the reflective beam would shift after binding of one protein to the bound protein [45, 47]. The advantage of the SPR method is that it can detect both on and off rates in real-time at high sensitivity, leading to the determination of protein interaction affinity. However, the SPR method has some caveats. First, proteins immobilized on the metal sensor surface may change their conformation, and the different orientations of immobilized ligands on the metal surface may result in decreased affinity determination [48, 49]. Second, the surface-immobilized ligands would lead to a higher local concentration than that of the solution, resulting in the binding kinetic difference from ideal pseudo-first-order binding due to the mass transfer effect [49, 50]. Third, the SPR method for KD determination might not be valid when a binding event does not follow a simple Langmuir-type binding model [50]. Finally, the re-binding effect and nonspecific binding to the sensor chip may occur; thus, often leading to significant variations of determined parameters [51]. Moreover, the SPR requires two binding proteins in the assay to be highly pure.
In addition to SPR, the ITC is also often used for KD determination [40, 52]. These methods offer experimental convenience, but also have some disadvantages. ITC directly measures heat release that are caused by the endothermic or exothermic reaction between two interacting molecules [53, 54]. The ITC often requires environmentally unfriendly labeling and cannot determine impure protein interactions. ITC requires relatively large amounts (i.e., millimolar range) of highly purified proteins, which often are hard to obtain. Systematic errors in heart calibration, cell volume, and other errors (e.g., baseline and gas bubbles), can lead to inaccuracies in KD values [55,56,57]. In addition, ITC requires expensive specialized instrumentation. Other methods, such as radioactive labeling, ultracentrifugation, and fluorescence polarization, are also used to determine molecular interaction affinity with some limitations. For the ultracentrifugation assay, the elongated centrifugation in a non-physiological buffer can shift the equilibrium between bound and free proteins, especially for those proteins with fast dissociation rates; thus, the determined KD values may not represent true equilibrium constants. In addition, large amounts of proteins, which can be nonspecifically adsorbed on the walls of centrifuge tubes during high-speed centrifugation, are required for ultracentrifugation. In principle, the FP assay, which measures the changed of polarized light from one interactive partner after binding, can potentially address the above issues [58]. The FP assay has successfully determined peptide or nucleic acid interaction dissociation constants and an HTS platform for small molecule drug screening. However, the FP method determines polarized light changes of free-rotated fluorescence molecules; therefore, one interactive partner must be small and attached to a fluorophore, significantly affecting the interaction affinity. When measuring large proteins, the FP assay becomes less sensitive [59]. It also typically requires sensitive instruments for accurate quantification, such as fluorescence microscopy, preventing its high-throughput application.
Recently, several new attempts have been made to develop the FRET assay for protein interaction affinity determination. The three-cube or ratiometric analyses of the FRET signal can be used to elucidate the sensitized FRET signal, which can be used to determine protein interaction affinity (KD). However, the three-cube analysis approach neecds to measure the molar extinction coefficients of fluorophores and FRET efficiency, which are instrument-dependent and variable parameters, making it difficult to turn the approach into a general and robust methodology [60, 61]. The recent FRET analysis approach measures the sensitized FRET signal using a point-to-point subtraction strategy and the ratiometric approach, which does not exclude signals from direct emissions of donor and acceptor and is not mathematically rigorous [31, 32]. The ratiometric FRET assay to determine protein interaction affinity usually generates KD values higher than those determined by the SPR or the ITC due to the above reasons. The lack of accuracy and robustness of these FRET analysis approaches for KD determinations is mainly due to the estimations of multiple mathematic and fluorescence parameters, resulting in the inaccuracy of sensitized FRET signal determination.
Acceptor emission-based qFRET assay for K D determination
The newly developed qFRET analysis can accurately and robustly determine the sensitized FRET signal, which is determined by the subtraction of the total fluorescence signal at FRET wavelength with the fluorescence signals of the free donor and the free acceptor at the FRET wavelength, which are obtained from the donor or acceptor emissions multiplied by their cross-wavelength correlation coefficiencies, α and β, respectively [32, 62] (Fig. 1B and C). The FRET donor and acceptor interactions lead to both acceptor emission and donor quenching (Fig. 3A) as demonstrated in the case of CyPet-SUMO1 and YPet-Ubc9 interaction, which are the small Ubiquitin-like peptide and E2 conjugating enzyme in the SUMOylation cascade (Fig. 3B). According to the fundamental mass action law for molecular interaction, the sensitized FRET signal can be correlated with protein-protein interaction events for the KD determination if both fluorescence emissions of free FRET donor and acceptor can be determined (Fig. 3C).

The qFRET assay for both acceptor emission and donor quenching based-protein–protein interaction dissociation constant KD determination. A. FRET spectrum diagram of both acceptor sensitized emission and donor quenching when two fluorophores are close to each other. B. FRET signals increase as the acceptor concentration increases. C. Diagram of fluorescence signals of protein–protein interaction dissociation constant KD determination. D. KD values of SUMO1-Ubc9 determined by the qFRET at four donor concentrations in both acceptor emission and donor quenching modes, as compared with the KD values determined by the SPR and ITC
The qFRET signal analysis method uses the cross-wavelength correlation coefficiency for both donor and acceptor to determine the emissions of free donor and acceptor and, therefore, extrapolate sensitized FRET signal from the emissions of free donors and acceptors (Fig. 1). In this approach, the first step is to determine the direct emissions of the FRET donor and acceptor. In order to measure the direct emissions of the donor and acceptor, a cross-wavelength coefficiency method has been developed. First, a correlation coefficiency constant ratio, α, which is defined as the direct emission of the donor at the acceptor emission or FRET wavelength (FLDA) is divided its emission at 475 nm when excited at 414 nm (FLDD), FLDA/FLDD, and the α can be predetermined using pure donor fluorescent protein (Fig. 1B). Therefore, the emission of the donor at the FRET emission wavelength (530 nm) can be calculated as α*FLDD. A similar but mathematically different approach for the acceptor direct emission determination. The emission of the free acceptor at the FRET emission wavelength (530 nm) when excited at the donor excitation wavelength (414 nm) (FLAD) is proportional to its emission at the FRET emission wavelength (530nm) when excited at 475 nm (FLAA), which can be extrapolated with a constant, β defined as FLAD/FLAA, and could be predetermined using a pure acceptor protein (Fig. 1B). Second, after α and β are determined, when two interactive proteins are mixed, the emissions of the FRET donor at 475 nm when excited at 414 nm and the FRET acceptor at 530 nm when excited at 475 nm is determined, the sensitized FRET emission signal can be calculated by subtracting the sum emissions of free donor and acceptor,
Once the EmFRET is determined, following the general mass action law for molecular interaction,
where Protein 1-FRET donor indicates Protein1 tagged to the FRET donor and Peotein2-FRET acceptor indicates Protein 2 tagged to the FRET acceptor.
The equilibrium constant, KD, can be defined as in Eq. (3) or Fig. 1C.
After rearrangement,
where [Protein2-Acceptor]boundmax is the maximal Protein2-Acceptor bound to Protein1-donor in theory. [Protein2-Acceptor]bound is proportional to the FRET signal from the bound proteins,
Then, Eq. (4) can be converted into Eq. (6) using the relationship shown in Eq. (5):
where the A is the fixed concentration of Protein1-Donor and X is the different titration concentrations of Protein2-Acceptor added into the assay. The KD and EmFRETmax values can be determined through the multivariable regression using Eq. (6) after the EmFRET values are determined using Eq. (1) at various Protein2–Acceptor concentrations.
Many protein–protein interaction affinity KD values were determined using the FRET acceptor emission approach. For example, the KD value between SUMO1 peptide and Ubc9 was determined at 0.41 ± 0.02 μM, which is compatible with the value of 0.35 μM from the SPR, or the value of 0.25 ± 0.07 μM from ITC [38, 39, 52] (Fig. 3D).
Donor quenching-based qFRET assay for K D determination
When FRET happens between a donor and an acceptor, the transferred energy from the donor to the acceptor leads to a reduced fluorescence emission of the donor, called fluorescence quenching (Fig. 3A). As the FRET signal emitted by the FRET acceptor is proportional to the amount of FRET donor and acceptor bound complex, the decrease of the FRET donor signal, also called the quenching of donor, is proportional to the bound complex, and therefore could be used for KD determination [63]. In contrast to the FRET acceptor emission method, fluorescence quenching is a more general approach than fluorescence emission, as all FRET acceptors can quench donor fluorescence, especially in small molecule FRET pairs, while not all acceptors can emit light during the FRET assay [64]. The fluorescence quenching energy is positively proportional to the amount of acceptor bound to the donor, or the concentrations of the protein–protein interaction complex (Fig. 3B). The fluorescence quenching approach was first demonstrated in characterizing antibody–hapten binding by Velick et al. and later macromolecules by Liu and Schultz [65, 66].
The emission intensity of Protein 1–FRET donor decreases as FRET occurs, following the law of mass action for the protein-protein interaction in Eq. (2) (Fig. 3B). Because the quenching energy of Protein 1–FRET donor when FRET occurs is proportional to the number of bound protein complexes, the relationship of the decreased donor light emission and the concentration of bound protein can be represented as follows:
where ΔEmdonor is the decrease of emission intensity at the donor emission wavelength when excited at its wavelength at each specific concentration of Protein 2–FRET acceptor, n is a constant related to the FRET efficiency, affinity, and distance between Protein 1–FRET donor and Protein 2–FRET acceptor, and n can be experimentally determined; [Protein 2–FRET acceptor]bound is the concentration of bound Protein 2–FRET acceptor. As follows,
where x is the concentration of the FRET acceptor in the FRET assay.
To simplify the mathematic procedure, we set the total concentration of Protein 1–FRET donor as a constant A, the concentrations of Protein 2–FRET acceptor as a variable X, and ΔEmdonor as the variable Y, we can convert the concentration of bound and free Protein 1–FRET or Protein 2–FRET acceptor proteins in Eq. (2) to:
and
and
Therefore, we can derive KD as following using Eqs. (9, 10, 11),
We can determine the value of KD and the constant n using the multi-variable regression with Eq. (12) by fitting ΔEm bound (Y) and the total Protein 2–FRET acceptor concentration, X.
The qFRET quenching method has been used to determine the protein–protein interaction disassociation constant KD value of the same protein pair of SUMO1–Ubc9 as before. The KD value was determined as 0.47 ± 0.03μM, which is very compatible with the value of 0.41 ± 0.02 μM determined using the acceptor emission approach as well as the KD values determined with the SPR and the ITC (Fig. 3D).
Both acceptor emission and donor quenching qFRET measurements are carried out in the 384-well plates, and fluorescence signals are determined using fluorescence plate readers, which can quickly assess fluorescence intensity. Therefore, this approach can be implemented in a high-throughput assay mode for large-scale measurement.
The qFRET methods for high-throughput enzymatic kinetics determinations
Current approaches for protease endopeptidase kinetics determinations
Enzyme catalytic efficiency and specificity for a substrate are generally best described by its ratio of the turnover number vs Michaelis constant, kcat/KM, and so-called enzyme kinetics. Many biochemical and biophysical methods are used to examine enzyme kinetics, including Western blotting, radioactive-labeled substrate, colorimetric assays, fluorescence-labeled substrate, and substrate mimics. However, most current enzymatic kinetics methods have some limitations and drawbacks due to non-natural substrates, poor sensitivity or slow kinetics of detection methods. For example, the protease kinetics of the sentrin-specific protease 1(SENP)1 for maturations of pre-SUMO, 2 and 3 by was determined in solution for product formation using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) [67, 68]. This approach showed SENP1’s preference but did not detail quantitative kinetics due to the limitations of slow detection of product formation. In another study of SENP, an organic fluorophore, 7-amino-4-carbamoylmethylcoumarin (ACC), was used to label a tetrapeptide substrate (QTGG), which mimicked the substrate terminal amino acid sequence. The ACC moiety was quenched in the labeled substrate and became highly fluorescent upon cleavage by SENPs. The kcat/KM determined for SENP1 for the QTGG peptide was ~ 300 M−1 s−1, which was at least two orders of magnitude lower than the natural substrates due to the distorted binding affinity of the small peptide substrate by the enzymes [68, 69]. Another study produced an assay-tagged 7-amino-4-methylcoumarin (AMC) with mature full-length SUMO peptide at its C-terminus. The values of kcat/KM were 2.4 × 106 M−1 s−1 for SUMO1 and 5.6 × 105 M−1 s−1 for SUMO2, But these values are generally lower than traditionally considered enzyme kinetics numbers. Moreover, this system could not differentiate the activities between SENPs’ isopeptidase and endopeptidase as there is no specific sequence of either SUMO tail or SUMO-specific conjugated substrate after the AMC moiety [70].
Recently, a TR-FRET assay has been developed for protease kinetics determinations due to the nature of delayed fluorescence determination in TR-FRET and strong fluorescent signals from metals. For example, in the studies of SENPs or deubiquitinating enzymes (DUBs), the FRET pair Eu-cryptate and allophycocyanin (APC) were conjugated to the anti-Myc and anti-FLAG antibodies, which interacted with pre-Nedd8 with Myc and FLAG fusions on the N- and C-terminuses, respectively [71]. In another TR-FRET assay for SENP kinetics study, FRET pair, Terbium (Tb) and YFP (yellow fluorescence protein), were tagged on SUMO and anti-RanGAP, respectively, to study SENP isopeptidase kinetics during SENP’s deconjugation [72]. However, these FRET-based assays require additional steps for FRET assay development, including the conjugation of immune antibodies or the chemical conjugation of thiol-reactive Tb chelate to ubiquitin-AC or other fluorophores. The variable conjugation efficiencies and the indirect measurement may add additional variables during the quantitative analysis. Furthermore, all of the above FRET-based protease assays use the traditional ratio metric method to quantify the FRET signal, and thus emissions from free FRET donor and acceptor that would lead to an inaccurate FRET signal determination are not considered in these assays [23, 73].
qFRET method for protease endopeptidase kinetics determination, k cat , K M , and k cat /K M
The qFRET assay can also track the sensitized FRET signal changes during the protease digestion reaction in real-time (Fig. 4A). The initial efforts focused on development of qFRET analysis to differentiate donor, acceptor, and sensitized FRET signal to correlate the absolute FRET signal with molecular interaction events for the protein interaction affinity KD by calibration curve or direct estimation of absolute FRET signal via the correlation coefficiency constant [32, 74]. A similar methodology was then modified for the protease endopeptidase kinetics determinations. The first development was to determine the sensitized absolute FRET signal, which corresponded to the undigested substrate, such as CyPet-preSUMO1-YPet, and direct emissions of the digested free donor and acceptor using two external standard curves of the digested substrate with donor, CyPet-SUMO, and the acceptor, YPet, and substrate, CyPet-preSUMO-YPet, at different concentrations, respectively [75]. Then, emissions of digested YPet and CyPet-SUMO1 and the remaining substrate in the protease digest reaction were determined. The sensitized FRET signals were converted into substrate concentrations by pre-established fluorescence protein standard curves. This method needs several steps to determine the sensitized FRET signal and convert it to protein concentrations, and during the conversions, variability is introduced in each step of measurements. Therefore, a more straightforward method to determine the sensitized FRET signal by taking advantage of our recent development of “cross-talk” ratio or correlation coefficiency methodology for qFRET analysis [32, 37].

qFRET assays for protease endopeptidase and Isopeptidase kinetics determinations. A Schematic diagram of qFRET-based protease endopeptidase assay. B Schematic diagram of qFRET-based protease isopeptidase assay involves two steps-isopeptide substrate preparation and protease digestion. C The FRET spectrum change of FRET-protease substrate before and after protease digestion
The idea of a “cross-talk” ratio or correlation coefficiency method to determine a fluorophore’s direct emission is to use two pre-determined ratios for donor and acceptor, α and β, respectively (Fig. 1A and B). Then, during the kinetics assay, the reaction was monitored to determine three emissions simultaneously, the total fluorescence emission at the FRET wavelength, the emission of the donor at its maximum emission wavelength, such as 475 nm, when excited at the FRET excitation wavelength, such as 414 nm, and the emission of acceptor at its maximum emission wavelength, such as 535 nm, when excited at its maximum excitation wavelength, such as 475 nm.
Similarly to the correlation coefficiency for protein-protein interaction dissociation constant determination using qFRET, the correlation coefficiency constant of donor self-fluorescence (α) is determined using FLDA/FLDD, and the correlation coefficiency constant of the acceptor self-fluorescence (β) is determined using FLDA/FLAA
Then, we can differentiate the total fluorescence signal at 530 nm when excited at 414 nm (FLDA) as the contributions of three components: sensitized FRET emission(EmFRET) the donor’s direct emission (Id530/414) and the acceptor’s direct emission (Ia530/414):
or
where Id475/414 is the donor emission at 475 nm when excited at its excitation wavelength, 414 nm, and Ia530/475 is the acceptor emission at 530 nm when excited at its own excitation wavelength, 475 nm.
After digestion by the protease, such as SENP1, the fluorescence signal at 530 nm decreased, and the fluorescence signal at donor emission wavelength, 475 nm, increased because of substrate digestion (Fig. 4B). The remaining fluorescence emission at 530 nm (FL’DA) could still be divided into similar three components:
where FL’DA is the remaining emission at FRET or acceptor wavelength, FLDD’ is the fluorescence emission of the FRET donor during digestion, which consists of two parts: the undigested substrate, CyPet-(pre-SUMO)-YPet, and the digested donor, CyPet-SUMO, and FLDD’ is the fluorescence emission of the acceptor, YPet, which is constant whether the substrate is digested or not.
The amount of digested substrate is proportional to the decrease of sensitized FRET signal. Hence, during digestion of protease SENP1, the remaining substrate-induced acceptor’s emission EmFRET',
where C is the original concentration of substrate, CyPet-(Pre-SUMO)-YPet, and x is the concentration of digested substrate, CyPet-SUMO and -YPet, at the specific measurement time.
Therefore, the fluorescence emission at the FRET wavelength can be expressed as
In the internal calibration method, FLDA’, FLDD’ FLAA’ can be directly measured during the experiment, whereas FLDA, FLDD, FLAA, α and β are predetermined. So, the amounts of the digested substrate are determined during the experiment in real-time using the fluorescence signals determined during the reaction. The enzyme kinetics were determined using the internal calibration method, or a “cross-talk” ratio or correlation coefficient method, and compared to those determined by the standard curve method. The kinetics parameters determined by the internal calibration method were very similar to those determined by the standard curve method but with significantly less variation [34]. The internal calibration method eliminates the need for standard curves and reduces experimental procedures.
qFRET method for protease isopeptidase specificity and kinetics determination
A unique application of qFRET assay for proteins is protease isopeptidase kinetics determination. Protease isopeptidase activity refers to the breakage of a peptide bond between a peptide and a side chain of amino acids in a protein. Isopeptidase activity of proteases plays critical roles in protein degradation and activity regulations of physiological and pathological processes in living organisms, such as cancers and protein activity in infectious diseases. However, due to a lack of methodology, the kinetics of protease isopeptidase activity has yet to be explored.
Similar to the FRET-based protease assay design to study the endopeptidase activity of SENPs [34, 75, 76], the qFRET has also been developed for the deconjugation of peptide SUMO1 from the conjugated substrate RAnGap1c by the protease SENPs [77] (Fig. 4C). In this approach, the first step is to develop a pure peptide conjugated substrate with an isopeptide bond. The His-tagged FRET donor, CyPet, and the GST-tagged FRET acceptor, GST-YPet, were first genetically tagged to the SUMO1/2 and RanGAP1c, respectively. In the presence of SUMO activating E1 enzyme, Aos1/Uba2, and conjugation E2 enzyme, Ubc9, and ATP, His-CyPet-SUMO1/2 was covalently linked to GST-YPet-RanGAP1c to form the conjugated substrate, resulting a substrate with FRET signal. After SUMOylation, the reaction mixture was first flown through glutathione agarose beads to recover His-CyPet-SUMO1/2-GST-YPet-RanGAP1c and un-SUMOylated GST-YPet-RanGAP1C from the other proteins. The eluted proteins were then run through a Ni-NTA agarose beads, to capture the His-CyPet-SUMO1/2-GST-YPet-RanGAP1c as a pure substrate for isopeptidase kinetics determination.
The SUMO-conjugated substrate provided a unique reagent for the unique studies in both qualitative and quantitative analysis of protease SENP isopeptide activities. The FRET substrate, His-CyPet-SUMO-GST-YPet-RanGAP1C, was then incubated with different SENPs for isopeptidase kinetics study. In this study, the ratio matric (FL530/FL475) method of FRET analysis was used to characterize the changes in FRET signals for the digested substrate determination during the reaction. The results show that different specificities of SENPs towards different SUMO deconjugation: both SENP1C and SENP2C can deconjugate SUMO1 and SUMO2 from target substrate, RanGAP1C, at high efficiency, while as SENP5, SENP6, and SENP7 prefer SUMO2 but not SUMO1 for de-conjugation. All of these are in agreement with previous observations in biochemical assays [67, 78,79,80].
The kinetics parameter determinations for both isopeptidase and endopeptidase of protease SENP1 were performed using the qFRET method as described above. The digested substrate was determined (Fig. 4 right). The results indicated that SENP1 exhibited higher activity toward SUMO deconjugation than pre-SUMO maturation (especially for SUMO2). The initial velocity and kinetics parameters were then determined. This is the first time to determine protease isopeptidase kinetics accurately.
qFRET method for transfer reaction kinetics k cat, K M, and k cat /K M determination
The flexibility of the qFRET method to track product formation either through FRET signal generation or disappearance quantitatively enables it to measure many biochemical reactions, which are normally difficult for other methods. For example, a very common peptide transfer reaction in the cells is the activation immediately followed with a transfer reaction of Ubiquitin or other Ubls, such as SUMO and NEDD8. Ubiquitin and Ubls play important roles in protein degradation and activity regulations, and recent new technology developments related to Ubl, such as Proteolysis Targeting Chimeric (PROTAC) or Molecular Glu, are very promising for the next wave of therapeutics for cancers, infectious diseases, and others. Both Ubiquitination and SUMOylation processes involve an enzymatic cascade that includes E1 activating enzyme, E2 conjugating enzyme, and E3 ligase [81]. In the SUMOylation, the E1 activating enzyme, a heterodimer of Aos1 and Uba2 (or SUMO activating enzyme, SAE1 and SAE2), plays a vital role in initiating the cascade by using ATP to form a high-energy thioester bond intermediate [82].
The traditional methods for determining the kinetics of the E1 in ubiquitination and ubiquitination-like protein pathways need radioisotope or organic fluorescent dye-labeling of peptides in the enzyme reactions. Radioisotope measurement for the adenylation activating step was performed as an ATP:PPi exchange assay, and the [32P]ATP concentrations were measured on activated charcoal powder [83, 84] or paper [85]. The adenylation process of Ubiquitin required [3H]labeled substrate in adenylate assay [84]. Oregon green (Og) label was used in a steady-state kinetic analysis of human ubiquitin E1 [86]. In all above assays, the procedures are not only very time- and labor-consuming but also not environmentally friendly.
The transfer reaction kinetics of SUMOylation E1 activity was based on the pseudo-first-order association kinetics with a qFRET tracking for the product formation, CyPet-SUMO1•YPet-Ubc9 (Fig. 5A). The E1 activating enzyme in SUMOylation has three substrates: ATP, SUMO, and Ubc9. In the kinetics assay, one substrate concentration, such as CyPetSUMO1, was changed while maintaining saturated concentrations of the other two substrates, such as ATP and YPet Ubc9, as various substrate concentrations and vice versa. The product formation was tracked with the fluorescence signal (FLDA) (Fig. 5B), and initial velocity was calculated in the first 30 sec of the reaction (Fig. 5B). The Michelis-Menton curve was generated to determine KM and VMAX, for example of CyPet-SUMO1 as the substrate. The kinetics parameters for SUMO1,2,3 and ATP were determined (Fig. 5C). For the first time, we detailed the steady-state kinetic for the E1 activating enzyme of the SUMOylation pathway, and the approach can be used for other peptide transfer reaction characterizations. In addition, the quantitative FRET assay was conducted in a 384-well plate platform that can increase the efficiency of the steady-state kinetics assays by reading multiple samples simultaneously [87].

Peptide SUMO1 activation transfer kinetics assay. A The scheme of SUMO1 peptide activation and conjugation to its E2 conjugating enzyme Ubc9 in the presence of E1 heterodimer Aos1/Uba2 and ATP. B The FRET signal increases as CyPet-SUMO1 are conjugated to YPet-Ubc9. C Determined kinetics parameters of SUMOs peptide transfer reaction
The qFRET method for high-throughput screening (HTS) for chemical biology and drug discovery
Numerous small chemical compounds or biological molecules, which involved in modulations of other molecule activities, such as proteins, have been used as regulators used in biological research and drug discovery. Most bioactive molecules target enzymes and function by activating or inhibiting enzymatic activities. Another class of bioactive molecules activates receptors, such as GPCRs, or disrupt protein-protein interactions. Those bioactive molecules are usually discovered through high-quality HTS against a large collection of chemical or degenerative biological libraries. A good HTS assay must have good reproducibility, a high signal-to-noise ratio, low variation, and low cost. Due to the high sensitivity and potential low cost, the qFRET assays have also been developed into HTS assays for discovering protein-protein interaction inhibitor, protease inhibitor, and enzyme-catalyzed cascade inhibitor.
qFRET-based HTS for protein-protein interaction inhibitor discovery
Although many technologies have been developed to determine protein–protein interaction and affinity, disrupting these interactions with chemical compounds has been challenging partially because of the large and often flat interaction interfaces of two interactive molecules and as a result, few drugs have been developed to disrupt protein–protein interactions [88]. However, there has been significant efforts have been put into the search for small chemical compounds that could disrupt protein–protein interactions resulting in an increasing number of small molecules with reasonable potency have been identified. Those include nutlin-3, an inhibitor of p53-Mdm2 interaction [89], chelerythrine, an apoptosis-inducing compound that disrupts Bak/Bcl interaction [90], and Memoquin, an anti-Alzheimer drug that disrupts the aggregation of Amyloid-β [91]. The discovery of the inhibitors disrupting protein-protein interaction could open a new avenue to dissect biological processes in physiology and diseases.
A HTS assay seeking for inhibitors that can disrupt the interaction of SUMOyaltion E2 conjugating enzyme, Ubc9, and E3 ligase, PIAS1, was developed [36] (Fig. 6A). The FRET donor, CyPet, and the FRET acceptor were fused with Ubc9 and PIAS1, respectively. Then the fusion proteins were expressed in the bacterial cells and purified. To optimize the assay with a higher signal-to-background (S/B) ratio, the CyPet-Ubc9 and Ypet-PIAS1 ratio was set up to 1:1 and increased the concentrations of both proteins from 0.25 μM to 2 μM in order to achieve the strongest signal with EmFRET as readout. For quality assessment of HTS, a statistic factors, Z’, have been developed,
where \({\sigma }_{p}\) is the standard deviation of the positive control, \({\sigma }_{n}\) is the standard deviation of the negative control, \({\mu }_{p}\) is the mean of the positive control, and \({\mu }_{n}\) is the mean of the negative control in the HTS assay development and campaign. The Z’ values between 0.5 and 1 are regarded as excellent, values between 0 and 0.5 may be acceptable, and values less than 0 indicate that the assay is unable to perform in a high-throughput context.

The qFRET-based high-throughput screening(HTS) assays. A Design of qFRET-based HTS for discovering protein–protein interaction inhibitor discovery. B The signal-to-noise (S/B) and Z’ of the qFRET-based SUMO E2 Ubc9 – E3 PIAS1 protein–protein interaction inhibitor discovery. C qFRET-based SUMOyaltion full-pathway(except E3) HTS, including SUMOylation and deSUMOylation. D The qFRET signal changes during the HTS assay of SUMOylation and deSUMOylation reactions
The Z’ factor of the HTS assay for CyPet-Ubc9 and YPet-PIAS1 interaction screening was 0.76 with an S/B ratio of 6.88, suggesting a high quality and well suited to HTS (Fig. 6B). The HTS screening campaign for disrupting CyPet-Ubc9 and YPet-PIAS1 interaction inhibitor was carried out in 384-well plates. Since no Ubc9-PIAS1 interaction inhibitor were reported at the time, Sodium lauryl sulfate, which nonspecifically disrupts protein interactions, was used as the positive control. After the addition of compounds (0.5 μL in 1 mg/mL), the plates were incubated at room temperature for 30 min, and the fluorescence intensity signals, FLDA, FLDD and FLAA were measured by an EnSpire® multimode plate reader (PerkinElmer). A hit rate of 1.1% was achieved after secondary and confirmatory assays, in good agreement with general HTS assays. A compound with KD value of 1.93 ± 0.62 μM was finally confirmed with the SPR assay, indicating a reliable HTS assay. Another HTS assay to discover potential inhibitors disrupting SUMO1 and E2 Ubc9 was also developed based on the qFRET [35]. The optimization of CyPet-SUMO1 and YPet-Ubc9 ratio (12 pmole of CyPetSUMO1 and 21.6 pmole of YPetUbc9) was optimized leading to the Z’ factors above 0.7, indicating a reliable and robust HTS assay.
The qFRET-based HTS for full-pathway inhibitor discovery
Although extensive efforts have been taken to discover potent and specific SUMOylation inhibitors, the discovery of SUMOylation inhibitors has been very challenging because most of these inhibitors are either not potent or have no cellular SUMOylation inhibition activity [92,93,94,95,96,97,98,99,100,101]. Almost all other SUMOylation inhibitors reported in public have been tested but without potent activity in cells. Another strategy to discover the SUMOyaltion initiator was mimicking ATP and its analogs as the SUMOylation pathway utilizes it to activate SUMO peptides. However, the ATP analog design strategy is challenging for potential side activities and cellular permeability. The unavailability of potent and cellular active SUMOyaltion inhibitors is probably partially due to the need for robust HTS assay.
A unique capability of qFRET-based HTS format is a full SUMOylation/deSUMOylation pathways screening, which carries the peptide SUMO1 to the substrate conjugation in the presence of SUMOylation E1, E2 enzymes and then deconjugation with the addition of protease SENP1 (Fig. 6C). Even with this very complicated multi-enzyme involved HTS, the Z’ factor of both screening assay and got 0.56 and 0.61 for the SUMOylation and deSUMOylation inhibitor screening, respectively, suggesting a robust HTS assay. After an HTS campaign for screening more than 220,000 compounds, STE025 was discovered with high potency(Ki=0.6μM) and good cellular activity (IC50=1.6μM) was discovered with excellent specificity against Ubiquitination.
This full SUMOylation cycle screening has the potential to discover any inhibitor(s) in the cascade reactions, including protein-protein interactions, adenylation, transfer reaction, thioester formation, and isopeptidase digestion. So it would be a very efficient inhibitor discovery approach for SUMOylation and other Ubls. Due to the sensitivity and throughput, the potent SUMOyaltion inhibitor, STE025, and other inhibitors could be discovered. This is the first HTS assay for Ubl full-pathway screening, which can provide a powerful tool for Ubl inhibitor discovery in the future.
Future perspectives for the qFRET as a universal method in quantitative systems biology and medicine
The qFRET methodology and technology can be expanded to more comprehensive applications and new fields, such as protein interaction affinity determination without purification, induced protein degradation, and antibody discovery and development. For example, because the qFRET method only requires the fluorescence signal to monitor the interaction complex, the surrounding molecules, such as proteins and metabolites, as long as they do not interfere with the FRET fluorescence signals, the qFRET can determine protein interaction affinities in mixtures, such as cell extract or not very pure samples. The qFRET assay can provide data closer to physiological data in vivo as it can determine protein interaction affinity ex vivo directly in cell extracts without purification. These data may provide a better understanding of physiological and pathological processes, such as protein post-translational modification-induced activity changes, and opportunities for drug discovery modulating these processes.
Although the qFRET technology can be used for many biochemical and pharmacological parameter determination and high-throughput screenings that cannot be done with current technologies (Fig. 2), it also has some limitations. First, the biomolecules must be labeled with fluorescence proteins, which may change protein properties, such as protein interaction affinity. We had validated the interaction affinities of fluorescence protein-labeled SUMO1 and Ubc9 vs unlabeled SUMO1 and Ubc9. We did not find any significant differences between labeled and unlabeled proteins. But this can be protein dependent. Second, fluorescence-labeled proteins used in the assay need to be full-length proteins, as truncated proteins may either interfere with the fluorescent signals or act as a dominant-negative inhibitor. The full-length fluorescence-labeled proteins can be obtained through HPLC purifications. Third, although we observed that fluorescence protein may help most protein expressions, the fluoresce-labeled protein increases protein sizes and may affect protein expressions, especially in bacterial cells. Different expression systems, such as yeast, insect cells, or mammalian cells, can be used to express fluorescence-fused proteins.
The induced protein degradation, such as PROTAC and Molecular Glue, has become an exciting drug discovery and basic research approach. The induced protein degradation utilizes enhanced Ubiquitin E3 ligase-substrate interactions or other protein degradation pathways by small molecules or biologics for rapid substrate protein degradations, such as oncoproteins. The qFRET technology can be used to monitor and determine the E3-substrate interactions. In addition, the qFRET method can discover small molecules or biologics that induce or enhance the E3-substrate approximately or affinity. inter and screenings for molecules to enhance E3-substrate interactions.
The qFRET assays are carried out in a solution without a high purity requirement, and this condition is very similar to the physiological environment. Therefore, the qFRET-based measurements are closer to physiological events in living cells. In addition, the qFRET assay is very sensitive. Depending on the photonic detectors of the PMT or CCD camera, the concentration of fluorescence-tagged proteins required in the qFRET assay can be as low as nM to pM; therefore, a minimal amount of proteins is needed for the kinetics determinations. Third, the qFRET assay is environmentally friendly without any chemicals involved. Fourth, the genetic labeling of the fluorescence method is universal for any protein. Applications of the qFRET method as a universal approach to various protein-protein interactions, enzymatic kinetics, and enzymatic catalyzed pathways should provide high-quality protein interaction and catalytic affinities of systems, networks, and proteomes and provide comprehensive quantitative biological and biomedical maps without the need of laborious protein purification, especially for those difficult-to-be-expressed proteins. Finally, the qFRET assays are conducted in a 384-well or even higher-density plate format, which can be adapted to high-throughput large-scale network kinetics determination and quantitative systems biology.
Availability of data and materials
The formula and data generated data using the qFRET are available upon request.
References
Lippincott-Schwartz J. Emerging in vivo analyses of cell function using fluorescence imaging (*). Annu Rev Biochem. 2011;80:327–32. https://doi.org/10.1146/annurev-biochem-121010-125553.
Patterson G, Davidson M, Manley S, Lippincott-Schwartz J. Superresolution imaging using single-molecule localization. Annu Rev Phys Chem. 2010;61:345–67. https://doi.org/10.1146/annurev.physchem.012809.103444.
Specht EA, Braselmann E, Palmer AE. A Critical and Comparative Review of Fluorescent Tools for Live-Cell Imaging. Annu Rev Physiol. 2017;79:93–117. https://doi.org/10.1146/annurev-physiol-022516-034055.
Lakowicz J. Principles of Fluorescence Spectroscopy, 3rd Edition. New York City: Springer; 2006.
Zhou X, Herbst-Robinson KJ, Zhang J. Visualizing dynamic activities of signaling enzymes using genetically encodable FRET-based biosensors from designs to applications. Methods Enzymol. 2012;504:317–40. https://doi.org/10.1016/B978-0-12-391857-4.00016-1.
Datta R, Heaster TM, Sharick JT, Gillette AA, Skala MC. Fluorescence lifetime imaging microscopy: fundamentals and advances in instrumentation, analysis, and applications. J Biomed Opt. 2020;25:1–43. https://doi.org/10.1117/1.JBO.25.7.071203.
Ishikawa-Ankerhold HC, Ankerhold R, Drummen GP. Advanced fluorescence microscopy techniques–FRAP, FLIP, FLAP. FRET and FLIM Molecules. 2012;17:4047–132. https://doi.org/10.3390/molecules17044047.
Huang R, et al. Development of a pH-activatable fluorescent probe and its application for visualizing cellular pH change. Analyst. 2012;137:4418–20. https://doi.org/10.1039/c2an35671f.
Tarrant MK, Cole PA. The chemical biology of protein phosphorylation. Annu Rev Biochem. 2009;78:797–825. https://doi.org/10.1146/annurev.biochem.78.070907.103047.
Pluth MD, Tomat E, Lippard SJ. Biochemistry of mobile zinc and nitric oxide revealed by fluorescent sensors. Annu Rev Biochem. 2011;80:333–55. https://doi.org/10.1146/annurev-biochem-061009-091643.
Carter KP, Young AM, Palmer AE. Fluorescent sensors for measuring metal ions in living systems. Chem Rev. 2014;114:4564–601. https://doi.org/10.1021/cr400546e.
Owicki JC. Fluorescence polarization and anisotropy in high throughput screening: perspectives and primer. J Biomol Screen. 2000;5:297–306. https://doi.org/10.1177/108705710000500501.
Hendrickson OD, Taranova NA, Zherdev AV, Dzantiev BB, Eremin SA. Fluorescence Polarization-Based Bioassays: New Horizons. Sensors (Basel). 2020;20:33. https://doi.org/10.3390/s20247132.
Selvin PR. Principles and biophysical applications of lanthanide-based probes. Annu Rev Biophys Biomol Struct. 2002;31:275–302. https://doi.org/10.1146/annurev.biophys.31.101101.140927.
Lea WA, Simeonov A. Fluorescence polarization assays in small molecule screening. Expert Opin Drug Discov. 2011;6:17–32. https://doi.org/10.1517/17460441.2011.537322.
Forster T. Zwischenmolekulare energiewanderung und fluoreszenz. Ann Phys Berlin. 1948;2:21.
Hires SA, Zhu Y, Tsien RY. Optical measurement of synaptic glutamate spillover and reuptake by linker optimized glutamate-sensitive fluorescent reporters. Proc Natl Acad Sci U S A. 2008;105:4411–6. https://doi.org/10.1073/pnas.0712008105.
Verveer PJ, Wouters FS, Reynolds AR, Bastiaens PI. Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane. Science. 2000;290:1567–70.
Saucerman JJ, et al. Systems analysis of PKA-mediated phosphorylation gradients in live cardiac myocytes. Proc Natl Acad Sci U S A. 2006;103:12923–8.
Tron L, et al. Flow cytometric measurement of fluorescence resonance energy transfer on cell surfaces. Quantitative evaluation of the transfer efficiency on a cell-by-cell basis. Biophys J. 1984;45:939–46. https://doi.org/10.1016/S0006-3495(84)84240-X.
Zal T, Gascoigne NR. Photobleaching-corrected FRET efficiency imaging of live cells. Biophys J. 2004;86:3923–39. https://doi.org/10.1529/biophysj.103.022087.
Chen H, Puhl HL 3rd, Koushik SV, Vogel SS, Ikeda SR. Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophys J. 2006;91:L39-41. https://doi.org/10.1529/biophysj.106.088773.
Gordon GW, GB, Liang XH, Levine B, Herman B. Quantitative Ffluorescence Resonance Energy Transfer Measurements using Fluroescnece miscroscope. Biophys J. 1998; 74: 2702–2713.
Tsuji A, et al. Direct observation of specific messenger RNA in a single living cell under a fluorescence microscope. Biophys J. 2000;78:3260–74. https://doi.org/10.1016/S0006-3495(00)76862-7.
Wallrabe H, Periasamy A. Imaging protein molecules using FRET and FLIM microscopy. Curr Opin Biotechnol. 2005;16:19–27. https://doi.org/10.1016/j.copbio.2004.12.002.
Kenworthy AK. Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods. 2001;24:289–96. https://doi.org/10.1006/meth.2001.1189.
Valentin G, et al. Photoconversion of YFP into a CFP-like species during acceptor photobleaching FRET experiments. Nat Methods. 2005;2:801. https://doi.org/10.1038/nmeth1105-801.
Van Munster EB, Kremers GJ, Adjobo-Hermans MJ, Gadella TW Jr. Fluorescence resonance energy transfer (FRET) measurement by gradual acceptor photobleaching. J Microsc. 2005;218:253–62. https://doi.org/10.1111/j.1365-2818.2005.01483.x.
Elangovan M, et al. Characterization of one- and two-photon excitation fluorescence resonance energy transfer microscopy. Methods. 2003;29:58–73. https://doi.org/10.1016/s1046-2023(02)00283-9.
Mehta K, Hoppe AD, Kainkaryam R, Woolf PJ, Linderman JJ. A computational approach to inferring cellular protein-binding affinities from quantitative fluorescence resonance energy transfer imaging. Proteomics. 2009;9:5371–83. https://doi.org/10.1002/pmic.200800494.
Lam AD, et al. Mapping dynamic protein interactions to insulin secretory granule behavior with TIRF-FRET. Biophys J. 2010;99:1311–20. https://doi.org/10.1016/j.bpj.2010.06.014.
Song Y, Madahar V, Liao J. Development of FRET Assay into Quantitative and High-throughput Screening Technology Platforms for Protein-Protein Interactions. Ann Biomed Eng. 2011;39:1224–34. https://doi.org/10.1007/s10439-010-0225-x.
Song Y, Rodgers VG, Schultz JS, Liao J. Protein interaction affinity determination by quantitative FRET technology. Biotechnol Bioeng. 2012;109:2875–83. https://doi.org/10.1002/bit.24564.
Liu Y, Song Y, Jiang L, Liao J. Quantitative analysis of FRET assay in biology. Front Biol. 2012;7:57–64.
Song Y, Liao J. An in vitro Forster resonance energy transfer-based high-throughput screening assay for inhibitors of protein-protein interactions in SUMOylation pathway. Assay Drug Dev Technol. 2012;10:336–43. https://doi.org/10.1089/adt.2011.0394.
Wang YZ, et al. An in vitro Forster resonance energy transfer-based high-throughput screening assay identifies inhibitors of SUMOylation E2 Ubc9. Acta Pharmacol Sin. 2020;41:1497–506. https://doi.org/10.1038/s41401-020-0405-7.
Liao J, Madahar V, Dang R, Jiang L. Quantitative FRET (qFRET) Technology for the Determination of Protein-Protein Interaction Affinity in Solution. Molecules. 2021;26:9. https://doi.org/10.3390/molecules26216339.
Braun P, Gingras AC. History of protein-protein interactions: from egg-white to complex networks. Proteomics. 2012;12:1478–98. https://doi.org/10.1002/pmic.201100563.
Nibbe RK, Chowdhury SA, Koyuturk M, Ewing R, Chance MR. Protein-protein interaction networks and subnetworks in the biology of disease. Wiley Interdiscip Rev Syst Biol Med. 2011;3:357–67. https://doi.org/10.1002/wsbm.121.
Syafrizayanti, Betzen C, Hoheisel JD, Kastelic D. Methods for analyzing and quantifying protein-protein interaction. Expert Rev Proteomics. 2014; 11: 107–120. https://doi.org/10.1586/14789450.2014.875857.
Xing S, Wallmeroth N, Berendzen KW, Grefen C. Techniques for the Analysis of Protein-Protein Interactions in Vivo. Plant Physiol. 2016;171:727–58. https://doi.org/10.1104/pp.16.00470.
Huttlin EL, et al. Architecture of the human interactome defines protein communities and disease networks. Nature. 2017;545:505–9. https://doi.org/10.1038/nature22366.
Lu H, et al. Recent advances in the development of protein-protein interactions modulators: mechanisms and clinical trials. Signal Transduct Target Ther. 2020;5:213. https://doi.org/10.1038/s41392-020-00315-3.
Schuck P. Use of surface plasmon resonance to probe the equilibrium and dynamic aspects of interactions between biological macromolecules. Annu Rev Biophys Biomol Struct. 1997;26:541–66. https://doi.org/10.1146/annurev.biophys.26.1.541.
Douzi B. Protein-Protein Interactions: Surface Plasmon Resonance. Methods Mol Biol. 2017;1615:257–75. https://doi.org/10.1007/978-1-4939-7033-9_21.
Stahelin RV. Surface plasmon resonance: a useful technique for cell biologists to characterize biomolecular interactions. Mol Biol Cell. 2013;24:883–6. https://doi.org/10.1091/mbc.E12-10-0713.
Nguyen HH, Park J, Kang S, Kim M. Surface plasmon resonance: a versatile technique for biosensor applications. Sensors (Basel). 2015;15:10481–510. https://doi.org/10.3390/s150510481.
Helmerhorst E, Chandler DJ, Nussio M, Mamotte CD. Real-time and Label-free Bio-sensing of Molecular Interactions by Surface Plasmon Resonance: A Laboratory Medicine Perspective. Clin Biochem Rev. 2012;33:161–73.
Schuck P, Zhao H. The role of mass transport limitation and surface heterogeneity in the biophysical characterization of macromolecular binding processes by SPR biosensing. Methods Mol Biol. 2010;627:15–54. https://doi.org/10.1007/978-1-60761-670-2_2.
Schuck P, Minton AP. Analysis of mass transport-limited binding kinetics in evanescent wave biosensors. Anal Biochem. 1996;240:262–72. https://doi.org/10.1006/abio.1996.0356.
Nieba L, Krebber A, Pluckthun A. Competition BIAcore for measuring true affinities: large differences from values determined from binding kinetics. Anal Biochem. 1996;234:155–65. https://doi.org/10.1006/abio.1996.0067.
Zhou M, Li Q, Wang R. Current Experimental Methods for Characterizing Protein-Protein Interactions. ChemMedChem. 2016;11:738–56. https://doi.org/10.1002/cmdc.201500495.
Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–7.
Damian L. Isothermal titration calorimetry for studying protein-ligand interactions. Methods Mol Biol. 2013;1008:103–18. https://doi.org/10.1007/978-1-62703-398-5_4.
Tellinghuisen J. Volume errors in isothermal titration calorimetry. Anal Biochem. 2004;333:405–6. https://doi.org/10.1016/j.ab.2004.05.061.
Tellinghuisen J, Chodera JD. Systematic errors in isothermal titration calorimetry: concentrations and baselines. Anal Biochem. 2011;414:297–9. https://doi.org/10.1016/j.ab.2011.03.024.
Tellinghuisen J. Calibration in isothermal titration calorimetry: heat and cell volume from heat of dilution of NaCl(aq). Anal Biochem. 2007;360:47–55. https://doi.org/10.1016/j.ab.2006.10.015.
Raines RT. Fluorescence polarization assay to quantify protein-protein interactions: an update. Methods Mol Biol. 2015;1278:323–7. https://doi.org/10.1007/978-1-4939-2425-7_19.
Park SH, Raines RT. Fluorescence polarization assay to quantify protein-protein interactions. Methods Mol Biol. 2004;261:161–6. https://doi.org/10.1385/1-59259-762-9:161.
Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron. 2001;31:973–85. https://doi.org/10.1016/s0896-6273(01)00438-x.
Erickson MG, Liang H, Mori MX, Yue DT. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 2003;39:97–107. https://doi.org/10.1016/s0896-6273(03)00395-7.
Liao JY, Song Y, Liu Y. A new trend to determine biochemical parameters by quantitative FRET assays. Acta Pharmacol Sin. 2015;36:1408–15. https://doi.org/10.1038/aps.2015.82.
Jiang L, et al. Protein-Protein Affinity Determination by Quantitative FRET Quenching. Sci Rep. 2019;9:2050. https://doi.org/10.1038/s41598-018-35535-9.
Wu L, et al. Forster resonance energy transfer (FRET)-based small-molecule sensors and imaging agents. Chem Soc Rev. 2020;49:5110–39. https://doi.org/10.1039/c9cs00318e.
Velick SF, Parker CW, Eisen HN. Excitation energy transfer and the quantitative study of the antibody hapten reaction. Proc Natl Acad Sci U S A. 1960;46:1470–82.
Liu BL, Schultz JS. Equilibrium binding in immunosensors. IEEE Trans Biomed Eng. 1986;33:133–8. https://doi.org/10.1109/TBME.1986.325887.
Xu Z, Au SW. Mapping residues of SUMO precursors essential in differential maturation by SUMO-specific protease, SENP1. Biochem J. 2005;386:325–30. https://doi.org/10.1042/BJ20041210.
Mikolajczyk J, et al. Small ubiquitin-related modifier (SUMO)-specific proteases: profiling the specificities and activities of human SENPs. J Biol Chem. 2007;282:26217–24. https://doi.org/10.1074/jbc.M702444200.
Drag M, Mikolajczyk J, Krishnakumar IM, Huang Z, Salvesen GS. Activity profiling of human deSUMOylating enzymes (SENPs) with synthetic substrates suggests an unexpected specificity of two newly characterized members of the family. Biochem J. 2008;409:461–9. https://doi.org/10.1042/BJ20070940.
Kolli N, et al. Distribution and paralogue specificity of mammalian deSUMOylating enzymes. Biochem J. 2010;430:335–44. https://doi.org/10.1042/BJ20100504BJ20100504.
Engels IH, et al. A time-resolved fluorescence resonance energy transfer-based assay for DEN1 peptidase activity. Anal Biochem. 2009;390:85–7. https://doi.org/10.1016/j.ab.2009.03.035.
Carlson CB, Horton RA, Vogel KW. A Toolbox Approach to High-Throughput TR-FRET-Based SUMOylation and DeSUMOylation Assays. Assay Drug Dev Technol. 2009. https://doi.org/10.1089/adt.2008.188.
Berney C, Danuser G. FRET or no FRET: a quantitative comparison. Biophys J. 2003;84:3992–4010. https://doi.org/10.1016/S0006-3495(03)75126-1.
Martin SF, Tatham MH, Hay RT, Samuel IDW. Quantitative analysis of multi-protein interactions using FRET: Application to the SUMO pathway. Protein Sci. 2008;17:777–84. https://doi.org/10.1110/ps.073369608.
Liu Y, Song Y, Madahar V, Liao J. Quantitative Forster resonance energy transfer analysis for kinetic determinations of SUMO-specific protease. Anal Biochem. 2012;422:14–21. https://doi.org/10.1016/j.ab.2011.12.019S0003-2697(11)00790-1.
Jiang L, et al. Internal calibration forster resonance energy transfer assay: a real-time approach for determining protease kinetics. Sensors (Basel). 2013;13:4553–70. https://doi.org/10.3390/s130404553.
Liu Y, et al. Isopeptidase Kinetics Determination by a Real Time and Sensitive qFRET Approach. Biomolecules. 2021;11:16. https://doi.org/10.3390/biom11050673.
Shen L, et al. SUMO protease SENP1 induces isomerization of the scissile peptide bond. Nat Struct Mol Biol. 2006;13:1069–77. https://doi.org/10.1038/nsmb1172.
Lima CD, Reverter D. Structure of the human SENP7 catalytic domain and poly-SUMO deconjugation activities for SENP6 and SENP7. J Biol Chem. 2008;283:32045–55. https://doi.org/10.1074/jbc.M805655200.
Reverter D, et al. Structure of a complex between Nedd8 and the Ulp/Senp protease family member Den1. J Mol Biol. 2005;345:141–51. https://doi.org/10.1016/j.jmb.2004.10.022.
Walden H, et al. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol Cell. 2003;12:1427–37.
Haas AL, Warm JV, Herschko A, Rose IA. Ubiquitin-activating enzyme Mechanism and role in protein-ubiquitin conjugation. J Biol Chem. 1982;257:2543–8.
Wang J, Cai S, Chen Y. Mechanism of E1–E2 interaction for the inhibition of Ubl adenylation. J Biol Chem. 2010;285:33457–62. https://doi.org/10.1074/jbc.M110.135582.
Burch TJ, Haas AL. Site-Directed Mutagenesis of Ubiquitin. Differential Roles for Arginine in the Interaction with Ubiquitin-Activating Enzyme. Biochemistry. 1994;33:7300–8. https://doi.org/10.1021/bi00189a035.
Bruzzese FJ, et al. Development of a charcoal paper adenosine triphosphate:pyrophosphate exchange assay: kinetic characterization of NEDD8 activating enzyme. Anal Biochem. 2009;394:24–9. https://doi.org/10.1016/j.ab.2009.07.011.
Wee KE, et al. Steady-state kinetic analysis of human ubiquitin-activating enzyme (E1) using a fluorescently labeled ubiquitin substrate. J Protein Chem. 2000;19:489–98.
Bossis G, et al. A fluorescence resonance energy transfer-based assay to study SUMO modification in solution. Methods Enzymol. 2005;398:20–32. https://doi.org/10.1016/S0076-6879(05)98003-8.
Blazer LL, Neubig RR. Small molecule protein-protein interaction inhibitors as CNS therapeutic agents: current progress and future hurdles. Neuropsychopharmacology. 2009;34:126–41. https://doi.org/10.1038/npp.2008.151.
Vassilev LT, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. https://doi.org/10.1126/science.10924721092472.
Chan SL, et al. Identification of chelerythrine as an inhibitor of BclXL function. J Biol Chem. 2003;278:20453–6. https://doi.org/10.1074/jbc.C300138200.
Cavalli A, et al. A small molecule targeting the multifactorial nature of Alzheimer’s disease. Angew Chem Int Ed Engl. 2007;46:3689–92. https://doi.org/10.1002/anie.200700256.
Fukuda I, et al. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem Biol. 2009;16:133–40. https://doi.org/10.1016/j.chembiol.2009.01.009.
Fukuda I, et al. Kerriamycin B inhibits protein SUMOylation. J Antibiot (Tokyo). 2009;62:221–4. https://doi.org/10.1038/ja.2009.10.
Kim YS, Nagy K, Keyser S, Schneekloth JS Jr. An electrophoretic mobility shift assay identifies a mechanistically unique inhibitor of protein sumoylation. Chem Biol. 2013;20:604–13. https://doi.org/10.1016/j.chembiol.2013.04.001.
Brandt M, et al. Development of a high-throughput screen to detect inhibitors of TRPS1 sumoylation. Assay Drug Dev Technol. 2013;11:308–25. https://doi.org/10.1089/adt.2012.501.
Hirohama M, et al. Spectomycin B1 as a novel SUMOylation inhibitor that directly binds to SUMO E2. ACS Chem Biol. 2013;8:2635–42. https://doi.org/10.1021/cb400630z.
Kumar A, Ito A, Hirohama M, Yoshida M, Zhang KY. Identification of sumoylation activating enzyme 1 inhibitors by structure-based virtual screening. J Chem Inf Model. 2013;53:809–20. https://doi.org/10.1021/ci300618e.
Takemoto M, et al. Inhibition of protein SUMOylation by davidiin, an ellagitannin from Davidia involucrata. J Antibiot (Tokyo). 2014;67:335–8. https://doi.org/10.1038/ja.2013.142.
Suzawa M, et al. A gene-expression screen identifies a non-toxic sumoylation inhibitor that mimics SUMO-less human LRH-1 in liver. Elife. 2015;4:22. https://doi.org/10.7554/eLife.09003.
He X, et al. Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat Chem Biol. 2017;13:1164–71. https://doi.org/10.1038/nchembio.2463.
Li YJ. Allosteric Inhibition of Ubiquitin-like Modifications by a Class of Inhibitor of SUMO-Activating Enzyme. Cell Chem Biol. 2019;26:278-288 e276. https://doi.org/10.1016/j.chembiol.2018.10.026.
Acknowledgements
I am very grateful to all the previous and current members of Liao’s group for their contributions to the developments of qFRET described in this review paper over more than a decade. I want to thank Gary Ng at Alloy Therapeutics for some comments on technology comparisons in Fig. 2.
Funding
The work was partially supported by the National Institutes of Health Grant AI076504, the UCR Academic Senate Grant, and the Attaisina gift grant.
Author information
Authors and Affiliations
Contributions
JL developed the concept of qFRET methodology and supervised experimental procedures and applications of the qFRET, and write the manuscript. J.L is responsible the overall design and implementation of the qFRET technology and its applications.
Corresponding author
Ethics declarations
Competing interests
JL filed the qFRET trademark application.
The SUMOylation inhibitor discovered by the qFRET-mediated full SUMOylation pathway screening was patented by the University of California.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liao, J. Develop quantitative FRET (qFRET) technology as a high-throughput universal assay platform for basic quantitative biomedical and translational research and development. Med-X 1, 13 (2023). https://doi.org/10.1007/s44258-023-00014-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44258-023-00014-y