
Abstract
Stomach (gastric) cancer is one of the most prevalent and deadly cancers worldwide and most gastric cancers are adenocarcinomas. Based on prior research, there is an association between Helicobacter pylori (H. pylori) infection together with the frequency of duodenal ulcer, distal gastric adenocarcinoma, mucosa-associated lymphoid tissue (MALT) lymphoma, and antral gastritis. Helicobacter pylori virulence and toxicity factors have been identified before that significantly influence the clinical outcomes of H. pylori infection and gastric adenocarcinoma. However, it remains unclear exactly how different strains of H. pylori affect gastric adenocarcinoma. Current research suggests this involves tumor suppressor genes, like p27 but also H. pylori toxic virulence proteins. Therefore, we quantified known H. pylori genotypes within adenocarcinoma patients to establish the prevalence of known toxins that include cytotoxin-associated gene A (cagA) as well as vacuolating cytotoxin A (vacA) within patients of variable adenocarcinoma diagnosis. This analysis used gastrectomy samples validated for DNA viability. The incidence of H. pylori in adenocarcinoma patients in Jordan was established to be 54.5% positive (ureA gene positive) with cagA genotype occurrence at 57.1%, but also in this population study vacA gene ratios found to be 24.7%:22.1%:14.3%:14.3%. (vacAs1:vacAs2:vacAm1:vacAm2). Using immunohistochemistry (IHC), we confirmed with statistical significance that p27 was dysregulated and suppressed, within nearly all H. pylori vacA genotypes. In addition, within 24.6% of H. pylori samples analyzed was a different bacterial genotype, and curiously that p27 protein expression was retained in 12% of tested adenocarcinoma H. pylori samples. This is suggestive that p27 could be used as a prognostic indicator but also that an unknown genotype could be contributing to the regulatory effects of p27 protein within this bacterial and cellular environment that may include other virulence factors and unknown immune system regulatory changes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
1.1 Background
Stomach cancer (also known as gastric cancer) is the third most common cancer-related cause of death worldwide and 95% are adenocarcinomas [1]. Adenocarcinomas develop from glands of the stomach mucosa, or its most superficial layer [1]. However, there are additional cancers that can develop from the stomach, such as leiomyosarcomas, which develop from the muscles that surround the mucosa, and mucosal associated lymphoid tissues (MALT) lymphomas [1]. The MALT is composed of immune system cells that include B and T cells, but also those that present both pathogenic as well as tumor associated antigens (TAA). These include monocytes, macrophages and dendritic cells (DCs) that modulate the systemic immune response before, during and after disease. Cytokines (interleukins or IL) can be expressed and secreted within epithelial cell layers surrounding gastric tissues that include IL–8, tumor necrosis factor (TNF–α) and interferons (type I/II/III). Gastric cancers are frequently discovered at advanced stages, so the prognosis can be poor [2]. Helicobacter pylori was classified as a class 1 carcinogen by the World Health Organisation (WHO) in 1994, with epidemiological, clinical, and experimental data demonstrating a link between H. pylori infection and the progression of gastric adenocarcinoma [2]. Helicobacter pylori is considered to thrive in the acidic pH environment of the stomach. Prior reports suggest that three classifications existed for adenocarcinoma with a 2018 report defining five classifications. These include papillary, tubular, poorly cohesive, mucinous, weakly coherent, with various histological variants (e.g., squamous cell carcinoma) (see Supplementary Materials). Individuals with H. pylori infection are indicated to have a 6–fold higher chance of developing gastric cancer [3]. Prior reports suggest that greater than 50% of the global population are infected by H. pylori [4] The presence of this bacterium increases the risk of developing gastric adenocarcinoma [5]. Helicobacter pylori possess virulence factor genes that affect toxicity and pathogenicity including cytotoxin–associated pathogenicity island (cagPAI) and vacuolating cytotoxin A (vacA) through proteins affecting H. pylori virulence (see Fig. 1).

Perspectives of Helicobacter pylori genome
These encode proteins that include vacA toxin (molecular mass 140 kDa), initially formed from a protoxin. VacA was originally named after its ability to form vacuolar like membrane vesicles within gastric epithelial cells that enhance H. pylori colonization of the gastrointestinal (GI) tract. In addition, within vacA genes, approximately 30 genes encode cagPAI (molecular mass 40 kDa) proteins that affect components of the intestinal type IV secretion system (T4SSs) protein. These are present in other Gram negative (-ve) bacteria [6,7,8,9]. The T4SS proteins assemble through protein pili–like structures encoded by various other genes that code for other cagA proteins (e.g., CagI, CagY etc.), whilst delivering cagA protein into host cells upon bacterial attachment utilizing adhesion molecules [8, 10]. Other types of secretion systems have been elucidated as there are 6 types known [6,7,8,9]. Moreover, currently data on Uniprot indicates that cagPAI genes encode 378 proteins categorized now (see Supplementary Materials). These toxic virulence factors have been proven to be crucial in defining clinical outcomes of H. pylori infection and development of gastric adenocarcinoma [10]. Prior laboratory studies have demonstrated the cagA gene role in carcinogenesis through stimulation of aberrant cell proliferation [11]. Over 90% of H. pylori strains occur in Southeast Asia occur with more than 60% of strains from Europe and North America that are reported to possess the cagA gene [12]. In general, vacA strains encompass type s1a and type m1 that produce greater toxin amounts, followed by type s1b and type m2 strains, which generate toxin in moderate amounts, while vacA type s2 and type m2 strains exhibit less or no vacuolar toxin activity [13, 14]. The presence of various genotypes of vacA has been reported, including s1a, s1b, s2, ml, and m2 strains of H. pylori that display unusually variable toxicity [15]. Therefore, characterizing the toxicity of H. pylori in specific carcinomas allows further knowledge of virulence factors within clinically diverse and vulnerable patients.
1.2 Background and Molecular Mechanisms of Helicobacter pylori
Vacuolating cytotoxin A gene is considered to have diverse polymorphisms in allelic expression within the signal (s), middle (m), intermediate (i), deletion (d) and c regions [16, 17]. Current data on Uniprot are indicative of 3794 variations within H. pylori vacA genes (see Supplementary Materials). The vacA gene produces a toxin composed of two subunits (p33/p55) considered to target cellular mitochondria [4, 18]. These are known to bind to other cellular integrin receptors. VacA is suggested to enter phospholipid cell layers by forming a membrane pore, and then as vesicle like endosomes. Past research indicates that anion selective channels are created, with the p55 subunit required for membrane binding. As a result, increasing transport of chloride ions to change the mitochondrial inner membrane electrochemical potential [4, 18]. This homeostatic balance within the GI tract can therefore, affect the immune system with differential effects on not only regulatory T cells, but also helper T cell, cytotoxic T cell, and antigen presenting cells (APCs), as well as B cells together with natural killer cells. These are essential in immune systems of clinically diagnosed cancer patients. Either therapeutic or cancer related immunosuppression can therefore contribute to prolonged microbial infection thereby affecting disease outcomes [19]. Helicobacter pylori also secrete a urease enzyme (550kDa) that catalyzes hydrolysis into products of ammonia and carbonic acid. Urease is encoded by a fused ure gene cluster (ureA and ureB) that encodes other accessory proteins (UreE, UreF, UreI, etc) [20]. Urease is thought to elicit a strong B cell serum immunoglobulin response and facilitate host colonization [20]. Therefore, in combination with VacA toxin these contribute to prolonged infection in the GI tract. Concurrent initiation occurs by upregulation of nuclear transcription factor (NF–kB) in combination with upregulation of the cytokine interleukin–8 (IL–8) that are both crucial factors [21, 22] Recently it has also been suggested that IL-8 could also act as a chemoattractant (named CXCR8) for Epstein Barr Virus (EBV) infected B cells [21]. The p27 protein (often referred to as KIP1) is a member of the cyclin-dependent kinase inhibitor (CDKI) KIP family of tumor-suppressor proteins regulated by transforming growth factor beta (TGF–β) [20]. One proposed mechanism for the development of cancer is the inactivation of this p27KIP1 tumor suppressor gene, located on chromosome 12p13 through transcriptional regulation [23, 24]. Eukaryotic cells have a network of regulatory proteins that affect cell cycle control, which regulate and control the cell cycle to prevent malignant cell proliferation and cancer [23]. Activities of the cell cycle checkpoints are governed by cyclin-dependent kinases (CDKs), a family of protein kinases that bind to regulatory proteins known as cyclins [23, 25]. Inhibition of CDKs (e.g., CDK2) is one mechanism that p27 uses to inhibit cell cycle progression, although cell division, proliferation, and apoptosis are other roles of p27 [26]. Reduced p27 expression has been shown to be a marker of aggressive cancer and poor prognosis, including colon, breast, malignant melanoma, liver, stomach, lung, as well as brain tumors [27]. Furthermore, H. pylori infection in gastric cancer patients has been associated with reduced p27 expression [23, 28]. C–terminal phosphorylation regulates p27 function through known growth and survival factor phosphoinositide 3–kinases (PI3K) and protein kinase B (AKT) that regulate growth (G1) to synthesis (S) cell cycle progression. Helicobacter pylori and CagA protein may also bind to other cytosolic proteins like Csk, Src homology 2 domain-containing tyrosine phosphatase–2 (Shp–2) or c–jun. VacA is considered to bind to and enter gastric cells, as a pore forming toxin, utilizing receptors that include the epidermal growth factor (EGR) receptor, but also heparin sulphate amongst others. [4]. Within immune cell compartments as we discussed in our last paper cellular markers can clarify individual cell types [29]. Recent research is evocative that VacA toxin can utilize a cluster of differentiation molecules (CD18) β2 integrin adhesion subunit molecule expressed on T cells that forms part of the CD11a/CD18 transmembrane receptor leukocyte function associated antigen (LFA–1) complex as well as an enzyme like γ-glutamyl transferase (GGT) central to amino acid transfer and leukotriene synthesis [19, 30]. Little is known about the immunology of H. pylori infection; however, it is indicated that the cytokines, IFN–γ and TNF–α, play a protective role [31]. One study investigating H. pylori in vivo so far has suggested that cagA protein can suppress DC function [32]. However early research studies suggest that cagA can translocate into APCs (monocytes, macrophages and dendritic cells) [33]. More recently 889 disease enhanced genes (DEGs) were investigated in vitro during Helicobacter pylori infection [33]. In this study it was suggested that Toll-like receptor 4 (TLR4) together with chemokine CXCR3 expression could be modulated by H. pylori infection specifically by cagA protein [33].
1.3 Helicobacter pylori Epidemiology and Other Factors
In several prior studies conducted around the world, cagA, vacAs1, and vacAm1 genes, are considered to play a role in pathogenicity and linked to gastric cancer [14, 36, 37]. The geographic distribution of H. pylori strains varies. For instance, the most common strains in East Asia carry the cagA, vacAs1, and vacAm1 genes [38]. The prevalence of vacAs1b subtype was approximately 100% in South America, 80% in the Spain and Portugal strains, and low in East Asia [38]. The mechanism of the development of gastric adenocarcinoma in H. pylori-infected samples has not yet been fully determined [39]. Establishing causal genetic factors therefore may eventually clarify relationship with antimicrobial resistance. Some earlier reports between 2000 and 2005 quantify changes in antimicrobial resistance within H. pylori infection to metronidazole, clarithromycin, amoxicillin and tetracycline [40]. Other reviews consider antimicrobial resistance further in 2023 during H. pylori infection [41].
In this investigation, we therefore sought to identify the relationship between H. pylori infection and p27 expression in gastric cancer tissues in Jordanian patients. We utilized gastrectomy samples rather than tissue biopsies taken from patients with H. pylori. These samples were clinically diagnosed with variable adenocarcinoma grades from hospitals in Jordan. Helicobacter pylori genotypes of each clinical sample were characterized alongside p27 protein expression confirmation. To ascertain whether p27 protein was related to the development of gastric cancer in these patients, the level of p27 gene expression was also assessed. To our knowledge, this is the only study that has quantified variable H. pylori genotypes in adenocarcinoma with confirmation of expression of p27 protein using FFPE-gastrectomy samples obtained from patients in Jordan.
2 Materials and Methods
2.1 Sample Collection
In this study, archived histological samples (n=77) with linked patient demographic and clinical data were obtained within 8 years (2005 and 2013) from the pathology department medical records of King Abdullah University Hospital (KAUH) at Jordan University of Science and Technology (JUST) (Irbid, Jordan) and the Jordanian Royal Medical Services (JRMS) (Amman, Jordan) (Table 1). These clinical samples (n = 77) were examined using formalin-fixed paraffin-embedded tissue analysis (FFPE). All samples were classified as gastric adenocarcinomas and stored at 25 ℃. This study was carried out with consent from the Institutional Review Board (IRB), Ethics Committee, at Jordan University of Science and Technology (Ref: 20/51/201).
2.2 Clinical Samples Processing
Using a microtome, formalin-fixed tumor specimens were embedded in paraffin blocks and sectioned into 5 µm thick tissue slices (Energy beam science, East Granby, CT, USA). The microtome blade was changed and cutting surface both cleaned and sterilized with xylene and absolute ethanol (100%), respectively. Up to 10 tissue sections were collected in 1.5 ml sterile Eppendorf nuclease free tubes for DNA extraction, and 5 µm tissue slices were mounted on positively charged slides for IHC.
2.3 Immunohistochemistry (IHC) Staining
To evaluate p27KIP1 expression in tissue(s), all tissue section slides were treated with a mouse monoclonal anti-human IgG p27Kip1 antibody (Clone SX53G8.5 dilution 1:100; Code M7203; Dako Cytomation, Denmark) that cross-reacts with p27kip1 using a protocol described previously [42]. Slides were deparaffinized with xylene twice, for 5 minutes each, rehydrated through a series of graded alcohol washes, 2 times, for 3 minutes each, and then transferred once through 95%, 70%, and 50% alcohols for 3 minutes each). Endogenous peroxidase activity was blocked by incubating sections in 3% H2O2 solution in methanol at room temperature for 10 min. Then slides were rinsed with PBS twice, for 5 min each. To reveal the antigenic epitope, we performed antigen retrieval by pouring 300 mL of 10 mM citrate buffer, pH 6.0 into the staining container containing arranged slides, and incubating at 95 °C for 23 minutes using a pretreatment (PT) system (Dako, Agilent, Glostrup, Denmark). We removed the staining container at room temperature and allowed the slides to cool for 20 min. This was followed by washing in Dulbecco’s phosphate-buffered saline (PBS) twice, for 5 min each (Sigma Aldrich, St. Louis, MO, USA). Then, blocking buffer was drained from the slides. All slides were treated with p27 monoclonal antibody (H-1): sx-53G8.5 (Dako Cytomation, Agilent), diluted 1:100 in Dako antibody diluent, for 45 min at room temperature. Slides were washed with PBS twice, for 5 min each, according to manufacturer recommendation [42]. The detection was carried out using Dako EnVision®+ Dual Link System-HRP (DAB+) (Dako, Agilent). The Liquid DAB+ Substrate Chromogen System (Dako, Agilent) was used to view the slides, and Mayer’s hematoxylin (Polysciences) was used as a counterstain. As a negative control, primary antibodies were excluded. Tonsil and lymphoma patient tissue was used as a positive control. More than 5% of neoplastic cells should exhibit prominent brown nuclear staining for samples to be deemed positive for p27 protein expression. The scoring system used to evaluate the results of immunostaining was described previously [43]. To verify the initial diagnosis, all samples were examined and confirmed by a licensed pathologist at the pathology division of KAUH. All cells were counted on the slides using a high-power field scanner, and pictures were taken with a digital camera (Olympus model C-5060, Olympus, Tokyo, Japan).
2.4 DNA Extraction
For extracting genomic DNA, 5–10 sections from each block were placed into sterile, nuclease-free Eppendorf tubes. Prior to genomic DNA extraction, xylene and paraffin residues from paraffin sections were removed in the pathology lab in accordance with standard operating procedures [44]. To quickly and effectively deparaffinize the tissue, 1 mL of xylene was added to each tube, with tubes vortexed violently for 10 s. The mixture was centrifuged at a full speed of 4000 rpm for 2 min at room temperature, with pellet remaining in the tube. This procedure was performed twice. One mL of 96% ethanol was added to each pellet, vortexed and centrifuged at full speed for 2 min at room temperature, and supernatant removed by pipetting. To remove any leftover xylene, this step was repeated. Next, Eppendorf tubes containing the tissue pellets were opened and incubated at 37 °C for 15–30 min until the ethanol evaporated. Each pellet was then subjected to genomic DNA extraction using a QIAamp DNA FFPE Tissue kit (Qiagen, Hilden, Germany) according to manufacturer instructions. To ensure DNA purity, the extracted DNA was eluted, and concentration assessed using a NanoDrop ND-1000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA). For later use, all DNA extracts were kept in storage at −20 °C. Using an antibody to Ki-67, adjacent sections were immune-stained to detect proliferating cells, and apoptotic cells were detected using a terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling assay. Proliferation and apoptotic indices were calculated as previously described [42].
2.5 Detection of H. pylori Virulence Genes
Helicobacter pylori virulence genes were detected by PCR amplification. The primers used here are described below (see Tables 1 and 2). Primers targeted amplification of H. pylori genes and the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene as an indicator of DNA extraction validity and viability [45].
2.6 PCR for the GAPDH Gene
One of the universal housekeeping genes used to assess the integrity of DNA samples is GAPDH and is used as an internal control. This gene was subjected to PCR. Each PCR reaction with a total volume of 25 µL contained 12.5 µL of PCR master mix (Promega; Madison, WI, USA), 8.0 µL of nuclease free water, 1.0 µL (5 pmol/µL for GAPDH) of each, and 2.5 µL of DNA (100 ng/µL). A negative control reaction (containing all components except DNA template) was included. Amplification protocol was run with the thermal profile recommended by the master mix manufacturer; initial denaturation at 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 1 min, annealing at 54 °C for 1 min, and extension at 72 °C for 1 min. The amplification ended with a final extension at 72 °C for 5 min.
2.7 PCR ureA Gene to Confirm H. pylori Identity
PCR for detection of the H. pylori ureA gene was performed in 25 µL volumes as described above, using specific H. pylori ureA primers along with positive and negative controls. The amplification protocol was run with the thermal profile recommended by the manufacturer; initial denaturation at 95 ℃ for 10 min, then followed by 35 cycles of denaturation at 94 ℃ for 1 min, annealing at 47 ℃ for 1 min, and extension at 72 ℃ for 1 min.
2.8 PCR for H. pylori Virulence Genes
Samples positive for ureA gene underwent five PCR cycles to detect the presence of virulence genes of H. pylori (cagA, vacAs1, vacAs2, vacAm1, and vacAm2). PCR reactions were performed in 25 µL volumes (see Table 2 for temperature profile for each gene PCR cycle. The resulting amplicons and their corresponding lengths were visualized and documented via gel electrophoresis and analysis (Quantity 1 software, Bio-rad, CA, USA).
2.9 Statistical Analysis
All data were analyzed using SPSS version 19.0 software (SPSS, Inc., Chicago, IL, USA). Pearson’s chi-square (χ2 or Fisher test was used to analyze the statistical relationship between H. pylori infection, virulence genes, as well as p27 gene expression in the gastric cancer patients analyzed. At p ≤ 0.05, the results were considered significant.
3 Results
3.1 Demographic and Clinical Data
Patient ages ranged from 30 to 98 years at diagnosis with two groups, females (n=35) and males (n=42). Results showed that the gastric cancer cases (n=77), 39 were differentially diagnosed with diffuse type cancer and 38 were diagnosed with intestinal type (see Table 3 for summary of the demographic and clinical information for the two groups.)
3.2 Immunohistochemistry Results
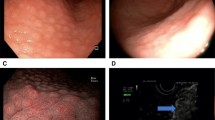
Demonstrations of positive IHC control, and negative and positive staining performed using antibodies against p27kip1 low power (×10), are presented in Figs. 2, 3, 4. Immunohistochemistry indicated that 56/77 (72.7%) of the gastric cancer samples were negative for p27 gene expression. Among the 56 negative samples, 27 were from females and 29 from males. Thirty cases of the negative IHC samples were of diffuse-type gastric cancer, while the remaining 26 were intestinal type. The cellular staining between p27 expression and H. pylori genes is shown below.

Immunohistochemistry staining of positive controls for p27. A-1: Positive control from tonsil tissue section showing cell staining (brown) of tonsil tissue sample (×10). A-2: Positive control from lymphoma tissue showing cell nuclei staining (brown) in lymphoma sample (×40). Primary antibody anti-p27, secondary antibody anti-Ki67 with peroxidase, DAB chromogen and hematoxylin stain

Immunohistochemistry of negative control for P27 expression in cancer tissue. B-1: Negative control from H. pylori uninfected gastric intestinal adenocarcinoma sample showing nuclei staining only. Low power (×10) view. B-2: Negative control from H. pylori un-infected gastric diffuse adenocarcinoma showing blue nuclear stain around gastric diffuse adenocarcinoma sample. Low power (×10) view. No primary antibody, secondary antibody, anti-Ki67 with peroxidase, DAB chromogen and hematoxylin stain

Immunohistochemical Expression of p27 and Ki67 in Cancer Cells. C-1: Immunohistochemical detection of p27 and Ki67 expression. Low power (×10) view of clear positive (brown stain) in epithelium of H. pylori infected gastric intestinal adenocarcinoma sample. C-2: Immunohistochemical detection of p27 and Ki67 expression. Low power (×10) view of clear positive (brown stain) in epithelium of H. pylori infected of gastric diffuse adenocarcinoma sample. Primary antibody anti-p27, secondary antibody anti-Ki67 with peroxidase, DAB chromogen and hematoxylin stain
3.3 PCR Results
All samples were positive for GAPDH, indicating that the test samples contained high-quality DNA and were suitable for further analysis. Regarding the ureA gene, 42 out of 77 samples were positive for the H. pylori ureA gene; however, no correlations between the ureA gene presence and the cancer type or gender were found. The majority of samples (37/42) positive for the ureA gene were negative for p27 protein expression (see Table 4), with statistical significance (p<0.05) indicative that the p27 protein was suppressed in almost all gastric cancer samples that were positive for H. pylori infection and ureA gene. The presence of virulence genes and their combinations are summarized (see Table 4).
No statistically significant correlations between gene occurrence, gender, or type of gastric cancer were found in this histological analysis; however, all virulence genes demonstrated significance (p-value < 0.05) correlation with protein p27 expression based on IHC and PCR data (Table 6).
4 Discussion
Helicobacter pylori is one of the most common bacterial infections in humans that may progress to gastritis, duodenal ulcers, peptic ulcer disease (PUD), gastric adenocarcinoma, and MALT associated lymphoma [2]. In the current study, we investigated the potential correlation between H. pylori infection and gastric adenocarcinoma in a Jordanian population. Although several studies have been conducted in Jordan to investigate the relationship between H. pylori infection and gastric cancer [50,51,52], to the best of our knowledge, this is the first study of its kind in the Middle East to look at the prevalence of H. pylori in patients with gastric cancer using gastrectomy samples rather than biopsies. Seventy-seven samples of adenocarcinoma gastrectomy, formalin-fixed, paraffin-embedded tissue samples were collected from the archives of the Pathology Department at JRMS and KAUH (Amman and Irbid cities, respectively) (Table 1). The samples were examined for the presence of past H. pylori infection, the prevalence of cagA and vacA allelic subtypes, and their correlation with one another, as well as with the expression of the tumor suppressor protein, p27.
Our data confirmed the presence of H. pylori in 42 out of 77 (54.5%) gastrectomy samples, using PCR amplification of the ureA gene (see Table 5), while the cagA virulence gene was detected in these samples in 57.1% (24/42) of cases. A total of 42 samples positive for the ureA gene were further processed for the amplification of cagA and vacA alleles (s1, s2, m1, m2) (Table 6).
According to our study, the predominant vacA genotypes of patients with H. pylori infection in the Jordanian population were s1: 24.7%, s2: 22.1%, m1: 14.3%, and m2 positive: 14.3%. On the other hand, we investigated the link between the expression of p27 and H. pylori infection. Our results showed that 56/77 (72.7%) of gastric adenocarcinomas tested negative for p27 expression (see Figure 3 and Table 6). Furthermore, 37 of 42 gastric adenocarcinoma tissue samples that were positive for ureA were also negative for p27 expression, with a significant correlation (p-value 0.05) (Table 4). Studies showed that cagA positivity rates and clinical outcomes differ between countries and population groups [53]. In general, the cagA prevalence rate has been found to be 50–60% in Middle Eastern countries [54, 55]. In our study, the cagA prevalence rate was 57.1%, whereas 100% of H. pylori strains in East Asian countries are cagA-positive [56]. From our results, we can demonstrate that virulent H. pylori strains associated with adenocarcinoma often carry cagA, as more than half of the cases under study were positive for the H. pylori strain carrying the cagA gene. Our results agree with those of a study performed in Japan, Korea, the United States, and Colombia that reported predominance of the cagA genotypes [53,54,55,56,57,58,59]. The different combinations of vacA s and m regions identify virulence characteristics of the H. pylori strain. It has been shown that type s1m1 strains can produce higher cytotoxin activity in vitro than type s1m2 strains, whereas the s2m2 strains do not produce detectable amounts of the cytotoxin and thus are considered less virulent [60]. Therefore, it is significant to identify the vacA profiles of the isolated strains and then evaluate the subtype combinations, together with the clinical outcome of the patient. According to the results of our study, the predominant genotype was vacAm1s1—unlike in other Middle Eastern countries, for example where the predominant subtype was reported to be vacAm1s2 in Turkey [55]; but similar to the results of other countries like South Africa and Mexico [58, 61]. The study of genotypes in four different countries reported that the vacAs1ml genotypes were predominant in Japan, Korea, the United States, and Colombia [54]. The same study reported a higher prevalence of the vacA s1 than the vacA s2 genotype. A study from Germany showed the most frequent allelic combinations were s1m2: 47.7%, s1m1: 35.4%), and s2m2: 15.4% [60]. Our results showed that 72.7% of gastric adenocarcinoma samples were negative for p27 expression, which means that the lack of p27 expression can be an essential change during gastric carcinogenesis. More recent research indicates differential resistance of anti-microbial therapeutics worthy of further investigation between cagA positive and vacA positive H. pylori [62].
Our results confirm a strong correlation between the presence of H. pylori in gastric adenocarcinoma and a lack of p27 expression as 37 out of 42 samples with positive ureA were negative for p27 expression with a significant association (p = 0.001). This result is consistent with a study done in vitro by Shirin et al., who found a strong correlation between the presence of H. pylori and the inhibition of p27 immunoexpression [63]. Although prior studies have found a correlation between different carcinoma types and expression of p27, we believe this is the first study to document this using validated viable gastrectomy samples. Therefore, this method of ascertaining individual phenotypes is viable to determine historical H. pylori strains. Furthermore, all samples were collected between 2003 and 2013 and validated by PCR and immunohistochemical analysis occurring between 2005 and 2013. Existing protocols at the time of project were indicative that longer PCR primers could potentially be analyzed for up to 20 years and antigenic degradation can occur of immunohistochemical samples similarly [64,65,66,67]. Whereas at that time we utilized an additional GAPDH assay to quantify DNA quality. In our combined analysis we show that FFPE could potentially be valid in combination with PCR utilizing FFPE immunohistochemistry on gastrectomy samples between 8 and 10 years. In addition, we utilized a well characterized ki67 protein marker IHC as a standard known marker of cell proliferation.
5 Limitations
The limitations are: PCR testing of unknown phenotypes, testing of gastric adenocarcinoma precursor condition tissues to assess p27 expression and other results, stratified or regression analyses by demographics, cancer sub-type, and prior treatment of H. pylori with antibiotics.
6 Conclusion
Based on these results, we concluded that the suppression of p27 expression and H. pylori infection in men and women of all ages were equally correlated and may be associated with the occurrence of gastric adenocarcinomas. Additionally, the expression of the vacA genotype does not however, appear to be associated with the inhibition of p27. The absence of p27 expression was found to have a propensity for the cagA (+) H. pylori genotype, despite the negative correlation. Additional research would help to firmly establish this association and pave the way for new perspectives for diagnosis, prognostic biomarkers, and targeted or individualized therapeutics. As p27 may represent a key diagnostic marker and predictor of adenocarcinoma prognosis in such individuals and may find use for guiding subsequent treatment decisions.
Availability of Data and Materials
Data contained within the article or supplementary materials are also available upon request. Suhaila A. Al-Sheboul ORCiD (0000-0001-9001-3232). Further information on paraffin and formalin embedding; Paraffin Embedding Technique; A Review of Preanalytical Factors Affecting Molecular, Protein, and Morphological Analysis of Formalin-Fixed, Paraffin-Embedded (FFPE) Tissue: How Well Do You Know Your FFPE Specimen? (2014). Archives of Pathology & Laboratory Medicine (allenpress.com); Immunofluorescence Staining of Paraffin Sections Step by Step (2020); Adenosquamous carcinoma; PowerPoint Presentation (esmo.org); cagPAI in UniProtKB search (378)|UniProt; vacA helicobacter pylori in UniProtKB search (3794)|UniProt.
Abbreviations
- IHC :
-
Immunohistochemistry
- H. pylori :
-
Helicobacter pylori
- FFPE:
-
Formalin-fixed, paraffin-embedded
- CagPAI :
-
Cytotoxin-associated pathogenicity island
- VacA :
-
Vacuolating cytotoxin
- JUST :
-
Jordan University of Science and Technology
- KAUH :
-
King Abdullah University Hospital
- JRMS :
-
Jordanian Royal Medical Services:
- GAPDH Gene:
-
Glyceraldehyde 3-phosphate dehydrogenase gene
References
Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. 2019;14:89–103. https://doi.org/10.5114/pg.2018.81072.
Eslick G-D. Helicobacter pylori infection causes gastric cancer? A review of the epidemiological, meta-analytic, and experimental evidence. World J Gastroenterol. 2006;12:2991–9. https://doi.org/10.3748/wjg.v12.i19.2991.
Yousefi B, Mohammadlou M, Abdollahi M, Salek Farrokhi A, Karbalaei M, Keikha M, Kokhaei P, Valizadeh S, Rezaiemanesh A, Arabkari V, et al. Epigenetic changes in gastric cancer induction by Helicobacter Pylori. J Cell Physiol. 2019;234:21770–84. https://doi.org/10.1002/jcp.28925.
Palframan SL, Kwok T, Gabriel K. Vacuolating cytotoxin A (VacA), a key toxin for helicobacter pylori pathogenesis. Front Cell Infect Microbiol. 2012. https://doi.org/10.3389/fcimb.2012.00092.
Zhang F, Chen C, Hu J, Su R, Zhang J, Han Z, Chen H, Li Y. Molecular mechanism of Helicobacter pylori-induced autophagy in gastric cancer. Oncol Lett. 2019;18:6221–7. https://doi.org/10.3892/ol.2019.10976.
Korotkov KV, Sandkvist M, Hol WGJ. The type II secretion system: biogenesis, molecular architecture and mechanism. Nat Rev Microbiol. 2012;10:336–51. https://doi.org/10.1038/nrmicro2762.
Meuskens I, Saragliadis A, Leo JC, Linke D. Type V secretion systems: an overview of passenger domain functions. Front Microbiol. 2019. https://doi.org/10.3389/fmicb.2019.01163.
Costa TRD, Harb L, Khara P, Zeng L, Hu B, Christie PJ. Type IV secretion systems: advances in structure, function, and activation. Mol Microbiol. 2021;115:436–52. https://doi.org/10.1111/mmi.14670.
Costa TRD, Felisberto-Rodrigues C, Meir A, Prevost MS, Redzej A, Trokter M, Waksman G. Secretion systems in gram-negative bacteria: structural and mechanistic insights. Nat Rev Microbiol. 2015;13:343–59. https://doi.org/10.1038/nrmicro3456.
Backert S, Tegtmeyer N, Fischer W. Composition, structure and function of the Helicobacter pylori Cag pathogenicity island encoded type IV secretion system. Future Microbiol. 2015;10:955–65. https://doi.org/10.2217/fmb.15.32.
Hatakeyama M. Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93:196–219. https://doi.org/10.2183/pjab.93.013.
Correa P, Piazuelo MB. Natural history of Helicobacter pylori infection. Dig Liver Dis. 2008;40:490–6. https://doi.org/10.1016/j.dld.2008.02.035.
Atrisco-Morales J, Martínez-Santos VI, Román-Román A, Alarcón-Millán J, De Sampedro-Reyes J, Cruz-Del Carmen I, Martínez-Carrillo DN, Fernández-Tilapa G. VacA S1m1 genotype and CagA EPIYA-ABC pattern are predominant among Helicobacter pylori strains isolated from Mexican patients with chronic gastritis. J Med Microbiol. 2018;67:314–24. https://doi.org/10.1099/jmm.0.000660.
Román-Román A, Martínez-Carrillo DN, Atrisco-Morales J, Azúcar-Heziquio JC, Cuevas-Caballero AS, Castañón-Sánchez CA, Reyes-Ríos R, Betancourt-Linares R, Reyes-Navarrete S, Cruz-Del Carmen I, et al. Helicobacter Pylori VacA S1m1 genotype but Not CagA or BabA2 increase the risk of ulcer and gastric cancer in patients from Southern Mexico. Gut Pathog. 2017;9:18. https://doi.org/10.1186/s13099-017-0167-z.
Diaz MI, Valdivia A, Martinez P, Palacios JL, Harris P, Novales J, Garrido E, Valderrama D, Shilling C, Kirberg A, et al. Helicobacter pylori VacA S1a and S1b alleles from clinical isolates from different regions of Chile show a distinct geographic distribution. World J Gastroenterol. 2005;11:6366–72. https://doi.org/10.3748/wjg.v11.i40.6366.
Soyfoo DM, Doomah YH, Xu D, Zhang C, Sang H-M, Liu Y-Y, Zhang G-X, Jiang J-X, Xu S-F. New genotypes of Helicobacter pylori VacA D-region identified from global strains. BMC Mol Cell Biol. 2021;22:4. https://doi.org/10.1186/s12860-020-00338-2.
Bakhti SZ, Latifi-Navid S, Mohammadi S, Zahri S, Bakhti FS, Feizi F, Yazdanbod A, Siavoshi F. Relevance of Helicobacter pylori Vac A 3ʹ-end region polymorphism to gastric cancer. Helicobacter. 2016;21:305–16. https://doi.org/10.1111/hel.12284.
Foo JH, Culvenor JG, Ferrero RL, Kwok T, Lithgow T, Gabriel K. Both the P33 and P55 subunits of the Helicobacter pylori VacA toxin are targeted to mammalian mitochondria. J Mol Biol. 2010;401:792–8. https://doi.org/10.1016/j.jmb.2010.06.065.
Djekic A, Müller A. The Immunomodulator VacA promotes immune tolerance and persistent Helicobacter pylori infection through its activities on T-cells and antigen-presenting cells. Toxins (Basel). 2016;8:187. https://doi.org/10.3390/toxins8060187.
Mobley H. The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment Pharmacol Ther. 1996;10:57–64. https://doi.org/10.1046/j.1365-2036.1996.22164006.x.
Domínguez-Martínez DA, Fontes-Lemus JI, García-Regalado A, Juárez-Flores Á, Fuentes-Pananá EM. IL-8 secreted by gastric epithelial cells infected with Helicobacter pylori CagA positive strains is a chemoattractant for Epstein-Barr virus infected B lymphocytes. Viruses. 2023;15:651. https://doi.org/10.3390/v15030651.
Audibert C, Janvier B, Grignon B, Salaüna L, Burucoa C, Lecron J-C, Fauchère J-L. Correlation between IL-8 induction, CagA status and VacA genotypes in 153 French Helicobacter pylori isolates. Res Microbiol. 2000;151:191–200. https://doi.org/10.1016/S0923-2508(00)00139-X.
Razavipour SF, Harikumar KB, Slingerland JM. P27 as a transcriptional regulator: new roles in development and cancer. Cancer Res. 2020;80:3451–8. https://doi.org/10.1158/0008-5472.can-19-3663.
Helal NS, Omran Z, Aboushousha T, Youssef M, Badawy A, Aboul-Ezz MA, Moussa M. Prognostic significance of P27 and survivin in H. Pylori gastritis and gastric cancer. Asian Pac J Cancer Prev. 2021;22:3553–9. https://doi.org/10.31557/APJCP.2021.22.11.3553.
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–66. https://doi.org/10.1038/nrc2602.
Abbastabar M, Kheyrollah M, Azizian K, Bagherlou N, Tehrani SS, Maniati M, Karimian A. Multiple functions of P27 in cell cycle, apoptosis, epigenetic modification and transcriptional regulation for the control of cell growth: a double-edged sword protein. DNA Repair. 2018;69:63–72. https://doi.org/10.1016/j.dnarep.2018.07.008.
André AR, Ferreira MVP, Mota RMS, Ferrasi AC, de Pardini MIMC, Rabenhorst SHB. Gastric adenocarcinoma and Helicobacter Pylori: correlation with P53 mutation and P27 immunoexpression. Cancer Epidemiol. 2010;34:618–25. https://doi.org/10.1016/j.canep.2010.05.005.
Al-Moundhri MS, Nirmala V, Al-Hadabi I, Al-Mawaly K, Burney I, Al-Nabhani M, Thomas V, Ganguly SS, Grant C. The prognostic significance of P53, P27kip1, P21waf1, HER-2/Neu, and Ki67 proteins expression in gastric cancer: a clinicopathological and immunohistochemical study of 121 Arab patients. J Surg Oncol. 2005;91:243–52. https://doi.org/10.1002/jso.20324.
Brown B, Ojha V, Fricke I, Al-Sheboul SA, Imarogbe C, Gravier T, Green M, Peterson L, Koutsaroff IP, Demir A, et al. Innate and adaptive immunity during SARS-CoV-2 infection: biomolecular cellular markers and mechanisms. Vaccines. 2023;11:408. https://doi.org/10.3390/vaccines11020408.
Link A, Langner C, Schirrmeister W, Habendorf W, Weigt J, Venerito M, Tammer I, Schlüter D, Schlaermann P, Meyer TF, et al. Helicobacter pylori VacA genotype is a predominant determinant of immune response to Helicobacter pylori CagA. World J Gastroenterol. 2017;23:4712. https://doi.org/10.3748/wjg.v23.i26.4712.
Yamamoto T, Kita M, Ohno T, Iwakura Y, Sekikawa K, Imanishi J. Role of tumor necrosis factor-alpha and interferon-gamma in Helicobacter pylori infection. Microbiol Immunol. 2004;48:647–54. https://doi.org/10.1111/j.1348-0421.2004.tb03474.x.
Tanaka H, Yoshida M, Nishiumi S, Ohnishi N, Kobayashi K, Yamamoto K, Fujita T, Hatakeyama M, Azuma T. The CagA protein of Helicobacter pylori suppresses the functions of dendritic cell in mice. Arch Biochem Biophys. 2010;498:35–42. https://doi.org/10.1016/j.abb.2010.03.021.
Chichirau BE, Scheidt T, Diechler S, Neuper T, Horejs-Hoeck J, Huber CG, Posselt G, Wessler S. Dissecting the Helicobacter Pylori-Regulated Transcriptome of B Cells. Pathog Dis. 2020. https://doi.org/10.1093/femspd/ftaa049.
D’Elios MM, Manghetti M, Almerigogna F, Amedei A, Costa F, Burroni D, Baldari CT, Romagnani S, Telford JL, Del Prete G. Different cytokine profile and antigen-specificity repertoire in Helicobacter pylori-specific T cell clones from the antrum of chronic gastritis patients with or without peptic ulcer. Eur J Immunol. 1997;27:1751–5. https://doi.org/10.1002/eji.1830270723.
Taylor JM, Ziman ME, Canfield DR, Vajdy M, Solnick JV. Effects of a Th1- versus a Th2-biased immune response in protection against Helicobacter pylori challenge in mice. Microb Pathog. 2008;44:20–7. https://doi.org/10.1016/j.micpath.2007.06.006.
Keikha M, Ali-Hassanzadeh M, Karbalaei M. Association of Helicobacter pylori VacA genotypes and peptic ulcer in Iranian population: a systematic review and meta-analysis. BMC Gastroenterol. 2020;20:266. https://doi.org/10.1186/s12876-020-01406-9.
Carlosama-Rosero YH, Bolaños-Bravo H, Sierra-Tórres CH, Rosero EA. Association of the Helicobacter pylori CagA, VacA, and IceA genotypes with chronic follicular gastritis in a Colombian population at high risk for gastric cancer. Rev Gastroenterol Mex. 2019;84:158–64. https://doi.org/10.1016/j.rgmxen.2018.03.012.
Xue Z, Yang H, Su D, Song X, Deng X, Yu C, Sun C, He L, You Y, Gong Y, et al. Geographic distribution of the CagA, VacA, IceA, OipA and DupA genes of Helicobacter pylori strains isolated in China. Gut Pathog. 2021;13:39. https://doi.org/10.1186/s13099-021-00434-4.
Díaz P, Valenzuela Valderrama M, Bravo J, Quest AFG. Helicobacter Pylori and gastric cancer: adaptive cellular mechanisms involved in disease progression. Front Microbiol. 2018. https://doi.org/10.3389/fmicb.2018.00005.
Chisholm SA, Teare EL, Davies K, Owen RJ. Surveillance of primary antibiotic resistance of Helicobacter pylori at centres in England and Wales over a six-year period (2000–2005). Eurosurveillance. 2007;12:3–4. https://doi.org/10.2807/esm.12.07.00721-en.
Dascălu RI, Bolocan A, Păduaru DN, Constantinescu A, Mitache MM, Stoica AD, Andronic O. Multidrug resistance in Helicobacter Pylori infection. Front Microbiol. 2023. https://doi.org/10.3389/fmicb.2023.1128497.
Shirin H, Sordillo EM, Kolevska TK, Hibshoosh H, Kawabata Y, Oh SH, Kuebler JF, Delohery T, Weghorst CM, Weinstein IB, et al. Chronic Helicobacter pylori infection induces an apoptosis-resistant phenotype associated with decreased expression of P27(Kip1). Infect Immun. 2000;68:5321–8. https://doi.org/10.1128/IAI.68.9.5321-5328.2000.
Li L, Wu W, Zheng W, Hui Q, Zhao C. Analysis of the correlation between P27 expression and Helicobacter pylori infection in gastric cancer. APMIS. 2021;130:21–5. https://doi.org/10.1111/apm.13193.
Patel PG, Selvarajah S, Boursalie S, How NE, Ejdelman J, Guerard K-P, Bartlett JM, Lapointe J, Park PC, Okello JBA, et al. Preparation of formalin-fixed paraffin-embedded tissue cores for both RNA and DNA extraction. J Vis Exp. 2016. https://doi.org/10.3791/54299.
Kosova AA, Khodyreva SN, Lavrik OI. Role of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in DNA repair. Biochem Mosc. 2017;82:643–54. https://doi.org/10.1134/s0006297917060013.
Bulajic M, Stimec B, Jesenofsky R, Kecmanovic D, Ceranic M, Kostic N, Schneider-Brachert W, Lowenfels A, Maisonneuve P, Löhr J-M. Helicobacter pylori in colorectal carcinoma tissue. Cancer Epidemiol Biomark Prev. 2007;16:631–3. https://doi.org/10.1158/1055-9965.epi-06-1031.
Khamis AS, Fadhil Al-Jibouri L, Al-Marzoqi AH, Shalan AA, Al-Taee ZM, Al Morshdi SF, Al Hajam RA, Al Dulamy KW. Helicobacter pylori genotype as predicts risk of (ulcer disease, gastric cancer, non-ulcer dyspepsia); role of some genes mediated signaling in infection. J Pharm Sci Res. 2018;10(6):1373–6.
Ruzsovics A, Molnar B, Unger Z, Tulassay Z, Pronai L. Determination of Helicobacter pylori CagA, VacA genotypes with real-time PCR melting curve analysis. J Physiol-Paris. 2001;95:369–77. https://doi.org/10.1016/s0928-4257(01)00050-x.
Dechwongya P, Limpisood S, Boonnak N, Mangmool S, Takeda-Morishita M, Kulsirirat T, Rukthong P, Sathirakul K. The intestinal efflux transporter inhibition activity of Xanthones from Mangosteen pericarp: an in silico, in vitro and ex vivo approach. Molecules. 2020;25:5877. https://doi.org/10.3390/molecules25245877.
Abdul-Razzak KK, Bani-Hani KE. Increased prevalence of Helicobacter pylori infection in gastric cardia of patients with reflux esophagitis: a study from Jordan. J Dig Dis. 2007;8:203–6. https://doi.org/10.1111/j.1751-2980.2007.00306.x.
Bani-Hani K-E, Yaghan R-J, Heis H-A, Shatnawi N-J, Matalka I-I, Bani-Hani A-M, Gharaibeh K-A. Gastric malignancies in northern Jordan with special emphasis on descriptive epidemiology. World J Gastroenterol. 2004;10:2174–8. https://doi.org/10.3748/wjg.v10.i15.2174.
Nimri LF, Matalka I, Bani Hani K, Ibrahim M. Helicobacter pylori genotypes identified in gastric biopsy specimens from Jordanian patients. BMC Gastroenterol. 2006;6:27. https://doi.org/10.1186/1471-230X-6-27.
Yamaoka Y, Kodama T, Gutierrez O, Kim JG, Kashima K, Graham DY. Relationship between Helicobacter pylori IceA, CagA, and VacA status and clinical outcome: studies in four different countries. J Clin Microbiol. 1999;37:2274–9. https://doi.org/10.1128/JCM.37.7.2274-2279.1999.
Benenson S, Halle D, Rudensky B, Faber J, Schlesinger Y, Branski D, Rabinowitz N, Wilschanski M. Helicobacter pylori genotypes in Israeli children: the significance of geography. J Pediatr Gastroenterol Nutr. 2002;35:680–4. https://doi.org/10.1097/00005176-200211000-00018.
Saribasak H, Salih BA, Yamaoka Y, Sander E. Analysis of Helicobacter pylori genotypes and correlation with clinical outcome in Turkey. J Clin Microbiol. 2004;42:1648–51. https://doi.org/10.1128/JCM.42.4.1648-1651.2004.
Tserentogtokh T, Gantuya B, Subsomwong P, Oyuntsetseg K, Bolor D, Erdene-Ochir Y, Azzaya D, Davaadorj D, Uchida T, Matsuhisa T, et al. Western-type Helicobacter pylori CagA are the most frequent type in Mongolian patients. Cancers. 2019;11:725. https://doi.org/10.3390/cancers11050725.
Nilsson C, Sillén A, Eriksson L, Strand M-L, Enroth H, Normark S, Falk P, Engstrand L. Correlation between Cag pathogenicity island composition and Helicobacter pylori-associated gastroduodenal disease. Infect Immun. 2003;71:6573–81. https://doi.org/10.1128/IAI.71.11.6573-6581.2003.
Letley DP, Lastovica A, Louw JA, Hawkey CJ, Atherton JC. Allelic diversity of the Helicobacter pylori vacuolating cytotoxin gene in South Africa: rarity of the VacA S1a genotype and natural occurrence of an S2/M1 allele. J Clin Microbiol. 1999;37:1203–5. https://doi.org/10.1128/JCM.37.4.1203-1205.1999.
Rudi J, Kolb C, Maiwald M, Kuck D, Sieg A, Galle PR, Stremmel W. Diversity of Helicobacter pylori VacA and CagA genes and relationship to VacA and CagA protein expression, cytotoxin production, and associated diseases. J Clin Microbiol. 1998;36:944–8. https://doi.org/10.1128/JCM.36.4.944-948.1998.
Chattopadhyay S, Datta S, Chowdhury A, Chowdhury S, Mukhopadhyay AK, Rajendran K, Bhattacharya SK, Berg DE, Nair GB. Virulence genes in Helicobacter Pylori strains from west bengal residents with Overt H. Pylori-associated disease and healthy volunteers. J Clin Microbiol. 2002;40:2622–5. https://doi.org/10.1128/JCM.40.7.2622-2625.2002.
Morales-Espinosa R, Castillo-Rojas G, Gonzalez-Valencia G, Ponce de León S, Cravioto A, Atherton JC, López-Vidal Y. Colonization of Mexican patients by multiple Helicobacter Pylori strains with different VacA and CagA genotypes. J Clin Microbiol. 1999;37:3001–4. https://doi.org/10.1128/JCM.37.9.3001-3004.1999.
Karbalaei M, TalebiBezminAbadi A, Keikha M. Clinical relevance of the CagA and VacA S1m1 status and antibiotic resistance in Helicobacter Pylori: a systematic review and meta-analysis. BMC Infect Dis. 2022;22:573. https://doi.org/10.1186/s12879-022-07546-5.
Shirin H, Kravtsov V, Shahmurov M, Petchenko P, Boaz M, Moss SF, Avni Y, Avinoach I. P27<SUP>Kip1</SUP> expression is inversely related to the grade of gastric MALT lymphoma. Int J Gastrointest Cancer. 2005;35:025–32. https://doi.org/10.1385/ijgc:35:1:025.
Gillio-Tos A, De Marco L, Fiano V, Garcia-Bragado F, Dikshit R, Boffetta P, Merletti F. Efficient DNA extraction from 25-year-old paraffin-embedded tissues: study of 365 samples. Pathology. 2007;39:345–8. https://doi.org/10.1080/00313020701329757.
Nagahashi M, Shimada Y, Ichikawa H, Nakagawa S, Sato N, Kaneko K, Homma K, Kawasaki T, Kodama K, Lyle S, et al. Formalin-fixed paraffin-embedded sample conditions for deep next generation sequencing. J Surg Res. 2017;220:125–32. https://doi.org/10.1016/j.jss.2017.06.077.
Grillo F, Bruzzone M, Pigozzi S, Prosapio S, Migliora P, Fiocca R, Mastracci L. Immunohistochemistry on old archival paraffin blocks: is there an expiry date? J Clin Pathol. 2017;70:988–93. https://doi.org/10.1136/jclinpath-2017-204387.
Sun X, Kaufman PD. Ki-67: more than a proliferation marker. Chromosoma. 2018;127:175–86. https://doi.org/10.1007/s00412-018-0659-8.
Funding
This work was supported by a grant funded by the Deanship of Research at the Jordan University of Science and Technology (Research ID: 20120158). However, Dr. Suhaila A. Al-Sheboul solely covered the entire publication expenses and no co-author contributed in publication fees.
Author information
Authors and Affiliations
Contributions
Conceptualization, SA-S.; methodology, AA-RM.; software, SAS and AA-RM.; validation, SA-S and AA-RM.; formal analysis, SA-S. and AA-RM.; investigation, SA-S and AA-RM.; resources, SA-S, AA-RM.; curation, SA-S, YS and BB; writing—original draft preparation, SA-S; writing—review and editing, SA-S, YS, BB; visualization, SA-S; BB and IM, supervision; SA-S.; project administration, SA-S; funding acquisition, SA-S. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors have no conflict of interest to declare.
Ethical Approval and Consent to Participate
Informed consents were waived because the data was anonymous at point of laboratory sample collection after clinical treatment. The study was authorized by the institutional review board (IRB) of ethics committee at Jordan University of Science and Technology (20/51/2012). This study was carried out in compliance with the Helsinki Declaration.
Consent for Publication
Clinical samples were obtained and donated from within stored clinical settings and therefore, no data were stored regarding sample identification.
Additional information
In Jordan, this is the first retrospective study to use validated viable gastrectomy tissue samples rather than tissue biopsies to characterize each of the currently defined H. pylori strains in patients with variable gastric cancer diagnosis whilst also comparing results with p27 expression within 8 years using these laboratory techniques.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Al-Sheboul, S.A., Mohammad, A.AR., Shboul, Y. et al. A Genetic and Immunohistochemical Analysis of Helicobacter pylori Phenotypes and p27 Expression in Adenocarcinoma Patients in Jordan. J Epidemiol Glob Health 13, 212–225 (2023). https://doi.org/10.1007/s44197-023-00099-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s44197-023-00099-z