
Abstract
Fumarate hydratase-deficient renal cell carcinoma (FH-RCC) is an independent pathological subtype of renal cell carcinoma with a clear driver gene and a high degree of malignancy. Recent studies have found that patients with somatic FH mutations have similar clinico-biological behavior and poor prognosis to patients with germline FH mutations. FH-RCC has the characteristics of early age of onset, atypical imaging manifestations, variable pathological patterns, difficult clinical diagnosis and poor effect on traditional drug treatment, thus greatly endangering the life and health of patients. Under the organization of the Rare Kidney Cancer Collaborative Group, Genitourinary Cancer Committee, China Anti-Cancer Association, this guideline was developed based on basic research, clinical cohort and evidence-based medicine evidence, including imaging manifestations, pathological diagnosis, genetic testing, surgical and systemic treatment options, and provided recommendations and references for the diagnosis and treatment norms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction of FH-RCC
As we are continually deepening our understanding of the molecular characteristics of renal carcinogenesis, more and more renal cell carcinomas with defined tumor driver genes are being recognized as independent pathological subtypes recognized by the world health organization (WHO). Hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCC) is a hereditary disease caused by germline mutations in the fumarate hydratase (FH) gene, which can manifest as skin, uterine smooth muscle disease, and renal malignancies. It is a separate subtype in the 2016 WHO classification of renal tumors because of its unique clinical pathology and molecular biology [1]. Somatic FH mutations may also lead to the development of renal cell carcinoma and have a very similar biological behavior to HLRCC caused by germline FH mutations. Therefore, the new WHO renal tumor classification 2022 has collectively referred to renal cell carcinoma caused by FH germline or somatic mutations as FH-deficient renal cell carcinoma (FH-RCC).
The early age of onset, atypical imaging, and variable pathology, collectively contribute to the difficult clinical diagnosis of FH-RCC and the lack of a standardized diagnostic process as well. On the other hand, FH-RCC is highly invasive, prone to early distant metastasis such as lymph node and bone metastasis, and lacks effective systemic treatment. Therefore, it is a major challenge for urologists to detect the disease early by imaging, diagnose it accurately by histopathology, molecular pathology, or even genetic testing, and formulate the best treatment strategy after diagnosis.
This collaboration group systematically reviewed the current research results of FH-RCC worldwide, and synthesized multidisciplinary physicians’ diagnostic and treatment knowledge of FH-RCC. The following clinical consensus on clinical imaging forewarning, pathologic precision diagnosis, and life-cycle management of FH-RCC is proposed through expert discussion and voting. The percentage of expert agreement is indicated in parentheses after the clinical recommendations. This consensus was developed to optimize the early diagnosis and treatment of this disease and to improve patient survival and prognosis.
2 Radiography of FH-RCC
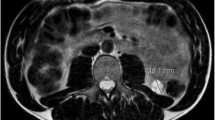
The average age of onset of FH-RCC is 40–46 years. Most tumors appear as cystic solid lesions on CT (Fig. 1), but occasionally they may appear as pure solid lesions [2, 3], with progressive enhancement on enhanced scans. The initial diagnosis is often combined with retroperitoneal lymph nodes and/or distant metastases [2,3,4], including bone, mediastinal and cervical lymph nodes, liver, adrenal glands, lung, and pleura (Fig. 1). Enhanced CT scans of the chest and whole abdomen are recommended for patients with a first clinical diagnosis of cystic renal occupancy, especially in younger patients. Suppose the solid component of the lesion shows progressive enhancement combined with retroperitoneal lymph node metastasis or distant metastasis, FH-RCC should be highly alerted. Additional CT-enhanced scans of the neck and SPECT should be performed in these patients to screen for enlarged lymph nodes and bone metastases in the neck and to identify systemic metastases. PET-CT is also of value in identifying systemic metastases. Some studies have shown that particular MR sequences such as blood oxygenation level-dependent MRI (BOLD-MRI) or magnetic resonance spectroscopy (MRS) can be used to identify metastases. It is valuable to distinguish FH-RCC from other cystic solid lesions that can show progressive enhancement and can be used as a direction to further suggest FH-RCC: FH-RCC has a significant aerobic glycolytic effect and low local ferritin content, and its solid component has a significantly lower R2* value in BOLD-MRI, unlike other renal cell carcinomas that show progressive enhancement in CT; FH-RCC tumors have a significant increase in yohimbezoic acid and a significantly higher waveform corresponding to yohimbezoic acid in MRS [5].

The typical CT scan of FH-RCC: A NECT, B arterial phase, C venous phase, D coronal reconstruction
Consensus 1: In patients with a first diagnosis of cystic renal lesions, especially in younger patients, CT enhancement of the chest and whole abdomen is recommended; if the lesion shows progressive enhancement, the possibility of FH-RCC should be alerted. If metastasis is detected after the initial diagnosis or confirmation of FH-RCC, the possibility of cervical lymph node metastasis should be alerted. Additional CT enhancement of the neck (75%) is recommended in addition to the usual CT enhancement of the chest and abdomen. Clinical studies are recommended to explore the value of BOLD-MRI, MRS, and PET-CT as adjunctive imaging tools for FH-RCC (75%).
3 Pathological diagnosis of FH-RCC
3.1 Morphological features
3.1.1 Gross morphology
FH-RCC tumors are predominantly cystic or solid. The solid area is grayish white or grayish brown, solid, and medium in texture; the cystic area has a smooth wall and clear fluid. Unlike other hereditary renal cancers, FH-RCC is mainly a unilateral, solitary lesion. The tumor mainly involves the renal parenchyma, but a few involve the renal medulla. The maximum diameter of the tumor was reported to be 0.9–18.0 cm by foreign researchers [6] and 3.0-16.9 cm by domestic researchers [7, 8].
3.1.2 Microscopic features
FH-RCC tumor cells are arranged in various ways, including papillary, sieve tube, glandular or solid arrangements, and in some cases, sarcoma-like or collecting duct-like patterns. The heterogeneity of tumor cell morphology is pronounced, and most cases show high-grade morphology: eosinophilic cytoplasm, large nuclei, and in some cases, inclusion body-like eosinophilic nuclei and perinuclear halo, which suggest the diagnosis of FH-RCC (Fig. 2G). In a few cases, eosinophilic low-grade morphology is seen (Fig. 2H), and these tumors tend to have a better prognosis. This morphologic heterogeneity leads to a high rate of underdiagnosis and misdiagnosis in cases without clinical suspicion or family history, which complicates the clinical work [6]. The diagnosis of FH-RCC is still difficult if based on morphological features alone.

Histopathologic characters of FH-RCC(HE): A cystic-solid, B papillary, C cribiform, D adenoid, E solid, F mixed papillary and solid, G inclusion body& perinuclear halos, H low grade eosinophilic tumor
Consensus 2: The morphologic diagnosis of FH-RCC is difficult and requires a high level of diagnostic experience for pathologists. It is recommended to combine relevant immunohistochemical markers to assist pathological diagnosis in suspected cases (75% of experts agree, same below). Also, it is still necessary to confirm relevant genetic testing after pathological diagnosis (100%).
3.2 Immunohistochemistry and FH detection
3.2.1 FH gene and protein defects
Most FH-RCC can be detected by immunohistochemical methods, but a few patients have positive FH protein expression. FH mutations are the most common cause of FH protein expression deficiency or loss of function (see “Section 4” for details).
FH germline mutations are specific to HLRCC, while FH somatic mutations alone are previously designated as FH-RCC [8, 9]. Since RCC due to FH germline and somatic mutations or large segmental deletions have the same pathological and biologic behavior, rare renal cell carcinomas with germline and somatic FH mutations will be uniformly classified as FH-RCC in the forthcoming new WHO pathological classification 2022.
3.2.2 S -(2-succino)cysteine (2SC)
FH is a key metabolic enzyme of the tricarboxylic acid cycle, and its defect leads to intracellular accumulation of ferredoxin, which in turn triggers a stable chemical modification of intracellular proteins: abnormal succination, which can be detected by 2SC antibodies. The positive rate of 2SC in FH-RCC is 100%, and the positive expression is diffusely positive for nuclei and cytoplasm (Fig. 3B), which can be used as an auxiliary diagnosis of FH-RCC. However, a small number of renal clear cell carcinomas and papillary renal cell carcinomas can also express 2SC weakly positively.

IHC of FH-RCC(×200): A deficiency of fumarate hydratase, B 2-succinate-Cysteine(2SC), 3C AKR1B10
3.2.3 AKR1B10
FH deficiency can lead to abnormalities in the KEAP1-NRF2 pathway [10, 11], and increased NRF2 expression further promotes AKR1B 10 expression [12]. AKR1B10 immunohistochemical positivity is strongly positive for diffuse cytoplasm and nucleus (Fig. 3C), which has high sensitivity and specificity for the diagnosis of FH-RCC and can be used as a potential marker for the diagnosis of FH-RCC.
Consensus 3: Immunohistochemical detection of FH protein is an important tool for diagnosing FH-RCC, but some FH-RCC tumor cells can still express FH protein. Combined detection of FH, 2SC, and AKR1B10 is recommended to improve the diagnostic accuracy of FH-RCC (70%). In the search for the cause of FH protein defects, if the corresponding FH mutation is not detected by genetic testing, multiplex ligation-dependent probeamplification (MLPA) is recommended in combination with clinicopathological features to determine the presence of FH large fragment deletion (86%).
3.3 Immune microenvironment characteristics
Due to their immunogenic nature, immune checkpoint inhibitor-based regimens have a pivotal role in treating advanced kidney cancer. The results showed that most FH-RCC had CD8 + and CD4 + T cell enrichment and PD-L1 expression (Fig. 4A and B) [8, 13, 14]; multiplex immunofluorescence also suggested increased immune infiltration in FH-RCC tumors (Fig. 5), suggesting that immunotherapy is one of the feasible treatment options for FH-RCC [8].

IHC of FH-RCC tumor immune microenvironment: A PD-L1, B CD8 + T infiltration

Representative immunofluorescence demonstrating the presence of overall CD8 + T cell, tumor-associated macrophage (CD68 and HLA-DR) and NK cell (CD56) infiltration in selected sample
Consensus 4: The degree of immune infiltration in FH-RCC is generally high. Assessment of the immune microenvironment (PD-L1 and CD8 + T cells) is necessary for FH-RCC to more precisely guide the development of clinical treatment strategies (60%).
3.4 Differential diagnosis
Prior to the discovery of FH mutations, most FH-RCC were described as papillary renal cell carcinoma type II, and in a few cases were diagnosed as collecting duct carcinoma or unclassified renal cell carcinoma. Morphologically, FH-RCC is mainly associated with papillary renal cell carcinoma type II, Xp11.2 translocation/TFE3 gene fusion-associated renal cell carcinoma, collecting ductal carcinoma, medullary carcinoma, and unclassified renal cell carcinoma types for differentiation, and immunohistochemistry and genetic testing are of great value for differential diagnosis.
Consensus 5: It is difficult for pathologists to make the differential diagnosis of FH-RCC from other pathological types of renal cell carcinoma based on histomorphology alone, and it needs to be combined with immunohistochemistry or even genetic testing of related molecules to maximize accurate diagnosis and differential diagnosis (100%).
4 FH-RCC gene detection
The FH gene is located on chromosome 1q42.3-43, which is about 22 kb long and consists of 10 exons. Theoretically, mutations occurring in the above functional domains or affecting their functions may reduce FH activity and induce FH-RCC. 60-80% of FH-RCC patients have germline FH mutations, and the presence of FH mutations can be clarified by testing germline samples (including blood or saliva). However, germline and tumor samples should be tested for patients with possible systemic FH mutations.
FH mutations are highly random and there are no clear high-frequency mutation hotspots. More than 280 pathogenic and potentially pathogenic FH mutations associated with the development of HLRCC have been identified, with missense mutations (50-60%) being the most common mutation type, followed by nonsense and shift mutations (together accounting for approximately 30%). It should be noted that FH large fragment deletions are present in 3 to 5% of patients. This type of mutation is a structural variant. Genetic testing based on common second-generation sequencing platforms (whole-exome sequencing and panel testing) and Sanger sequencing are not sensitive to this structural variant and therefore are prone to underdiagnosis. Thus, if HLRCC is clinically highly suspected and the genetic test shows negative, MLPA should be performed to clarify the presence of FH large segment deletion. If the above methods cannot clarify the mutation status, there may be promoter regions, intron regions, or more minor structural variants that require whole genome sequencing for clarification. In addition to the FH mutation, FH-RCC may harbour other oncogenic mutations, including NF2, ARID1B, KMT2D, SMARCC1, etc. FH-RCC combining these mutations may have a more malignant biological behavior. Therefore, single-gene assays for FH are insufficient to fully characterize the mutation profile of the FH-RCC system. In addition, loss-of-function FH mutations cause reduced FH activity, leading to a considerable accumulation of ferredoxin in cells and mitochondria, which can further mediate abnormal tumor transcription and epistasis regulation by inhibiting α-ketoglutarate-dependent dioxygenase family-related molecules. The above features are closely related to the patient’s clinical prognosis and drug therapy response. Therefore, whole-exome, transcriptome, and methylation sequencing for FH-RCC is an essential guide for patient prognosis assessment and treatment option selection.
Consensus 6: Simultaneous germline and systemic FH mutation testing are required for patients with suspected FH-RCC. For patients with high clinical suspicion of FH-RCC but negative mutation testing, MLPA is recommended to clarify the presence of FH large fragment deletion (85%). After the diagnosis of FH-RCC is confirmed, relevant genetic testing such as whole exome, transcriptome, and methylation sequencing is recommended to assist in the in-depth understanding of the disease and clinical decision-making (54%). Current genetic testing provides limited information for FH-RCC diagnosis and treatment strategies, and the relevant genetic testing tools and technologies after diagnosis should be determined according to the actual situation (27%).
5 Treatment options for FH-RCC
5.1 Local treatment of the primary lesion
For organ-confined FH-RCC, surgical resection of the primary lesion remains an important treatment. Because of FH-RCC’s aggressive nature and high risk of metastasis, the surgical strategy is also different from that of common kidney cancer. On the one hand, FH-RCC is often combined with cystic solid degeneration, which is highly prone to intraoperative cyst rupture and cyst fluid contamination of the operative area; on the other hand, FH-RCC is extremely aggressive, and the results of a cohort from West China Hospital of Sichuan University (West China Hospital) showed that the proportion of patients with tumor maximum diameter < 10 cm (≤ cT2a stage) who had a postoperative pathological upgrade (≥ pT3a stage) was as high as 50% (29/58). Literature also reported that a group of patients in North America presented with non-organ confined pathological escalation in 65% (15/23) [15]. Therefore, radical nephrectomy is preferentially recommended for patients with a high preoperative clinical diagnosis of FH-RCC, regardless of tumor size, while partial nephrectomy with wide margins of preserved renal units may be considered for patients with functional or anatomic isolated kidneys or with underlying renal dysfunction. When laparoscopic surgery is considered, whether by the transabdominal or retroperitoneal route, the possibility of channel implantation needs to be fully considered, the benefits and risks associated with it should be communicated to the patient, and the nephrectomy should be done strictly outside the renal fascia. There is a lack of reliable clinical evidence on whether hilar and retroperitoneal lymph node dissection is required for FH-RCC. However, since retroperitoneal lymph node metastasis often occurs early in this disease, a comprehensive preoperative imaging examination is recommended to determine whether to perform concurrent lymph node dissection and the extent of dissection based on preoperative imaging changes and intraoperative conditions. There is insufficient clinical evidence to determine whether patients diagnosed with FH-RCC after undergoing partial nephrectomy with preserved renal units need to undergo remedial radical nephrectomy as soon as possible. In patients with oligometastases who are in a stable stage of systemic therapy, reduction and metastasectomy may be considered after adequate case screening.
Although there is a lack of clinical evidence, given the highly malignant features of FH-RCC, and most of them have a more active tumor immune microenvironment, postoperative adjuvant immune checkpoint inhibitor therapy can be considered for high-risk patients regarding renal clear cell carcinoma [16], and the specific drugs and treatment courses can be decided after considering the relevant patient conditions. In patients with de novo metastatic FH-RCC, there is also a lack of clinical evidence on whether primary site reduction is worthwhile. The feasibility of reduction is recommended to be fully evaluated on a patient-by-patient basis.
Consensus 7: In patients with high preoperative suspicion of FH-RCC, it is recommended to prioritize radical surgical treatment in the absence of an absolute indication for renal preservation and to carefully evaluate the imaging presentation to decide whether to complete retroperitoneal lymph node dissection at the same time (70%). After a comprehensive assessment of tumor size, location, and other specific conditions, partial nephrectomy can be chosen (26%). Due to the high malignancy of FH-RCC, postoperative adjuvant therapy is necessary despite the lack of sufficient evidence (100%). It is recommended that adjuvant therapy be implemented after the completion of the surgery, as determined by multidisciplinary discussion (64%). In patients with metastatic FH-RCC, a primary focal reduction is worth trying (75%), and a reduction treatment plan should be developed based on a comprehensive evaluation by a multidisciplinary urologic oncology team (54%).
5.2 Systemic treatment
FH-RCC is less common, thus standardized diagnosis is difficult. There is a lack of data from multicenter clinical trials with large sample sizes; also there is significant heterogeneity in the available real-world treatment outcomes, and no consistent standardized treatment regimen is available.
5.2.1 Bevacizumab combined with erlotinib (E-B regimen)
In 2014, the results of a phase II clinical trial of the E-B regimen for patients with HLRCC showed that the combination had good clinical efficacy (objective remission rate of 60% and median progression-free survival time of 24.2 months) [17]. Subsequently, data from a single-center study at the National Cancer Institute were reported at the 2020 American Society of Clinical Oncology annual meeting, again showing good efficacy of the E-B regimen (objective remission rate 64% and median progression-free survival time of 21.1 months) [18]. However, due to the inclusion of patients with hereditary FH-RCC in this study, long enrollment time, single-center nature, and lack of clinical validation, the E-B regimen was delayed in gaining approval by the US Food and Drug Administration. Subsequently, data from a multicenter study from France and the Renji Hospital of Shanghai Jiaotong University School of Medicine (Renji Hospital) showed similar limitations [7, 19]. However, given the lack of effective systemic treatment options for FH-RCC, the E-B regimen is still recommended for treating FH-RCC in the NCCN kidney cancer treatment guidelines and the 2021 Chinese Society of Clinical Oncology guidelines [19, 20].
5.2.2 Anti-vascular targeted drugs combined with immune checkpoint inhibitors
According to the results of the FH-RCC histology study in this center, immunotherapy combined with targeted therapy was proposed in West China Hospital, and the relevant clinical data showed that the median progression-free survival time of sindilizumab combined with axitinib for metastatic FH-RCC reached 18.7 (unpublished data), which is significantly higher than the 5.5 months of tyrosine kinase inhibitor monotherapy regimen [8]. The center is also conducting sindilizumab in combination with axitinib. A prospective phase II clinical trial (NCT01130519) for the treatment of metastatic FH-RCC, evaluated as of May 2022, showed a disease control rate of 90% (18/20) and a median progression-free survival time of 22.7 months in the 20 patients included. A national multicenter retrospective study led by Renji Hospital included 77 patients with FH-RCC; 26 of them with advanced FH-RCC applied anti-vascular targeted drugs combined with immune checkpoint inhibitors as first-line treatment and had an objective remission rate of 27%, disease control rate of 85%, median progression-free survival time 18.0 months, and median overall survival time was not reached; 12 cases applied E-B regimen had an objective remission rate of 25%, disease control rate of 67%, median progression-free survival time of 10.0 months, and median overall survival time of 16.0 months; 29 cases applied anti-vascular targeted drugs as first-line treatment had an objective remission rate of 10%, disease control rate of 62%, and median progression-free survival time of 12.0 months and median overall survival time of 30.0 months [7]. In the literature, it was reported that one patient with metastatic HLRCC had survived more than 58 months with a combination of axitinib and nabumab by tumor reduction [20]. Another literature reported that one patient with HLRCC achieved complete remission after 31 weeks of treatment with nabumab combined with epitumumab [21]. These findings suggest that combination therapy based on immune checkpoint inhibitors may be effective in metastatic FH-RCC.
5.2.3 mTOR inhibitor combined with anti-vascular targeting regimen
Data from Memorial Sloan-Kettering Cancer Center showed that 18 of 26 patients with FH-RCC received the combination of mTOR inhibitors and anti-vascular targeting drugs, with an objective remission rate of 44%, which was significantly better than anti-vascular targeting drug monotherapy (objective remission rate of 20%) and mTOR inhibitor monotherapy (objective remission rate of 0) [22]. Based on this result, it seems that the mTOR inhibitor combined with anti-vascular targeting drug regimen can also benefit patients.
5.2.4 Other regimens
Real-world data from multiple centers in China and abroad suggest that neither anti-vascular targeted drug monotherapy, mTOR monotherapy, nor immune checkpoint inhibitor monotherapy can achieve an adequate therapeutic response, and therefore, monotherapy is not recommended.
Consensus 8: There is a lack of definitive systemic treatment options for FH-RCC. Patients are recommended to participate in relevant clinical trials (85%) as a priority. Considering the tumor immune microenvironment characteristics and drug accessibility, we recommend also prioritizing treatment with immune checkpoint inhibitors in combination with anti-vascular targeting agents (89%). In addition, patients with FH germline mutations may also be treated with E-B regimens (62%).
5.3 Follow-up
Given the highly aggressive and metastatic nature of FH-RCC, imaging review is recommended every 3 months for 2 years and every 6 months thereafter for patients who underwent radical resection with or without adjuvant therapy. Patients with metastatic FH-RCC receiving systemic therapy are recommended to undergo safety examinations every month and effectiveness examinations every 3 months. Since FH-RCC can metastasize to the neck, mediastinum, retroperitoneal lymph nodes, and abdominal organs such as lung and liver, bone metastases are relatively common, routine CT-enhanced scans covering the neck, chest and whole abdomen are recommended, together with SPECT bone scans. MRI has apparent advantages over CT in showing liver, brain, and bone metastases. If metastases are suspected in the areas mentioned above and CT and bone scan cannot confirm the diagnosis, MRI can be added to assist in the diagnosis. Since some patients with FH-RCC have cystic metastases, which may appear as small cyst-like changes in the early stage, patients with FH-RCC should undergo MRI to identify new cystic lesions, especially those located in the liver, during CT follow-up. In addition, since most bone metastases in FH-RCC show osteolytic bone destruction, SPECT and PET-CT show false negatives, MRI can provide diagnostic evidence for such bone metastases. Finally, PET-CT can also be used to help identify small lesions that are difficult to determine their nature.
6 Summary
FH-RCC is a rare type of renal cell carcinoma with an extremely aggressive and metastatic nature, which is challenging to diagnose and lacks effective systemic treatment. Clinical work needs to deepen our understanding of this disease, make clinical recognition as early as possible with imaging manifestations and medical history, and choose reasonable radical surgery or systemic treatment plan to improve patient prognosis.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on request.
References
Moch H, Cubilla AL, Humphrey PA, et al. The 2016 WHO classification of tumours of the urinary system and male genital organs-part a: renal, penile, and testicular tumours. Eur Urol. 2016;70(1):93–105. https://doi.org/10.1016/j.eururo.2016.02.029.
Nikolovski I, Carlo MI, Chen YB, et al. Imaging features of fumarate hydratase-deficient renal cell carcinomas: a retrospective study. Cancer Imaging. 2021;21(1):24. https://doi.org/10.1186/s40644-021-00392-9.
Yang L, Li XM, Hu YJ, et al. Multidetector CT characteristics of fumarate hydratase-deficient renal cell carcinoma and papillary type renal cell carcinoma[J]. Korean J Radiol. 2021;22(12):1996–2005. https://doi.org/10.3348/kjr.2021.0212.
Paschall AK, Nikpanah M, Farhadi F, et al. Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome: spectrum of imaging findings. Clin Imaging. 2020;68:14–9. https://doi.org/10.1016/j.clinimag.2020.06.010.
Casey RT, McLean MA, Challis BG, et al. Fumarate metabolic signature for the detection of reed syndrome in humans. Clin Cancer Res. 2020;26(2):391–6. https://doi.org/10.1158/1078-0432.CCR-19-1729.
Trpkov K, Hes O, Agaimy A, et al. Fumarate Hydratase-deficient renal cell carcinoma is strongly correlated with fumarate hydratase mutation and hereditary leiomyomatosis and renal cell carcinoma syndrome. Am J Surg Pathol. 2016;40(7):865–75. https://doi.org/10.1097/PAS.0000000000000617.
Xu Y, Kong W, Cao M, et al. Genomic Profiling and response to immune checkpoint inhibition plus tyrosine kinase inhibition in FH-deficient renal cell carcinoma. Eur Urol. 2022:S0302-2838(22)02406-X. https://doi.org/10.1016/j.eururo.2022.05.029. https://www.sciencedirect.com/science/article/abs/pii/S030228382202406X. Published online ahead of print June 14, 2022.
Sun G, Zhang X, Liang J, et al. Integrated molecular characterization of fumarate hydratase-deficient renal cell carcinoma[J]. Clin Cancer Res. 2021;27(6):1734–43. https://doi.org/10.1158/1078-0432.CCR-20-3788.
Pan X, Zhang M, Yao J, et al. Fumaratehydratase-deficient renal cell carcinoma: a clinicopathological and molecular study of 13 cases[J]. J Clin Pathol. 2019;72(11):748–54. https://doi.org/10.1136/jclinpath-2019-205924.
Clerici S, Boletta A. Role of the KEAP1-NRF2 axis in renal cell carcinoma. Cancers (Basel). 2020;12(11):3458. https://doi.org/10.3390/cancers12113458.
Kansanen E, Kuosmanen SM, Leinonen H, et al. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1(1):45–9. https://doi.org/10.1016/j.redox.2012.10.001.
Ooi A, Wong JC, Petillo D, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20(4):511–23. https://doi.org/10.1016/j.ccr.2011.08.024.
Furuya M, Iribe Y, Nagashima Y, et al. Clinicopathological and molecular features of hereditary leiomyomatosis and renal cell cancer-associated renal cell carcinomas. J Clin Pathol. 2020;73(12):819–25. https://doi.org/10.1136/jclinpath-2020-206548.
Alaghehbandan R, Stehlik J, Trpkov K, et al. Programmed death-1 (PD-1) receptor/PD-1 ligand (PD-L1) expression in fumarate hydratase-deficient renal cell carcinoma. Ann Diagn Pathol. 2017;29:17–22. https://doi.org/10.1016/j.anndiagpath.2017.04.007.
Lau HD, Chan E, Fan AC, et al. A clinicopathologic and molecular analysis of fumarate hydratase-deficient renal cell carcinoma in 32 patients. Am J Surg Pathol. 2020;44(1):98–110. https://doi.org/10.1097/PAS.0000000000001372.
Choueiri TK, Tomczak P, Park SH, et al. Adjuvant pembrolizumab after nephrectomy in renal-cell carcinoma[J]. N Engl J Med. 2021;385(8):683–94. https://doi.org/10.1056/NEJMoa2106391.
Srinivasan R, Su D, Stamatakis L, et al. 5 mechanism based targeted therapy for hereditary leiomyomatosis and renal cell cancer (HLRCC) and sporadic papillary renal cell carcinoma: interim results from a phase 2 study of bevacizumab and erlotinib. Eur J Cancer. 2014;50(Suppl 6):8. https://doi.org/10.1016/S0959-8049(14)70131-5.
Srinivasan RS, Gurram M, Al Harthy EA, et al. Results from a phase study of bevacizumab and erlotinib in subjects with advanced hereditary leiomyomatosis and renal cell cancer (HLRCC) or sporadic papillary renal cell cancer. J Clin Oncol. 2020;38(15 Suppl):5004. https://doi.org/10.1200/JCO.2020.38.15_suppl.5004.
Carril-Ajuria L, Colomba E, Cerbone L, et al. Response to systemic therapy in fumarate hydratase-deficient renal cell carcinoma[J]. Eur J Cancer. 2021;151:106–14. https://doi.org/10.1016/j.ejca.2021.04.009.
Yonese I, Ito M, Takemura K, et al. A case of metastatic hereditary leiomyomatosis and renal cell cancer syndrome-associated renal cell carcinoma treated with a sequence of axitinib and nivolumab following cytoreductive nephrectomy. J Kidney Cancer VHL. 2020;7(2):6–10. https://doi.org/10.15586/jkcvhl.2020.148.
Iribe Y, Furuya M, Shibata Y, et al. Complete response of hereditary leiomyomatosis and renal cell cancer (HLRCC)-associated renal cell carcinoma to nivolumab and ipilimumab combination immunotherapy by: a case report. Fam Cancer. 2021;20(1):75–80. https://doi.org/10.1007/s10689-020-00195-0.
Gleeson JP, Nikolovski I, Dinatale R, et al. Comprehensive molecular characterization and response to therapy in fumarate hydratase-deficient renal cell carcinoma. Clin Cancer Res. 2021;27(10):2910–9. https://doi.org/10.1158/1078-0432.CCR-20-4367.
Acknowledgements
The authors thank all the patients and their families involved in this study. We thank the Rare Kidney Cancer Collaborative Group, Genitourinary Cancer Committee, China Anti-Cancer Association and West China Hospital, Sichuan University for their support.
Experts involved in the preparation of this consensus
The multidisciplinary team of West China Hospital, Sichuan University (in no particular order): Wei Qiang (urology), Li Hong (urology), Zeng Hao (urology), Chen Niobium (pathology), Yao Jin (radiology), Huang Leng (nuclear medicine), Liu Zhenhua (urology), Nie Ling (pathology), Shen Pengfei (urology), Sun Guangxi (urology), Chen Yuntian (radiology), Zhang Xingming (Urology), Jia-Yu Liang (Urology), and Pan Xiuyi (Department of Pathology) Shanghai Renji Hospital, School of Medicine, Shanghai Jiao Tong University (in no particular order): Xue Wei (Department of Urology), Zhang Jin (Department of Urology), Hu Jiong (Department of Oncology), Kong Wen (Department of Urology), Wu Guangyu (Department of Radiology), Xu Yunze (Department of Urology), Wang Zhaoyu (Department of Pathology).
(Department of Urology), Wang Zhaoyu (Department of Pathology).
Experts involved in the review of this consensus (in hanyu pinyin order by last name): Bi Jianbin (Shengjing Hospital, China Medical University), Chen Huiqing (Affiliated Cancer Hospital, Shanxi Medical University), Dong Pei (Center for Cancer Control, Sun Yat-sen Memorial Hospital, Sun Yat-sen University), Gong Kan (Peking University First Hospital), Gou Xin (The First Hospital, Chongqing Medical University), Guan Wei (Tongji Hospital, Huazhong University of Science and Technology), Wang Zhaoyu (Department of Oncology, China). (Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology), Guo Jun (Cancer Hospital, Peking University), Guo Gang (First Medical Center, PLA General Hospital), He Zhisong (First Hospital, Peking University), He Dalin (First Hospital, Xi’an Jiaotong University), Huang Jian (Sun Yat-sen Memorial Hospital, Sun Yat-sen University), Li Changling (Cancer Hospital, Chinese Academy of Medical Sciences), Liao Hong (Sichuan Provincial Cancer Hospital), Lin Tian-Xin (Cancer Hospital, Sichuan Province), and Lin Tian-Xin (Cancer Hospital, Sichuan Province).
(Sun Yat-sen Memorial Hospital of Sun Yat-sen University), Tian-Xin Lin (Sun Yat-sen Memorial Hospital of Sun Yat-sen University), Nan Liu (Affiliated Cancer Hospital of Chongqing University), Ji-Yan Liu (West China Hospital of Sichuan University), Xiaodong Liu (The First Affiliated Hospital of Kunming Medical University), Xin Ma (The First Medical Center of PLA General Hospital), Jun Qi (Xinhua Hospital of Shanghai Jiaotong University School of Medicine), Lin Qi (Xiangya Hospital of Central South University), Xing Qiu (Sichuan Provincial People’s Hospital) ), Sheng Xinan (Peking University Cancer Hospital), Shi Guohai (Cancer Hospital of Fudan University), Wang Dong (Cancer Hospital of Chinese Academy of Medical Sciences), Wang Shaogang (Tongji Hospital of Tongji Medical College of Huazhong University of Science and Technology), Xing Jinchun (The First Affiliated Hospital of Xiamen University), Xing Nianzeng (Cancer Hospital of Chinese Academy of Hospital Sciences), Yao Xin (Cancer Hospital of Tianjin Medical University), Ye Dingwei (Affiliated Hospital of Fudan University), Zhang Xu (Cancer Hospital of Fudan University), and Zhang X. Cancer Hospital of Fudan University), Zhang Xu (The First Medical Center of PLA General Hospital), Zhang Aili (The Fourth Hospital of Hebei Medical University), Zheng Song (Fujian Medical University), and Zheng Zeng (The First Affiliated Hospital of Xiamen University).
(Hospital), Zheng Song (Union Hospital, Fujian Medical University), Zheng Junhua (Renji Hospital, Shanghai Jiaotong University School of Medicine), Zhou Fangjian (Center for Cancer Control, Sun Yat-sen University), Zhou Liqun (Peking University First Hospital).
Funding
None.
Author information
Authors and Affiliations
Contributions
Study concept and design: Q. Wei, W. Xue, Z. Liu and Y. Shen. Acquisition, analysis and interpretation of data: Q. Wei, W. Xue, Z. Liu and Y. Shen. Drafting of the manuscript: Z. Liu and Y. Shen.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The Ethics Committee of West China Hospital of Sichuan University approved the study protocols and all patients or family members provided Informed consent.
Consent for publication
Not applicable.
Competing interests
All authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shen, Y., Liu, Z., Wei, Q. et al. Consensus on clinical diagnosis and treatment of fumarate hydratase-deficient renal cell carcinoma. Holist Integ Oncol 3, 7 (2024). https://doi.org/10.1007/s44178-024-00071-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44178-024-00071-2