
Abstract
Brain metastases are the most common central nervous system malignancies in adults. The popular view is that due to the existence of the blood–brain barrier, whether there are immune cells in the central nervous system has always been controversial. Current research shows that immune cells do exist in the central nervous system and play a vital role in the occurrence and development of brain metastasis. The central nervous system has a unique immune microenvironment, and the study of its mechanism is of great significance for the prediction and treatment of brain metastases. This article aims to discuss the components of the brain tumor microenvironment (TME) and immune mechanism of tumor brain metastasis, in the hopes of making better treatment through combination therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Brain metastases are the major malignant tumors in the central nervous system (CNS), outnumbering primary brain tumors [1]. The incidence of brain metastasis appears to be increasing in some tumor types, such as lung cancer is the most common source of brain metastasis, accounting for about 40–60% of all brain metastases [2, 3], followed by breast cancer(about 10%-15%) and melanoma(6%) [4, 5]. The cerebrum is the most common location of metastases, with infiltrating the brain parenchyma, dura, or leptomeninges. Other less common locations of metastases are cerebellar, brain stem, and spinal cord [4]. The prognosis of patients with brain metastases is very poor, with a one-year survival rate of less than 20% [6]. In addition to worsening the prognosis, brain metastases also have a negative impact on the life quality of patients, causing a series of central nervous system symptoms such as headaches, visual and language problems, cognitive impairment, and seizures [7].
The science of central nervous system (CNS) immunology has been enlivened in the last decade with the dismantling of the concept of “immune privilege” and the role of the immune system in maintaining tumor dormancy is increasingly being appreciated. In order to better control brain metastases, it is important to understand the Immuno-molecular mechanism of brain metastasis formation.
2 CNS immune microenvironment
The CNS-resident immune system is mainly composed of innate immune cells, and it has been shown that about 90% of the immune cells within the CNS are microglia [8]. The CNS-myeloid cells including microglia and border-associated macrophages (BAMs), contribute significantly to maintaining homeostasis in the CNS [9, 10]. These myeloid cells in the CNS parenchyma and on the peripheral interface of the CNS are highly specialized and extremely plastic cells that immediately respond to changes in the CNS microenvironment [11]. In the healthy CNS, only very few peripheral immune cells (T cells, B cells, and monocytes) can reach the perivascular, leptomeningeal, and ventricular spaces. Glial cell restriction factors strictly restrict these cells from entering the central nervous system parenchyma under normal conditions [12] (Fig. 1).

Immune microenvironment of brain metastases
Previous studies generally agree that the CNS lacks a classical lymphatic drainage system, which partially explains the immune privilege mechanism of CNS. In recent years, studies exploring how immune cells enter and exit the meninges led to the discovery of a functional lymphatic system in mice that expresses molecular markers of lymphatic endothelial cells at the dural sinuses, which transport immune cells and connect with cervical lymph nodes [13, 14]. This lymphatic system in CNS may remove hydrophilic and lipophilic compounds as well as waste products from the brain parenchyma into the cerebrospinal fluid (CSF) via the CSF-interstitial fluid (ISF) exchange system [15,16,17]. The macromolecules and other waste products may subsequently be removed from CSF by drainage into cervical lymph nodes via the nasal mucosa lymphatics, purportedly via the cribriform plate [18, 19]. These vessels also tend to play a role in regular immune surveillance of the brain, i.e., drainage of immune macromolecules and immune cells under physiological conditions [18, 20]. In addition, mice models have shown that antigens from the brain can induce an immune response in cervical lymph nodes [21]. Over the past decade, multiple studies have shown that the vertebrate brain possesses a specialized lymphatic transport pathway, described as the glymphatic system and meningeal lymphatic vessels, challenging the conventional notion of CNS immune privilege [22, 23]. The role of brain-specific lymph angiogenesis in the development of brain metastases, and whether patients with brain metastases would benefit from therapies targeted against these vessels will be critical to address the future.
The results of single-cell sequencing showed that the invasion frequency of immune cells (including T cells, mast cells, and macrophages) in the microenvironment of brain metastases was found significantly more frequently than that of primary brain tumors [8]. The CNS immune microenvironment changes with the occurrence of brain metastases. CNS-myeloid cells including microglia and non-parenchymal macrophages are activated and gathered from the border regions of the brain (such as meninges and lateral ventricles) to the brain parenchyma [24, 25], periphery blood lymphocytes such as tumor-infiltrating lymphocytes(TILs) [26], B lymphocytes, neutrophils [27], nature kill cells (NK cells) [28], and mast cells [29] can also enter the CNS when the BBB is damaged [30, 31] (Table 1). Tumor cells not only recruit and expand immunosuppressive cells to form the tumor microenvironment and pre-metastasis niche, but also regulate the function and phenotype of normal immune cells to reverse the process from a tumor initiation state to a tumor promotion state, resulting in immune escape [32]. In addition, astrocytes provide structural support in the brain and are thought to have antigen-presenting functions in addition to their pro-tumor functions by secreting neurotrophic factors that support tumor cell proliferation [33, 34]. Reactive astrocytes can also benefit metastatic cells through modulatory effects on the innate and adaptive immune systems [35]. Therefore, astrocytes are also important players in the immune microenvironment of brain metastasis.
3 Development and immune adaptations of brain metastasis
The main steps of tumor brain metastasis include the (1) invasion of the primary tumor to the stroma and basement membrane, (2) the dissociation of tumor cells, (3) the destruction of the blood–brain barrier (BBB), and (4) the colonization and invasion of the CNS [36], a series of steps previously termed the “metastatic cascade”. The first two steps are the common premise of tumor metastasis, while the latter two steps have their uniqueness in brain metastasis. Under normal circumstances, CNS is an immune-exempt organ with absent or less lymphocyte infiltration. The existence of the BBB and the high expression of negative regulatory mechanisms (such as Vista, PD-L1) in CNS myeloid cells also reflect the anti-inflammatory adaptation evolution characteristics of the brain [37,38,39]. Loss of BBB integrity as a result of neuroinflammation, upregulation of proteolytic enzymes, or direct disruption by tumor cells may all promote metastatic invasion [40].
Stephen Paget first proposed the "seed and soil" hypothesis in 1889, and believed that the success of metastasis is the result of the interaction between disseminated tumor cells (DTCs) and metastatic niche [41]. Emerging evidence claims that the interactions between DTCs and their niches in CNS are bidirectional. Primary cancer cells can induce the formation of a specialized environment before they reach the target organ, with cancer cell-derived factors making the brain resident cells and/or brain extracellular matrix (ECM) more conducive to DTCs colonization and survival, which is called a “pre-metastatic niche (PMN)” [42, 43]. Vascular leakiness is the earliest event in the metastatic cascade [44], followed by the alteration of local resident cells in the brain (such as microglia) and the recruitment of peripheral blood circulation (such as Tumor-associated macrophages [TAMs]), to these PMNs, subsequently attracting DTCs [42, 45]. The metastatic tumor cells that have colonized the brain are also famous for their ability to manipulate immune responses and escape immune surveillance by secreting a number of tumor-derived factors with immunosuppressive functions, such as cytokines, chemokines, hormones, immunosuppressive proteins, and a variety of extracellular vesicles [46,47,48,49].
After entering the brain, DTCs move slowly due to the narrow diameter of blood vessels, adhere to the vascular endothelium, and migrate to the outside of the blood vessel with the help of cytokines and/or cells such as vascular endothelial growth factor (VEGF), epidermal growth factor receptor (EGFR), Serpins, hypoxia-inducible factor (HIF), and TAMs, which will increase the chance of tumor cells passing through BBB and promote the formation of brain metastasis [50,51,52,53,54]. Most cancer cells will die immediately after entering the brain, and the brain microenvironment directly affects the results of brain metastasis [55].
4 Influence of primary tumor cells on the “premetastatic brain”
It is well established that instead of passive receivers of DTCs, metastasis organs are selectively and actively modified by the primary tumor before metastatic spread has even occurred [47], and successful metastatic colonization could occur only at certain organ sites [56]. The PMN represents an immune abnormal, pro-tumorigenic inflammatory milieu devoid of cancer cells. The inflammation-like state of PMNs accelerated the migration of primary tumor cells to PMNs [47, 57, 58]. Improved mouse and organoid models of metastasis are enabling novel insights into the mechanism of brain metastasis [59, 60]. Primary breast cancer has been shown to induce a strong inflammatory response in the “premetastatic brain”, by induce CD11b ( +) Gr1 ( +) myeloid cells to accumulate in the brain and upregulate the expression of inflammatory mediators such as S100 calnexin A (S100A)8, S100A9, serum amyloid A (SAA) 3, CCL2, COX2 and PGE2 in the premetastatic brain [61]. This inflammatory response can lead to the recruitment of additional myeloid cells such as neutrophils, which support subsequent metastatic seeding and colonization of breast-brain metastases [62] (Fig. 2). Liu et al. built a multi-organ microfluidic bionic chip platform, which consisting of two organ chip units: an upstream ‘‘lung” and a downstream ‘‘brain” unit characterized by a functional ‘‘BBB” structure, may hopefully help study the mechanism of immunity in brain metastasis in the future [60].

Mechanisms of primary tumor influence on the pre-metastatic brain. Primary tumor induces accumulation of bone marrow derived CD11bGr1 cells migrated in the premetastatic brain from the systemic circulation to form “premetastatic soil” and inflammation mediators (such as S100A8, S100A9, SAA3, and CCL2), that attract additional myeloid cells (such as neutrophils) as well as metastatic tumor cells
The dynamic changes of immune biomarkers of primary tumor also plays critical roles in this process, which can be identified as biomarkers for brain metastasis. Primary tumors with some immune biomarkers are prone to be metastatic to brain. Paratore S, et al. find that the immunoreactivity of CXCL12 and CXCR4 was significantly higher in NSCLC samples of patients with brain metastases compared to those without brain metastases, and advocate a possible role for the CXCL12/CXCR4 axis in the metastatic evolution of NSCLC, and its potential use as prognostic markers and drug targets [63]. Shih DJH, et al. identify three regions with significantly higher amplification frequencies in brain metastases from lung adenocarcinoma, including MYC, YAP1 and MMP13, and significantly more frequent deletions in CDKN2A/B, which reveals previously unknown metastatic drivers [64].
5 The bidirectional effects between metastatic cells, immune cells, and native cells
In the CNS tumor microenvironment, immune cells, tumor cells, and native cells establish connections through the secretion of cytokines, chemokines, proteases, and growth factors to affect tumor progression and metastasis [9].
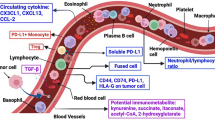
Brain metastatic cells can regulate immune cell activity by changing the intracranial immune microenvironment, thereby creating a "soil" suitable for self-colonization and growth and promoting the formation of brain metastases [65]. The ways in which brain metastatic cells manipulate immunity including (1) expressing immunosuppressive molecules such as indoleamine 2, 3-dioxygenase (IDO), programmed cell death-ligand 1 (PD-L1) [48], carbohydrate-binding protein, and galectin-1 [49]; (2) directly contact with intracranial immune cells to affect their function, for example, gap-junctional network between brain metastatic cancer cells and astrocytes was found in breast and lung brain metastases [66]; (3) recruits immunosuppressive cells into the brain by express chemokines such as granulocyte colony-stimulating factor (G-CSF) [67], ITGA3, and CXCL17 [68]. In the process of brain metastasis, the immune system of the CNS also plays a key role: (1) promote the occurrence of brain metastasis; (2) promote the local recruitment and colonization of brain metastasis by creating an immunosuppressive microenvironment [38] (Fig. 3).

The bidirectional effects between tumor cells and immune cells in microenvironment of brain metastases
5.1 CNS-myeloid cells
Myeloid origin cells comprise up to 32.7% of intra-tumoral cells in brain metastases [69]. They are not only recruited from the blood to the parenchyma, but are also derived from intraneural in situ [70].
5.1.1 Microglia
Microglia are the resident macrophages of the CNS [10], which develop from embryonic yolk sac progenitor cells [11], and are not replenished postnatally through peripheral mononuclear hematopoiesis [71, 72]. Microglia are characterized by low expression of CD45 on the cell surface and unique expression of P2RY12, SALL1, Cx3cr1, iglec-H, and Tmem119 [73, 74]. When activated, microglia can proliferate and migrate to areas of damage/inflammation [75, 76].
The immune role of microglia involves the presentation of antigens, promotes anti-tumor immune response by secrete cytokines such as tumor necrosis factor (TNF)-α, interferon (IFN)-γ, IL-1β, and IL-12, cytotoxicity via inducible nitric oxide lyase (iNOS) and superoxide, and phagocytic activity [75, 77, 78]. Microglia also express high mobility group box protein 1 (HMGB1), which facilitates antigen presentation and activation of the adaptive immune system. However, this effect is often offset by immunosuppressive microenvironment of brain metastatic, such as highly expressed of PD-L1 [79]. Microglia can also serve as active transporters and guiding rails for DTCs on their way into the brain tissue. An organotypic slice coculture model shows that microglia were attracted and accumulated at the site of the tumor cell plug, attaching themselves to the tumor cells and transporting them toward and into the tissue [80]. Microglia can also prepares the microenvironment for tumor cell invasion and colonization by upregulation of transforming growth factor (TGF)-β, IL-6 [81], IL-10 [75, 82] (modulates pro-inflammatory responses and induces local immunosuppression), C-X-C motif chemokine (CXCL)-5, CXCL-8 and CXCL-10 [38, 68] (recruit immunosuppressed neutrophils into the metastatic niche). Studies of melanoma brain metastasis have shown that microglia cells can act directly on melanoma cells to increase their malignant phenotype [83]. Microglia can also polarize into immunosuppressive M2-like phenotype and promote tumor growth and invasion by secreting growth factors or facilitating angiogenesis [84]. Microglia differentiated into M2-like phenotype can also form a dense protective layer around tumor cells to prevent the killing of brain metastases by TILs [85].
Brain metastases also exerted significant morphological changes on microglia cells. Melanoma cells can increase microglia cells proliferation, induced matrix metalloproteinase (MMP)-2 activation, and cell migration [83]. Metastatic breast cancer cells in the brain can produce Neurotrophin-3 (NT-3), which can downmodulate the cytotoxic response of microglia and promoting growth of brain metastases [86] (Fig. 4).

The dual mechanism of tumor suppressor and tumor promotion of microglia in brain metastasis
5.1.2 Tumor-associated macrophages
TAMs in the CNS immune microenvironment are highly branched, large, and motionless cells compared with small and high mobility blood-derived macrophages [87]. TAMs in the CNS immune microenvironment include embryo-derived macrophages and bone marrow-derived macrophages (BMDMs) recruited from the peripheral blood circulation and replenished through monocytosis [24, 88,89,90,91,92]. CD49D/ITGA4 was recently identified as a marker for BMDMs [93].
TAMs can phagocytose DTCs that enter the CNS and function as immune surveillance in the CNS [94]. However, the function of TAMs shows high plasticity, and mainly promoting brain metastasis [10, 95]. TAMs can co-expression of inhibitory molecules such as PD-L1 and PD-L2 and the release of inhibitory chemokines such as IL-10, TGF-β, CCL5, CCL20, and CCL22, which prevent the activation of adaptive immune responses in various cancers [9, 93, 96]. TAMs also secrete nitric oxide (NO), which inhibits the signaling of JAK3, STAT5, ERK, AKT, and as a result prevents the production of IL-2, thereby impairing the generation of cytotoxic and memory T cells [97]. TAMs can promote melanoma cell invasion in brain through the high expression of MMP-3 [98]. In breast cancer brain metastasis, TAMs can promote the formation of brain metastases by producing cathepsin S and the hydrolyzing the junctional adhesion molecule-B (JAM-B) [76].
Under the actions of different cytokines, macrophages are divided into different phenotypes, including M1 with antitumor properties and M2 with tumor-promoting properties [99]. However, activation of TAMs in situ are much more complex and does not necessarily conform to the M1/M2 paradigm [100, 101], so studies have used CD163 expression to separate putative "M2-like" from "M1-like" macrophages [100, 102]. The polarization of macrophages varies between different regions of the brain metastases. In a preclinical mouse model of intracranial breast cancer metastasis, flow cytometry analysis revealed significant differences in the activation state of metastasis-associated macrophages (MAMs) at the brain parenchyma as compared to the dura, and that those differences are causally linked to the metastatic site-specific cancer cell molecular profiles, such as inflammation-related pathways, NF-kB1 activity and cytokine profiles. The most significantly upregulated cytokine in brain parenchyma- versus dura-derived cancer cells was Lymphotoxin β and increased NF-κB1 activity, which may be directly involved in M2 polarization of parenchymal MAMs [103]. MPO + IBA1 + macrophages were also found in the microenvironment of brain tumors, and these cells were CD163 − and P2Y12 − , with signatures associated with reactive oxygen species biosynthesis and phagosome formation (indicative of cytotoxicity) and HIF1α signaling [6]. Furthermore, these MPO + IBA1 + macrophages were significantly associated with prolonged survival in patients [6]. Although rare in the TME, MPO + macrophages may have antitumor properties, as characteristics and are associated with a better prognosis.
5.1.3 Myeloid-derived suppressor cells
Medullary precursor cells (MPCs) mature blocked under tumor conditions, and thus stay at various stages of differentiation, become myeloid-derived suppressor cells (MDSCs) and are recruited to tumor sites and expanded to exert immunosuppressive functions. MDSCs are the precursor cells of dendritic cells (DCs), macrophages and granulocytes, and have the ability to significantly inhibit immune response [104]. CD11b is a marker of MDSCs. In a mouse model of breast cancer brain metastasis, primary breast tumors can induce CD11b + Gr1 + myeloid cell activation and express chemokines S100A8/9 in the brain, which can attract tumor cells, bone marrow-derived suppressor cells, and neutrophils for further recruitment into the brain [105]. Macrophage inflammatory factor-1γ (MIP-1γ) (also known as CCL9) is highly expressed in CD11b + Gr1 + myeloid cells of breast cancer and melanoma mice and promotes the survival and metastasis of tumor cells in a TGF-β-dependent manner [106].
5.1.4 Neutrophils
Significant accumulation containing neutrophils was also found in the immune microenvironment of brain metastases [27, 68]. It has been shown that S100 calbindin A (S100A)-8 and S100A9 are upregulated in the premetastatic niche in the brain, leading to the recruitment of neutrophils [105]. Brain metastases specific upregulation of CD177, ITGA3 and CXCL17, which modulates neutrophil recruitment and activation [68] and negative affects T cell proliferation [107, 108]. In breast cancer brain metastases, neutrophils can secrete the S100A8/9 to promote tumor cell colonization within the CNS [62, 105]. It has also been found that the receptor tyrosine kinase c-MET was upregulated in neutrophils in a brain metastases-specific manner [68], which is associated with the recruitment of immunosuppressive cell and pro-metastatic tumor microenvironment [108, 109].
Enhancer of zeste homolog 2 (EZH2) is highly expressed and phosphorylated in brain metastatic cells. Phosphorylation of EZH2 at Y696 switches EZH2's function from a methyltransferase to a transcription factor that increases c-JUN expression, and consequently up-regulates protumor genic inflammatory cytokines, including granulocyte colony-stimulating factor (G-CSF), which recruits immunosuppressive neutrophils into the brain to drive metastasis outgrowth [67]. Single-cell sequencing of lung cancer brain metastases revealed that a subset of ROS-producing neutrophils showed upregulation of the HIF-1 signaling pathway, expressing HIF1A, lactate dehydrogenase A (LDHA), and VEGF-A, which may be involved in tumor angiogenesis in brain malignancies [8].
In addition, neutrophils have prognostic value in patients with brain metastases, and a high ratio of neutrophils to lymphocytes in peripheral blood is associated with shorter survival time in patients with brain metastases. This was true in patients with surgically resected brain metastases [110,111,112].
5.2 Tumor-infiltrating lymphocytes
For primary brain tumors, such as glioblastoma (GBM), T cells account for less than 0.25% of the cells isolated in human GBM biopsies, with less than a quarter of TILs are identified as anti-tumor CD8( +) TILs [113]. Furthermore, these TILs cells often have impaired effector functions and exhausted phenotypes [68]. Studies have shown that the infiltration of brain metastases with high levels of effector CD3 + , cytotoxic CD8 + or memory CD45RO + T cells were significantly associated with better prognosis of glioma patients [114, 115]. Less abundant CD4 + central memory T (TCM) and CD8 + terminally differentiated effector T (Temra) and more NKT and CD8 + effector memory T (TEM) cells were found in lung cancer brain metastases compared with brain primary tumors [8]. CD8 + exhausted T (TEX) and CD4 + TEM cells were also observed in brain metastases. The differences in cellular proportions and genetic signatures between primary brain tumors and brain metastases indicates that T cells have been reprogrammed by metastatic tumors. The concentration of T cells in brain metastases can vary from zero to very high depending on the patient and the type of primary tumor [116, 117]. For example, melanoma brain metastases showed more abundant lymphocyte infiltrate than breast cancer brain metastases, with a sizeable CD8 + T cell fraction [68, 115]. Clinical studies have also shown that patients with melanoma brain metastases can benefit from immunotherapy, which may be related to the immune microenvironment of their brain metastases [118, 119].
The TILs of brain metastases are different from those of primary tumors, with fewer overall cells and an immunosuppressive phenotype in the tumor microenvironment [120]. Decreased lymphocyte infiltration and downregulation of vascular cell adhesion molecule 1 (VCAM1) were detected in brain metastases compared with primary lung cancer specimens [121]. Studies of brain metastases from lung cancer shown that brain metastases have significantly fewer T cell clones than paired primary lesions, despite the brain metastases had higher non-synonymous mutation burdens than primary lesions [122]. And the number of CD8-positive T cells in brain metastases were found not correlate with the variable amounts of MHC class I expression on brain metastases, suggesting that these T cells are not antigen-specific [123]. In addition, only a small proportion of T cells did demonstrate sings of cytotoxic activation such as release of granzyme-B [123]. Several studies showed that intra-tumoral CD8-positive T cells percentage was marginally associated with better prognosis. Zhou J, et al. demonstrate a discrepancy in PD-L1 expression and CD8-positive T cells density between primary lung cancers and their corresponding brain metastases, and find that low stromal CD8-positive T cells numbers in brain metastases were associated with significantly shorter overall survival (OS) compared to high stromal CD8-positive T cells counts [124]. Vilariño N, et al. report that Lower PD-L1 expression and less CD8-positive T cells infiltration are found in brain metastases compared with matched NSCLC primary tumors, suggesting an immunosuppressive microenvironment in the brain [120].
Regulatory T cells (Tregs) accumulation is one of the characteristics of TME in brain metastases [92]. Under normal circumstances, the body is in a state of T-helper 1 (Th1)/T-helper 2 (Th2) balance for correct functioning of the immune system. When tumors occur, the body effectively stimulates and mediates cellular immunity, activates cytotoxic T cells and macrophages, and produces antitumor effects by drifting to Th1 from the Th1/Th2 balance [125]. Tregs in the metastatic tumor microenvironment expression of inhibitory proteins and receptors, such as programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte associated protein 4 (CTLA4), T cell immunoglobulin and mucin-domain containing-3 (TIM-3), lymphocyte-activation gene 3 (LAG3), forkhead box p3 (FOXP3), inducible T-cell co-stimulator (ICOS), and T cell immunoreceptor with Ig and ITIM domains (TGIT) [126, 127]. Tregs can secreting immunosuppressive cytokines such as IL-4, IL-10, IL-35 and TGF-β [128,129,130].
5.3 Other immune cells
Clinical brain metastasis tumor samples show a more significant accumulation of lymphocytes relative to intracranial primary tumors, such as gliomas [68, 131]. In addition to T lymphocytes, immune cells such as NK cells [28], B lymphocytes, and mast cells (MCs) [29] are founded in the immune microenvironment of brain metastases. NK cells have the ability to lyse tumor cells in an antigen-independent manner. However, the anti-tumor activity of NK cells is usually inhibited due to the release of anti-inflammatory molecules (such as TGFβ, cyclooxygenase (COX), and prostaglandin E2, etc.) by tumor cells [132]. M0 and M2 cells express a membrane-bound form of IL-18, which involved in the acquisition of CCR7 by NK cells. The M0 and M2 cells, but not M1 macrophages (macrophages that adopt a protumor phenotype), have been shown to induced strong activation of resting NK cells [133]. However, a definitive link between TAM polarization and NK cell activation has not been demonstrated in brain metastases. MCs exert immunosuppressive effects by secreting the cytokines IL-8 and IL-10, as well as secreting VEGF and MMP-2 to promote metastasis and angiogenesis in brain metastases [134].
5.4 Astrocytes
Although astrocytes are not immune cells, studies have shown that they can function as antigen-presenting cells (APCs) in the immunological process of brain metastasis [33], and also modulate the innate and adaptive immune system to benefit metastatic tumor cells [35], which complete the immune microenvironment of metastatic tumor cells.
Astrocytes were originally thought to be the "glue" in the brain, account for 30% of CNS cells [135], provide the structural scaffold necessary for neuronal function, and help prevent the intracranial metastasis of tumor cells [136]. The vascular basement membrane (VBM) of the CNS may be the "soil" for the development of brain metastases as successful colonization of DTCs in brain tissue tends to enclose vascular growth by adhesion of L1 cell adhesion molecule (L1CAM) [50], which is called “vascular cooption” [53]. Studies have shown that astrocytes paly a site-specific role during anti-brain metastasis during this process. Astrocytes can release proenzyme activator, activate proenzyme released by neuronal cells and convert it into plasmin, which cleaves and degrades L1CAM and detaches tumor cells from the vascular wall to prevent brain metastasis. Plasmin can also cleave and activate Fas receptor released by astrocytes and activate the Fas-associated death domain protein (FADD) pathway in brain metastases causing tumor cell apoptosis [50]. However, brain metastatic cells from lung cancer and breast cancer can prevent this process by express high levels of anti-Plasmin serpins, including neuroserpin and serpin B2 [50].
Astrocytes can promote the invasion of brain metastases by producing extracellular matrix degradation factors such as heparinase and MMP [25, 137]. Studies have shown that astrocyte-derived exosomes mediate an intercellular transfer of miRNAs target PTEN, an important tumor suppressor, to metastatic tumor cells [138]. This result in an organ-specific phenomenon that primary tumor cells with normal expression of PTEN losing PTEN expression (both mRNA and protein level) after dissemination to the brain, but not to other organs [139].
The inflammatory environment caused by brain metastasis can activate astrocytes to further promote tumor growth. Astrocytes were found to undergo morphological changes in response to brain metastases, switching to a reactive phenotype [140]. Reactive astrocytes promote brain metastasis by altering the tumor microenvironment through signal transducer and activator of transcription 3 (STAT3) and are a promising target for the treatment of brain metastasis [141]. Metastatic breast tumor cells in the brain highly expressed IL-1β, which then ‘activated’ surrounding astrocytes and enhances Jagged1 (JAG1) expression in reactive astrocytes through NF-κB pathway. The activation of JAG1 in astrocytes triggers the Notch signaling in tumor cells through cell–cell interaction, then the Notch signaling promotes the self-renewal and metastatic growth of breast cancer stem-like cells (CSCs) through up-regulation of Hes Family BHLH Transcription Factor 5 (HES5) [142]. Breast and lung metastases in the CNS express protocadherin (PCDH)7, which promotes the assembly of gap-junctional network between brain metastatic cancer cells and astrocytes, and then activating the astrocytes by transferring the second messenger cGAMP. The activated astrocytes can production of inflammatory cytokines such as IFN-α and TNF, which activate the STAT1 and NF-κB pathways in brain metastatic cells, thereby supporting tumor growth and chemoresistance [66].
6 Progress in immunotherapy of brain metastases
Immune checkpoint inhibitors (ICIs) have made some breakthroughs in the treatment of brain metastases [143], and the nivolumab, ipilimumab, and pembrolizumab has been reported to improve the prognosis of some patients with brain metastases such as metastatic melanoma and lung cancer [144, 145]. In addition, CART cell therapy, oncolytic viruses, and tumor vaccines also have some room for development for the treatment of brain metastases. Some immune system activating cytokines are also being studied as potential therapeutic strategies for brain metastases.
6.1 Immune checkpoint inhibitors
Different from traditional chemotherapy and radiotherapy treatments, ICIs work or not has little correlation with whether the drug itself can penetrate the blood–brain barrier, but the efficacy to mobilize more cytotoxic T cells to the brain metastases [146].
In a randomized, open-label, phase 3 trial (OAK), in patients with a history of brain metastases, the median OS was longer (16.0 vs 11.9 months; hazard ratio = 0.74; 95% CI: 0.49–1.13) and the probability of developing new symptomatic brain lesions was lower with atezolizumab than with docetaxel [147]. Patients with brain metastases from NSCLC were also included in CHECKMATE-017, CHECKMATE-057, and KEYNOTE-010, but did not demonstrate a survival benefit following treatment with ICIs [148, 149].
Clinical studies of ICIs for first-line treatment of patients with brain metastases from NSCLC have also achieved some promising results. In the KEYNOTE-189 study, which involved the most patients with brain metastases (n = 108) so far, an OS benefit was observed in the pembrolizumab-combination group versus the placebo-combination group in the subgroups of patients with brain metastases. HR for OS with pembrolizumab-combination versus placebo-combination was improved in patients with (0.41; 95% CI, 0.24 to 0.67) than without (0.59; 95% CI, 0.46 to 0.75) brain metastases [150]. A pooled analysis of KEYNOTE-021, KEYNOTE-189, and KEYNOTE-407 shown that in patients with brain metastases, median OS was 18.8 months with pembrolizumab plus chemotherapy and 7.6 months with chemotherapy (HR 0.48 [95% CI, 0.32–0.70]), and median PFS was 6.9 months and 4.1 months (HR 0.44 [95% CI, 0.31–0.62]), respectively [151]. A phase 2 trial of pembrolizumab in patients with NSCLC or melanoma with untreated brain metastases (ClinicalTrials.gov NCT02085070) showed that 29.7% (11/37) of the patients with PD-L1 expression ≥ 1% (N = 37) had brain metastasis response, and no treatment response was observed in the group with PD-L1 less than 1% or non-evaluable patients. Furthermore, the 2-year OS rate was 34% in PD-L1-positive NSCLC patients with brain metastases (compared with 14.3% seen in previous observational studies), suggesting that pembrolizumab is safe and effective in the treatment of this challenging population [152]. In another phase II study, the disease control rate of ipilimumab in melanoma brain metastases (MBM) was 24% (12/51), intracranial response was consistent with extracranial response, and the two-year survival rate of the cohort of patients with brain metastases was 26% [153]. Brain metastases are common and associated with poor clinical outcomes in patients with SCLC. However, the role of ICIs in patients with SCLC brain metastases remains uncertain [154]. Subgroup analyses of IMpower133 shown that in brain metastases from SCLC, adding atezolizumab to chemotherapy as first-line therapy did not increase OS [155].
For patients with brain metastases, the combination of CTLA-4 and PD-1 inhibitors has also achieved good efficacy. An open-label, multicenter, phase 2 study, among 94 patients with a median follow-up of 14.0 months, the combination of ipilimumab and nivolumab (NIVO + IPI) in patients with untreated, asymptomatic MBM from the CheckMate-204 study showed the rate of intracranial clinical benefit was 57%, and the best benefit with an estimated 12-month survival reaching 81.5% [156]. A recent update analysis of the study reported (median follow-up of 20.6 months) an intracranial clinical benefit rate (CBR; proportion of pts with complete response [CR] + partial response [PR] + stable disease [SD] ≥ 6 months) of 58.4% [157]. Intracranial antitumor activity was observed with NIVO + IPI in pts with symptomatic MBM. Among 11 patients with untreated, symptomatic MBM, the intracranial objective response rate was 16.7% and the CBR was 22.2% at a median follow-up of 5.2 months [157]. 3-year outcomes of treatment with combination NIVO + IPI for patients with active, untreated MBM, with 101 asymptomatic pts and 18 symptomatic pts, shown the OS of 72% and 37%, respectively (median follow-up: 34 months, asymptomatic cohort; 7.5 months, symptomatic cohort) [158].
6.2 Combination of immunotherapies and radiation
Stereotactic radiosurgery (SRS) or whole-brain radiation therapy has been the mainstay for the treatment of patients with symptomatic brain metastases. Radiation can induce immunogenic cell death (ICD) of tumor cells, release of damage-associated molecular patterns or stress molecules and act as "tumor vaccine in situ" [159], further induce adaptive cellular immune response, and produce "distant effect" at the irradiated and non-irradiated sites [160]. Radiotherapy can up-regulate the expression of MHC molecules and immune costimulatory molecules in tumors [161], increase the expression of proinflammatory cytokines such as TNF-α, IL-1β and adhesion molecules, promote the infiltration of T cells in the tumor microenvironment [162], as well as remove Tregs cells [163]. Studies have shown that PD-L1 expression on tumor cells is found to be upregulated after 24 h of radiotherapy, which sensitizes them to anti-PD-L1 therapy [164], and the inflammatory response resulting from radiotherapy may persist for several days [165]. Therefore, radiation is also considered as an immune adjuvant, and the response varies with the time and the dose of fractionation regimen [166].
In 2012, a retrospective analysis of 77 patients with melanoma brain metastases showed that using ipilimumab in a supportive treatment paradigm of SRS was significantly improved the survival time to 21.3 months, as compared to 4.9 months in the non-ipilimumab group [167]. The benefit of combination of SRS and ICIs for melanoma brain metastases was subsequently demonstrated in multiple studies [168,169,170,171,172]. As the combination of SRS and ICIs is becoming more widely used in the treatment of brain metastases, there is a need to validate the optimal timing of these two therapies relative to one another. In a study of patients with melanoma brain metastases, SRS before or during ipilimumab treatment was associated with a better 1-year survival rate (65%/56% versus 40%) and a smaller risk of local intracranial recurrence within 1 year (64%/69% versus 92%) compared with SRS after ipilimumab [173]. Benefits of concurrent SRS and ICIs have also shown in the treatment of brain metastases from NSCLC [174]. An international meta-analysis compares the safety and efficacy of patients with brain metastases treated with SRS/ICI, either concurrently or non-concurrently. The result showed that the concurrently administration of SRS/ICI improved 1-year OS (64.6% versus 51.6%, p < 0.001) and 1-year regional brain control (38.1% and 12.3%, p = 0.049) versus sequential therapy [175].
Data from early clinical studies such as KEYNOTE-010, KEYNOTE-189, CHECKMATE-017 and CHECKMATE-057 suggest that ICIs may be an effective for patients with brain metastases [148, 149, 176]. The updated results of CheckMate-204 show a high rate of durable intracranial responses, further supporting NIVO + IPI as a first-line treatment in this challenging population [156,157,158]. SRS combined with ICIs has shown survival benefit in brain metastases from melanoma and NSCLC [167,168,169,170, 174]. However, further study is needed to explore the clinical efficacy of ICIs in the treatment of brain metastases, and ongoing trials using immunotherapy in the setting of brain metastases are outlined in Table 2.
6.3 Tumor vaccines
The anti-tumor vaccines including peptide vaccines and tumor antigen-activated DCs are engineered to discriminately target cancer by induce adaptive immune, and these immunotherapeutic approaches have demonstrated some efficacy against intracranial tumors [177,178,179].
DC vaccines are reinfused into patients by editing antigens and expanded culturing in vitro, which show the ability to evolve with tumors and increase the diversity of antigens presented over time [180]. In a phase I/II clinical study, 3 of 20 patients with metastatic melanoma were treated with DC incubated with irradiated melanoma cells and GM-CSF, and 1 of these patients was still in disease remission 3 years after treatment [178]. In another study of patients with metastatic renal cancer, one patient with four brain metastases showed complete and durable regression of brain metastases after treated with DC vaccine [179]. Due to the uncertain efficacy of cancer vaccines and the lack of large randomized controlled trial (RCT), only few sporadic reports can provide a reference for clinical decision-making. A phase IIa trial studies the efficiency of Anti-HER2/HER3 DC vaccine in the treatment of brain metastases from breast cancer is ongoing (NCT04348747).
6.4 Other potential immunotherapy strategies for brain metastases
CART cell therapy has been widely used in hematological tumors and gradually applied in solid tumors, but it is still challenging for the treatment of brain metastases. However, off-target and side effect such as cerebral edema pose serious challenges in the treatment of brain metastases using CAR T-cell therapy [181]. Local intracranial delivery of HER2-CARs has showed potent in vivo antitumor activity in orthotopic xenograft models [182]. A phase I trial studies the side effects and best dose of HER2-CAR T cells via intraventricular administration in treating patients with cancer that has spread to the brain or leptomeninges is ongoing (NCT03696030).
Studies on the immune mechanisms of tumor brain metastasis also provide some potential therapeutic targets. systemic prophylactic administration of a toll-like receptor (TLR) 9 agonist, CpG-C, in the brain in three tumor models in mice have shown therapeutic effects against brain metastases. Intravital imaging showed that CpG-C can be taken up by endothelial cells, astrocytes, and microglia without affecting BBB integrity and tumor brain extravasation. CpG-C-activated microglia cells then contact, kill, and phagocytize tumor cells in the early stages of tumor brain invasion more than nonactivated microglia, which may provide a new intervention against brain metastasis [183].
7 Conclusion and looking ahead
The occurrence and development of tumor brain metastasis is a complex biological process with multiple stages and multiple cell interactions. Primary tumors can influence and engineer the PMN of the CNS more conducive to DTCs colonization and survival, and the unique immune microenvironment of the CNS also plays an important role in the process of brain metastatic cascade. CNS-myeloid cells play a dominant role in promoting the development of brain metastases by creating an immunosuppressive microenvironment. Astrocytes and neutrophils may play a dual role of promotion and inhibition during the development of brain metastases. DTCs that successfully enter and colonize the CNS can further affect the intracranial immune microenvironment by secreting various cytokines and promote the progression of brain metastases.
With our growing understanding of immuno-molecular mechanism of brain metastasis, new therapies target immune system provide additional strategies toward the treatment of brain metastases. At present, the immunotherapy for brain metastases is still in the exploratory stage, mainly including many difficulties and challenges. Firstly, the establishment of animal model of brain metastasis is one of the biggest challenges. Some of the top upregulated (VIM, LGALS3, IFITM2, LYZ) and downregulated (GFP34, CD81) genes in mouse brain metastasis-associated CNS-myeloid cells showed similar trends in humans, which indicates that the mice model is clinically relevant [38]. However, studies in mice may not be applicable when human/tissue data were correlated. Application of a gating scheme used on mouse samples to human brain metastasis mass cytometry data showed that the human brain metastasis samples were likewise infiltrated by a diverse array of myeloid and T cell subsets compared to mice samples [38]. In addition, there are great technical difficulties in simulation of the BBB and the formation of intracranial metastases. Secondly, there are differences in the immune microenvironment of brain metastases from different primary tumors. Thirdly, the development of biomarkers to predict the efficacy of specific immunotherapies for patients with brain metastases is also a hot research direction. Better and more precise assessment of the immune microenvironment of brain metastases will provide a reference for precise treatment and individualized treatment. Fourthly, merging different therapies and evaluating their combined efficacy for tumor brain metastases is still needed.
In conclusion, the mechanism of the occurrence and development of tumor brain metastases are still largely unaware at the moment. Moving forward, more in-depth studies are needed to discover new therapeutic targets and expand the role of immunotherapy in the treatment of brain metastases.
Availability of data and materials
Not applicable.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA. 2020;70(1):7–30.
Goncalves P, Peterson S, Vigneau F, Shore R, Quarshie W, Islam K, et al. Risk of brain metastases in patients with nonmetastatic lung cancer: analysis of the metropolitan detroit surveillance, epidemiology, and end results (SEER) data. Cancer. 2016;122(12):1921–7.
Takahashi T, Yamanaka T, Seto T, Harada H, Nokihara H, Saka H, et al. Prophylactic cranial irradiation versus observation in patients with extensive-disease small-cell lung cancer: a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18(5):663–71.
Ostrom QT, Wright CH, Barnholtz-Sloan JS. Brain metastases: epidemiology. Handb Clin Neurol. 2018;149:27–42.
Habbous S, Forster K, Darling G, Jerzak K, Holloway CMB, Sahgal A, et al. Incidence and real-world burden of brain metastases from solid tumors and hematologic malignancies in Ontario: a population-based study. Neurooncol Adv. 2021;3(1):vdaa178.
Yuzhalin AE, Yu D. Brain metastasis organotropism. Cold Spring Harb Perspect Med. 2020;10(5):a037242.
Peters S, Bexelius C, Munk V, Leighl N. The impact of brain metastasis on quality of life, resource utilization and survival in patients with non-small-cell lung cancer. Cancer Treat Rev. 2016;45:139–62.
Sun HF, Li LD, Lao IW, Li X, Xu BJ, Cao YQ, et al. Single-cell RNA sequencing reveals cellular and molecular reprograming landscape of gliomas and lung cancer brain metastases. Clin Transl Med. 2022;12(11):e1101.
You H, Baluszek S, Kaminska B. Immune microenvironment of brain metastases-are microglia and other brain macrophages little helpers? Front Immunol. 2019;10:1941.
Madore C, Yin Z, Leibowitz J, Butovsky O. Microglia, lifestyle stress, and neurodegeneration. Immunity. 2020;52(2):222–40.
Prinz M, Erny D, Hagemeyer N. Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol. 2017;18(4):385–92.
Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci. 2017;20(2):136–44.
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337–41.
Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212(7):991–9.
Rangroo Thrane V, Thrane AS, Plog BA, Thiyagarajan M, Iliff JJ, Deane R, et al. Paravascular microcirculation facilitates rapid lipid transport and astrocyte signaling in the brain. Sci Rep. 2013;3:2582.
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4(147):147ra11.
Abbott NJ, Pizzo ME, Preston JE, Janigro D, Thorne RG. The role of brain barriers in fluid movement in the CNS: is there a “glymphatic” system? Acta Neuropathol. 2018;135(3):387–407.
Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of CNS immune privilege. Trends Immunol. 2015;36(10):569–77.
Kida S, Pantazis A, Weller RO. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol Appl Neurobiol. 1993;19(6):480–8.
Raper D, Louveau A, Kipnis J. How do meningeal lymphatic vessels drain the CNS? Trends Neurosci. 2016;39(9):581–6.
Song E, Mao T, Dong H, Boisserand LSB, Antila S, Bosenberg M, et al. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature. 2020;577(7792):689–94.
Li W, Chen D, Liu N, Luan Y, Zhu S, Wang H. Modulation of lymphatic transport in the central nervous system. Theranostics. 2022;12(3):1117–31.
Lan YL, Wang H, Chen A, Zhang J. Update on the current knowledge of lymphatic drainage system and its emerging roles in glioma management. Immunology. 2023;168(2):233–47.
Winkler F. The brain metastatic niche. J Mol Med (Berl). 2015;93(11):1213–20.
Shumakovich MA, Mencio CP, Siglin JS, Moriarty RA, Geller HM, Stroka KM. Astrocytes from the brain microenvironment alter migration and morphology of metastatic breast cancer cells. Faseb j. 2017;31(11):5049–67.
Bieńkowski M, Preusser M. Prognostic role of tumour-infiltrating inflammatory cells in brain tumours: literature review. Curr Opinion Neurol. 2015;28(6):647–58.
Sagiv J, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015;10(4):562–73.
Fares J, Fares MY, Fares Y. Natural killer cells in the brain tumor microenvironment: defining a new era in neuro-oncology. Surg Neurol Int. 2019;10:43.
Amit M, Laider-Trejo L, Shalom V, Shabtay-Orbach A, Krelin Y, Gil Z. Characterization of the melanoma brain metastatic niche in mice and humans. Cancer Med. 2013;2(2):155–63.
Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med. 2018;24(2):144–53.
Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. 2017;18(2):123–31.
Liu Y, Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med (Berl). 2016;94(5):509–22.
Rostami J, Fotaki G, Sirois J, Mzezewa R, Bergström J, Essand M, et al. Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson’s disease brain. J Neuroinflammation. 2020;17(1):119.
Sutter PA, Crocker SJ. Glia as antigen-presenting cells in the central nervous system. Curr Opin Neurobiol. 2022;77:102646.
Priego N, Valiente M. The potential of astrocytes as immune modulators in brain tumors. Front Immunol. 2019;10:1314.
van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res. 2011;728(1–2):23–34.
Deczkowska A, Amit I, Schwartz M. Microglial immune checkpoint mechanisms. Nat Neurosci. 2018;21(6):779–86.
Guldner IH, Wang Q, Yang L, Golomb SM, Zhao Z, Lopez JA, et al. CNS-native myeloid cells drive immune suppression in the brain metastatic niche through Cxcl10. Cell. 2020;183(5):1234–48.e25.
Sophie BA, Barbara K, Georg W, Orsolya R, Gerda R, Adelheid W, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015;17(8):1064–75.
Tiwary S, Morales JE, Kwiatkowski SC, Lang FF, Rao G, McCarty JH. Metastatic brain tumors disrupt the blood-brain barrier and alter lipid metabolism by inhibiting expression of the endothelial cell fatty acid transporter Mfsd2a. Sci Rep. 2018;8(1):8267.
Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8(2):98–101.
Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–7.
Carvalho R, Paredes J, Ribeiro AS. Impact of breast cancer cells´ secretome on the brain metastatic niche remodeling. Semin Cancer Biol. 2020;60:294–301.
Percy DB, Ribot EJ, Chen Y, McFadden C, Simedrea C, Steeg PS, et al. In vivo characterization of changing blood-tumor barrier permeability in a mouse model of breast cancer metastasis: a complementary magnetic resonance imaging approach. Invest Radiol. 2011;46(11):718–25.
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37.
Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. 2016;165(1):45–60.
Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. 2017;17(5):302–17.
Herrera-Rios D, Mughal SS, Teuber-Hanselmann S, Pierscianek D, Sucker A, Jansen P, et al. Macrophages/Microglia represent the major source of indolamine 2,3-dioxygenase expression in melanoma metastases of the brain. Front Immunol. 2020;11:120.
Sedgwick AJ, Ghazanfari N, Constantinescu P, Mantamadiotis T, Barrow AD. The Role of NK Cells and Innate Lymphoid Cells in Brain Cancer. Front Immunol. 2020;11:1549.
Valiente M, Obenauf A, Jin X, Chen Q, Zhang X, Lee D, et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014;156(5):1002–16.
Ni W, Chen W, Lu Y. Emerging findings into molecular mechanism of brain metastasis. Cancer Med. 2018;7(8):3820–33.
Voutouri C, Kirkpatrick ND, Chung E, Mpekris F, Baish JW, Munn LL, et al. Experimental and computational analyses reveal dynamics of tumor vessel cooption and optimal treatment strategies. Proc Natl Acad Sci U S A. 2019;116(7):2662–71.
Carbonell WS, Ansorge O, Sibson N, Muschel R. The vascular basement membrane as “soil” in brain metastasis. PLoS ONE. 2009;4(6):e5857.
Wenes M, Shang M, Di Matteo M, Goveia J, Martin-Perez R, Serneels J, et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab. 2016;24(5):701–15.
Pozzi S, Scomparin A, Ben-Shushan D, Yeini E, Ofek P, Nahmad A, et al. MCP-1/CCR2 axis inhibition sensitizes the brain microenvironment against melanoma brain metastasis progression. JCI insight. 2022;7(17):e154804.
Hart IR, Fidler IJ. Role of organ selectivity in the determination of metastatic patterns of B16 melanoma. Cancer Res. 1980;40(7):2281–7.
Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9(4):285–93.
Ordóñez-Morán P, Huelsken J. Complex metastatic niches: already a target for therapy? Curr Opin Cell Biol. 2014;31:29–38.
Francia G, Cruz-Munoz W, Man S, Xu P, Kerbel RS. Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat Rev Cancer. 2011;11(2):135–41.
Liu W, Song J, Du X, Zhou Y, Li Y, Li R, et al. AKR1B10 (Aldo-keto reductase family 1 B10) promotes brain metastasis of lung cancer cells in a multi-organ microfluidic chip model. Acta Biomater. 2019;91:195–208.
Liu Y, Kosaka A, Ikeura M, Kohanbash G, Fellows-Mayle W, Snyder LA, et al. Premetastatic soil and prevention of breast cancer brain metastasis. Neuro Oncol. 2013;15(7):891–903.
Yanagi H, Watanabe T, Nishimura T, Hayashi T, Kono S, Tsuchida H, et al. Upregulation of S100A10 in metastasized breast cancer stem cells. Cancer Sci. 2020;111(12):4359–70.
Paratore S, Banna G, D’Arrigo M, Saita S, Iemmolo R, Lucenti L, et al. CXCR4 and CXCL12 immunoreactivities differentiate primary non-small-cell lung cancer with or without brain metastases. Cancer Biomarkers : Sec A Dis Markers. 2011;10(2):79–89.
Shih D, Nayyar N, Bihun I, Dagogo-Jack I, Gill C, Aquilanti E, et al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nat Genet. 2020;52(4):371–7.
McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science (New York, NY). 2016;351(6280):1463–9.
Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533(7604):493–8.
Zhang L, Yao J, Wei Y, Zhou Z, Li P, Qu J, et al. Blocking immunosuppressive neutrophils deters pY696-EZH2-driven brain metastases. Sci Transl Med. 2020;12(545):eaaz5387.
Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell. 2020;181(7):1643–60.e17.
Andreou T, Rippaus N, Wronski K, Williams J, Taggart D, Cherqui S, et al. Hematopoietic stem cell gene therapy for brain metastases using myeloid cell-specific gene promoters. J Natl Cancer Inst. 2020;112(6):617–27.
Prodinger C, Bunse J, Krüger M, Schiefenhövel F, Brandt C, Laman JD, et al. CD11c-expressing cells reside in the juxtavascular parenchyma and extend processes into the glia limitans of the mouse nervous system. Acta Neuropathol. 2011;121(4):445–58.
Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–51.
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10(12):1538–43.
Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13(11):1118–28.
Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–9.
He BP, Wang JJ, Zhang X, Wu Y, Wang M, Bay BH, et al. Differential reactions of microglia to brain metastasis of lung cancer. Mol Med (Cambridge, Mass). 2006;12(7–8):161–70.
Sevenich L, Bowman R, Mason S, Quail D, Rapaport F, Elie B, et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol. 2014;16(9):876–88.
Medawar PB. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol. 1948;29(1):58–69.
Raza M, Prasad P, Gupta P, Kumar N, Sharma T, Rana M, et al. Perspectives on the role of brain cellular players in cancer-associated brain metastasis: translational approach to understand molecular mechanism of tumor progression. Cancer Metastasis Rev. 2018;37(4):791–804.
Poggi A, Musso A, Dapino I, Zocchi MR. Mechanisms of tumor escape from immune system: role of mesenchymal stromal cells. Immunol Lett. 2014;159(1–2):55–72.
Pukrop T, Dehghani F, Chuang HN, Lohaus R, Bayanga K, Heermann S, et al. Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia. 2010;58(12):1477–89.
Tsukamoto H, Fujieda K, Miyashita A, Fukushima S, Ikeda T, Kubo Y, et al. Combined blockade of IL6 and PD-1/PD-L1 signaling abrogates mutual regulation of their immunosuppressive effects in the tumor microenvironment. Cancer Res. 2018;78(17):5011–22.
Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. 2015;128:95–139.
Izraely S, Ben-Menachem S, Sagi-Assif O, Telerman A, Zubrilov I, Ashkenazi O, et al. The metastatic microenvironment: melanoma-microglia cross-talk promotes the malignant phenotype of melanoma cells. Int J Cancer. 2019;144(4):802–17.
Yuan A, Hsiao YJ, Chen HY, Chen HW, Ho CC, Chen YY, et al. Opposite effects of M1 and M2 macrophage subtypes on lung cancer progression. Sci Rep. 2015;5:14273.
Fitzgerald DP, Palmieri D, Hua E, Hargrave E, Herring JM, Qian Y, et al. Reactive glia are recruited by highly proliferative brain metastases of breast cancer and promote tumor cell colonization. Clin Exp Metas. 2008;25(7):799–810.
Louie E, Chen XF, Coomes A, Ji K, Tsirka S, Chen EI. Neurotrophin-3 modulates breast cancer cells and the microenvironment to promote the growth of breast cancer brain metastasis. Oncogene. 2013;32(35):4064–77.
Chen Z, Ross JL, Hambardzumyan D. Intravital 2-photon imaging reveals distinct morphology and infiltrative properties of glioblastoma-associated macrophages. Proc Natl Acad Sci U S A. 2019;116(28):14254–9.
Kiss M, Van Gassen S, Movahedi K, Saeys Y, Laoui D. Myeloid cell heterogeneity in cancer: not a single cell alike. Cell Immunol. 2018;330:188–201.
Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11(11):762–74.
Goldmann T, Wieghofer P, Jordão M, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. 2016;17(7):797–805.
Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12(9):623–35.
Friebel E, Kapolou K, Unger S, Núñez NG, Utz S, Rushing EJ, et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell. 2020;181(7):1626–42.e20.
Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. 2016;17(9):2445–59.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–8.
Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci. 2019;22(5):729–40.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61.
Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168(2):689–95.
Qiao S, Qian Y, Xu G, Luo Q, Zhang Z. Long-term characterization of activated microglia/macrophages facilitating the development of experimental brain metastasis through intravital microscopic imaging. J Neuroinflammation. 2019;16(1):4.
Sørensen M, Dahlrot R, Boldt H, Hansen S, Kristensen B. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol Appl Neurobiol. 2018;44(2):185–206.
Karimi E, Yu MW, Maritan SM, Perus LJM, Rezanejad M, Sorin M, et al. Single-cell spatial immune landscapes of primary and metastatic brain tumours. Nature. 2023;614(7948):555–63.
Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18(1):234.
Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol. 2013;15(8):1079–87.
Rippaus N, Taggart D, Williams J, Andreou T, Wurdak H, Wronski K, et al. Metastatic site-specific polarization of macrophages in intracranial breast cancer metastases. Oncotarget. 2016;7(27):41473–87.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68.
Liu Y, Kosaka A, Ikeura M, Kohanbash G, Fellows-Mayle W, Snyder LA, et al. Premetastatic soil and prevention of breast cancer brain metastasis. Neuro Oncol. 2013;15(7):891–903.
Yan HH, Jiang J, Pang Y, Achyut BR, Lizardo M, Liang X, et al. CCL9 Induced by TGFβ signaling in myeloid cells enhances tumor cell survival in the premetastatic organ. Cancer Res. 2015;75(24):5283–98.
Yang T-H, St John LS, Garber HR, Kerros C, Ruisaard KE, Clise-Dwyer K, et al. Membrane-Associated Proteinase 3 on Granulocytes and Acute Myeloid Leukemia Inhibits T Cell Proliferation. J Immunol (Baltimore, Md : 1950). 2018;201(5):1389–99.
Park J, Wysocki R, Amoozgar Z, Maiorino L, Fein M, Jorns J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Translatl Med. 2016;8(361):361ra138.
Glodde N, Bald T, van den Boorn-Konijnenberg D, Nakamura K, O’Donnell JS, Szczepanski S, et al. Reactive neutrophil responses dependent on the receptor tyrosine kinase c-MET limit cancer immunotherapy. Immunity. 2017;47(4):789–802.e9.
Mitsuya K, Nakasu Y, Kurakane T, Hayashi N, Harada H, Nozaki K. Elevated preoperative neutrophil-to-lymphocyte ratio as a predictor of worse survival after resection in patients with brain metastasis. J Neurosurg. 2017;127(2):433–7.
Chowdhary M, Switchenko JM, Press RH, Jhaveri J, Buchwald ZS, Blumenfeld PA, et al. Post-treatment neutrophil-to-lymphocyte ratio predicts for overall survival in brain metastases treated with stereotactic radiosurgery. J Neurooncol. 2018;139(3):689–97.
Li H, Wang W, Yang X, Lian J, Zhang S, Cao J, et al. The Clinical Prognostic Value of the Neutrophil-to-Lymphocyte Ratio in Brain Metastases from Non-Small Cell Lung Cancer-Harboring EGFR Mutations. Cancer Manag Res. 2020;12:5659–65.
Han S, Ma E, Wang X, Yu C, Dong T, Zhan W, et al. Rescuing defective tumor-infiltrating T-cell proliferation in glioblastoma patients. Oncol Lett. 2016;12(4):2924–9.
Han S, Zhang C, Li Q, Dong J, Liu Y, Huang Y, et al. Tumour-infiltrating CD4(+) and CD8(+) lymphocytes as predictors of clinical outcome in glioma. Br J Cancer. 2014;110(10):2560–8.
Berghoff AS, Fuchs E, Ricken G, Mlecnik B, Bindea G, Spanberger T, et al. Density of tumor-infiltrating lymphocytes correlates with extent of brain edema and overall survival time in patients with brain metastases. Oncoimmunology. 2016;5(1):e1057388.
Croft PK, Chittoory H, Nguyen TH, Saunus JM, Kim WG, McCart Reed AE, et al. Characterization of immune cell subsets of tumor infiltrating lymphocytes in brain metastases. Biology. 2021;10(5):425.
Gonzalez H, Mei W, Robles I, Hagerling C, Allen BM, Hauge Okholm TL, et al. Cellular architecture of human brain metastases. Cell. 2022;185(4):729–45.e20.
Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61(7):1019–31.
Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20(19):5064–74.
Vilariño N, Bruna J, Bosch-Barrera J, Valiente M, Nadal E. Immunotherapy in NSCLC patients with brain metastases. Understanding brain tumor microenvironment and dissecting outcomes from immune checkpoint blockade in the clinic. Cancer Treatment Rev. 2020;89:102067.
Kudo Y, Haymaker C, Zhang J, Reuben A, Duose DY, Fujimoto J, et al. Suppressed immune microenvironment and repertoire in brain metastases from patients with resected non-small-cell lung cancer. Ann Oncol. 2019;30(9):1521–30.
Mansfield AS, Ren H, Sutor S, Sarangi V, Nair A, Davila J, et al. Contraction of T cell richness in lung cancer brain metastases. Sci Rep. 2018;8(1):2171.
Berghoff AS, Lassmann H, Preusser M, Höftberger R. Characterization of the inflammatory response to solid cancer metastases in the human brain. Clin Exp Metastasis. 2013;30(1):69–81.
Zhou J, Gong Z, Jia Q, Wu Y, Yang Z, Zhu B. Programmed death ligand 1 expression and CD8 tumor-infiltrating lymphocyte density differences between paired primary and brain metastatic lesions in non-small cell lung cancer. Biochem Biophys Res Commun. 2018;498(4):751–7.
Kumar S, Saini R, Mahindroo N. Recent advances in cancer immunology and immunology-based anticancer therapies. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2017;96:1491–500.
Berghoff AS, Venur VA, Preusser M, Ahluwalia MS. Immune checkpoint inhibitors in brain metastases: from biology to treatment. Am Soc Clin Oncol Educ Book. 2016;35:e116–22.
Li Y, Hu X, Lin R, Zhou G, Zhao L, Zhao D, et al. Single-cell landscape reveals active cell subtypes and their interaction in the tumor microenvironment of gastric cancer. Theranostics. 2022;12(8):3818–33.
Jacobs JFM, Idema AJ, Bol KF, Grotenhuis JA, Vries IJMD, Wesseling P, et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J Neuroimmunol. 2010;225(1–2):195–9.
Chaudhary B, Elkord E. Regulatory T cells in the tumor microenvironment and cancer progression: role and therapeutic targeting. Vaccines (Basel). 2016;4(3):28.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nature Reviews Cancer Clinical Oncology. 2017;14(7):399–416.
Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. 2019;20(9):1100–9.
Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172(5):1022–37.e14.
Bellora F, Castriconi R, Dondero A, Reggiardo G, Moretta L, Mantovani A, et al. The interaction of human natural killer cells with either unpolarized or polarized macrophages results in different functional outcomes. Proc Natl Acad Sci USA. 2010;107(50):21659–64.
Roy A, Libard S, Weishaupt H, Gustavsson I, Uhrbom L, Hesselager G, et al. Mast cell infiltration in human brain metastases modulates the microenvironment and contributes to the metastatic potential. Front Oncol. 2017;7:115.
Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46(6):957–67.
Wilhelm I, Molnár J, Fazakas C, Haskó J, Krizbai IA. Role of the blood-brain barrier in the formation of brain metastases. Int J Mol Sci. 2013;14(1):1383–411.
Marchetti D, Li J, Shen R. Astrocytes contribute to the brain-metastatic specificity of melanoma cells by producing heparanase. Cancer Res. 2000;60(17):4767–70.
Lin, Zhang, Siyuan, Jun, Yao, Frank, et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature. 2015;527(7576):100–4.
Wikman H, Lamszus K, Detels N, Uslar L, Wrage M, Benner C, et al. Relevance of PTEN loss in brain metastasis formation in breast cancer patients. Breast Cancer Res. 2012;14(2):R49.
Sarmiento Soto M, Larkin JR, Martin C, Khrapitchev AA, Maczka M, Economopoulos V, et al. STAT3-mediated astrocyte reactivity associated with brain metastasis contributes to neurovascular dysfunction. Can Res. 2020;80(24):5642–55.
Priego N, Zhu L, Monteiro C, Mulders M, Wasilewski D, Bindeman W, et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat Med. 2018;24(7):1024–35.
Xing F, Kobayashi A, Okuda H, Watabe M, Pai SK, Pandey PR, et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med. 2013;5(3):384–96.
Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open. 2019;2(5):e192535.
Topalian S, Sznol M, McDermott D, Kluger H, Carvajal R, Sharfman W, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020–30.
Goldberg SB, Gettinger SN, Mahajan A, Chiang AC, Herbst RS, Sznol M, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016;17(7):976–83.
Taggart D, Andreou T, Scott KJ, Williams J, Rippaus N, Brownlie RJ, et al. Anti-PD-1/anti-CTLA-4 efficacy in melanoma brain metastases depends on extracranial disease and augmentation of CD8(+) T cell trafficking. Proc Natl Acad Sci USA. 2018;115(7):E1540–9.
Gadgeel SM, Lukas RV, Goldschmidt J, Conkling P, Park K, Cortinovis D, et al. Atezolizumab in patients with advanced non-small cell lung cancer and history of asymptomatic, treated brain metastases: Exploratory analyses of the phase III OAK study. Lung cancer (Amsterdam, Netherlands). 2019;128:105–12.
Borghaei H, Brahmer J, Horn L, Ready N, Steins M, Felip E, et al. P2.35: Nivolumab vs docetaxel in advanced NSCLC: CheckMate 017/057 2-Y update and exploratory cytokine profile analysis: track: immunotherapy. J Thora Oncol. 2016;11(10, Supplement):S237–8.
Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet (London, England). 2016;387(10027):1540–50.
Gadgeel S, Rodríguez-Abreu D, Speranza G, Esteban E, Felip E, Dómine M, et al. Updated analysis from KEYNOTE-189: pembrolizumab or placebo plus pemetrexed and platinum for previously untreated metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol. 2020;38(14):1505–17.
Powell SF, Rodríguez-Abreu D, Langer CJ, Tafreshi A, Paz-Ares L, Kopp HG, et al. Outcomes With Pembrolizumab Plus Platinum-Based Chemotherapy for Patients With Non-Small-Cell Lung Cancer and Stable Brain Metastases: Pooled Analysis of KEYNOTE-021, 189, and 407. J Thorac Oncol. 2021.
Goldberg SB, Schalper KA, Gettinger SN, Mahajan A, Herbst RS, Chiang AC, et al. Pembrolizumab for management of patients with NSCLC and brain metastases: long-term results and biomarker analysis from a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2020;21(5):655–63.
Margolin K, Ernstoff MS, Hamid O, Lawrence D, McDermott D, Puzanov I, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459–65.
Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2019;394(10212):1929–39.
Mansfield AS, Liu SV, Szczęsna A, Havel L, Kzrakowski M, Hochmair MJ, et al. Abstract CT199: IMpower133: Primary efficacy and safety + CNS-related adverse events in a Ph1/3 study of first-line (1L) atezolizumab (atezo) + carboplatin + etoposide in extensive-stage SCLC (ES-SCLC). Cancer Res. 2019;79(13 Supplement):CT199-CT.
Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379(8):722–30.
Tawbi HA-H, Forsyth PAJ, Hodi FS, Lao CD, Moschos SJ, Hamid O, et al. Efficacy and safety of the combination of nivolumab (NIVO) plus ipilimumab (IPI) in patients with symptomatic melanoma brain metastases (CheckMate 204). J Clin Oncol. 2019;37(15_suppl):9501-.
.A. Margolin HAT, P.A. Forsyth, F..S. Hodi, A. Algazi, O. Hamid, C.D. Lao, S.J. Moschos, M.B. Atkins, K. Lewis, M.A. Postow, R.P. Thomas, N.I. Khushalani, A.C. Pavlick, M.S. Ernstoff, D.A. Reardon, C. Chung, C. Lee, T. Bas, M. Askelson. 1039MO - CheckMate 204: 3-year outcomes of treatment with combination nivolumab (NIVO) plus ipilimumab (IPI) for patients (pts) with active melanoma brain metastases (MBM). Annals of Oncology. 2021;32(suppl_5):S867-S905.
Schmid TE, Multhoff G. Radiation-induced stress proteins - the role of heat shock proteins (HSP) in anti- tumor responses. Curr Med Chem. 2012;19(12):1765–70.
Rodriguez-Ruiz ME, Vitale I, Harrington KJ, Melero I, Galluzzi L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat Immunol. 2020;21(2):120–34.
Rudqvist NP, Pilones KA, Lhuillier C, Wennerberg E, Sidhom JW, Emerson RO, et al. Radiotherapy and CTLA-4 blockade shape the TCR repertoire of tumor-infiltrating T Cells. Cancer Immunol Res. 2018;6(2):139–50.
Thompson RF, Maity A. Radiotherapy and the tumor microenvironment: mutual influence and clinical implications. Adv Exp Med Biol. 2014;772:147–65.
Kachikwu EL, Iwamoto KS, Liao YP, DeMarco JJ, Agazaryan N, Economou JS, et al. Radiation enhances regulatory T cell representation. Int J Radiat Oncol Biol Phys. 2011;81(4):1128–35.
Yoneda K, Kuwata T, Kanayama M, Mori M, Kawanami T, Yatera K, et al. Alteration in tumoural PD-L1 expression and stromal CD8-positive tumour-infiltrating lymphocytes after concurrent chemo-radiotherapy for non-small cell lung cancer. Br J Cancer. 2019;121(6):490–6.
Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548(7668):466–70.
Lauber K, Dunn L. Immunotherapy mythbusters in head and neck cancer: the abscopal effect and pseudoprogression. Am Soc Clin Oncol Educ Book. 2019;39:352–63.
Knisely JP, Yu JB, Flanigan J, Sznol M, Kluger HM, Chiang VL. Radiosurgery for melanoma brain metastases in the ipilimumab era and the possibility of longer survival. J Neurosurg. 2012;117(2):227–33.
Stokes WA, Binder DC, Jones BL, Oweida AJ, Liu AK, Rusthoven CG, et al. Impact of immunotherapy among patients with melanoma brain metastases managed with radiotherapy. J Neuroimmunol. 2017;313:118–22.
Robin TP, Breeze RE, Smith DE, Rusthoven CG, Lewis KD, Gonzalez R, et al. Immune checkpoint inhibitors and radiosurgery for newly diagnosed melanoma brain metastases. J Neurooncol. 2018;140(1):55–62.
Diao K, Bian SX, Routman DM, Yu C, Ye JC, Wagle NA, et al. Stereotactic radiosurgery and ipilimumab for patients with melanoma brain metastases: clinical outcomes and toxicity. J Neurooncol. 2018;139(2):421–9.
Ahmed KA, Abuodeh YA, Echevarria MI, Arrington JA, Stallworth DG, Hogue C, et al. Clinical outcomes of melanoma brain metastases treated with stereotactic radiosurgery and anti-PD-1 therapy, anti-CTLA-4 therapy, BRAF/MEK inhibitors, BRAF inhibitor, or conventional chemotherapy. Ann Oncol. 2016;27(12):2288–94.
Choong ES, Lo S, Drummond M, Fogarty GB, Menzies AM, Guminski A, et al. Survival of patients with melanoma brain metastasis treated with stereotactic radiosurgery and active systemic drug therapies. Eur J Cancer. 2017;75:169–78.
Kiess AP, Wolchok JD, Barker CA, Postow MA, Tabar V, Huse JT, et al. Stereotactic radiosurgery for melanoma brain metastases in patients receiving ipilimumab: safety profile and efficacy of combined treatment. Int J Radiat Oncol Biol Phys. 2015;92(2):368–75.
Golden EB, Demaria S, Schiff PB, Chachoua A, Formenti SC. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol Res. 2013;1(6):365–72.
Lehrer EJ, Peterson J, Brown PD, Sheehan JP, Quiñones-Hinojosa A, Zaorsky NG, et al. Treatment of brain metastases with stereotactic radiosurgery and immune checkpoint inhibitors: an international meta-analysis of individual patient data. Radiother Oncol. 2019;130:104–12.
Garassino M, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, Speranza G, et al. OA04.06 Evaluation of TMB in KEYNOTE-189: pembrolizumab plus chemotherapy vs placebo plus chemotherapy for nonsquamous NSCLC. J Thorac Oncol. 2019;14:S216–7.
Nitschke NJ, Bjoern J, Iversen TZ, Andersen MH, Svane IM. Indoleamine 2,3-dioxygenase and survivin peptide vaccine combined with temozolomide in metastatic melanoma. Stem Cell Investig. 2017;4:77.
Dillman R, Selvan S, Schiltz P, Peterson C, Allen K, Depriest C, et al. Phase I/II trial of melanoma patient-specific vaccine of proliferating autologous tumor cells, dendritic cells, and GM-CSF: planned interim analysis. Cancer Biother Radiopharm. 2004;19(5):658–65.
Laurell A, Lönnemark M, Brekkan E, Magnusson A, Tolf A, Wallgren AC, et al. Intratumorally injected pro-inflammatory allogeneic dendritic cells as immune enhancers: a first-in-human study in unfavourable risk patients with metastatic renal cell carcinoma. J Immunother Cancer. 2017;5:52.
Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Sci. 2015;348(6236):803–8.
Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, et al. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. 2018;6(1):36–46.
Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, et al. Regional delivery of chimeric antigen receptor-engineered t cells effectively targets HER2(+) breast cancer metastasis to the brain. Clin Cancer Res. 2018;24(1):95–105.
Benbenishty A, Gadrich M, Cottarelli A, Lubart A, Kain D, Amer M, et al. Prophylactic TLR9 stimulation reduces brain metastasis through microglia activation. PLoS Biol. 2019;17(3):e2006859.
Acknowledgements
Not applicable.
Funding
This work was supported by Jilin Scientific and Technological Development Program (CN)(20190303146SF) (Jiuwei Cui), and General Program of National Natural Science Foundation of China (81874052) (Jiuwei Cui).
Author information
Authors and Affiliations
Contributions
HG (1) substantial contributions to conception and design, acquisition of data, and analysis and interpretation of data; (2) drafting the article; BW, WL and NC (1) contributions to data collection; (2) revising the article; JC (1) contributions to conception and design; (2) make important changes to the paper; and (3) final approval of the version to be published. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, H., Wang, B., Li, W. et al. Current landscape and challenges ahead of immuno-molecular mechanism and immunotherapy strategy of brain metastases. Holist Integ Oncol 2, 31 (2023). https://doi.org/10.1007/s44178-023-00053-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44178-023-00053-w