
Abstract
Background
Cystinosis, a rare autosomal recessive disease, stems from genetic alterations in the CTNS gene, leading to a malfunction of lysosomal ‘cystinosin’ protein. This dysfunction causes intracellular cystine accumulation, resulting in nephropathic and ocular abnormalities. Cystinosis is relatively rare in Asian countries, partly due to underreporting and lack of awareness, and cases often lack sufficient genetic evidence to support their diagnosis. This study presents a descriptive case series involving four Indian patients with cystinosis, elucidating clinical and genetic aspects.
Methods
All four patients underwent comprehensive ophthalmic evaluations. The corneal cystine crystal (CCC) score was determined using anterior segment optical coherence tomography (AS-OCT) and in vivo confocal microscopy (IVCM). Genetic testing was performed using whole exome sequencing (WES).
Results
Corneal crystal deposition, a hallmark of cystinosis, was evident in all cases. Systemic analysis revealed manifestations such as polyuria, bony abnormalities, growth retardation, hypothyroidism, and developmental delay. Genetic testing in two patients identified a homozygous pathogenic variant c.18_21delGACT (p.Thr7PhefsX7) in the CTNS gene, previously reported to cause cystinosis in different ethnic populations.
Conclusions
Our case series sheds light on underrepresented cases of cystinosis in the Indian population. The rarity of this condition poses diagnostic challenges, leading to delayed or inaccurate diagnoses. AS-OCT can serve as a viable alternative to IVCM for assessing corneal crystal density status in cystinosis. Timely recognition and management are crucial in preventing complications, and the inclusion of genetic testing can expedite cystinosis diagnosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cystinosis (OMIM 219800) is a rare autosomal recessive disease caused by genetic alterations in the CTNS gene (OMIM 606272), leading to dysfunction of the lysosomal membrane-specific carrier protein ‘cystinosin’. This protein is crucial for cystine transport, and its failure results in intracellular cystine accumulation. The disease primarily affects the kidneys and eyes, with potential impacts on other organs, including muscles, pancreas, thyroid gland, and white blood cells as well [1]. The incidence is 1 in 100,000–200,000 live births in the United States and Europe. The incidence is observed relatively lower in Asian countries, partly attributed to underreporting, influenced by the lack of awareness of the disease [2]. Cystinosis manifests in three forms: the severe ‘nephropathic infantile’ form, the milder ‘juvenile’ form, and the ‘non-nephropathic’ adult form [3]. Ocular manifestations, such as corneal cystine crystal (CCC) accumulation, are observed in all forms, leading to photophobia and complications like punctate keratopathy, filamentary keratitis, and band-shaped keratopathy, along with glaucoma, retinopathy, and photoreceptor degenerations [4]. Comprehensive diagnosis includes physical and clinical evaluation, renal tests, ophthalmic examination, genetic testing, and follow-up assessments [5]. Initial signs may include failure to thrive, growth retardation, thirst, polyuria, rickets, pale skin, and blonde hair. The polymorphonuclear (PMN) leukocyte cystine concentration is a pivotal early-stage biochemical test for cystinosis, while corneal cystine deposits, which typically appear in late infancy, can be visualized using AS-OCT (anterior segment optical coherence tomography) and IVCM (in vivo confocal microscopy) [4, 6, 7]. Given that there could be variability in PMN leukocyte cystine content, confirming the diagnosis through genetic testing of the CTNS gene can be more consistent [8]. Cystinosis management includes oral cysteamine to delay renal failure, cysteamine eye drops to reduce corneal crystals, providing supportive care such as electrolyte supplementation for symptom management and in severe end-stage renal failure cases, and haemodialysis or renal transplantation to restore renal function [9]. The reported cases of cystinosis, particularly those supported by genetic evidence, are scarce in Asian countries, including India. The rarity of the disease and the lack of awareness can result in misdiagnosis and maltreatment of patients, posing the need for studies and reports from different ethnic populations [10]. This case series describes four underrepresented Indian patients with comprehensive ocular and genetic analysis emphasizing the importance of early diagnosis and management.
Materials and methods
The study constitutes a descriptive and retrospective cystinosis case series carried out at L V Prasad Eye Institute, Hyderabad, adhering to the principles outlined in the Declaration of Helsinki. Four paediatric patients who presented with clinical signs of cystinosis were evaluated in our outpatient department (OPD) during follow-up between April 2021 and May 2023. Informed consents were procured. Their demographic information, ophthalmic clinical findings, genetic tests, and treatment measurements were accessed (Table 1).
Visual acuity was assessed individually for each eye, utilizing Snellen charts for three patients and Teller Acuity charts for the other patient, administered by a skilled optometrist. Visual acuity was quantified as a logarithm of the minimum angle of resolution (logMAR). All patients underwent slit lamp examination to assess the presence of conjunctival and corneal cysteine crystals, along with slit lamp photography to record these findings (Figs. 2 and 3). The density of central corneal crystals was evaluated by an experienced scorer comparing slit lamp corneal images with slit lamp reference library photographs of corneas containing cystine crystals at different densities. Scores were assigned on a scale ranging from 0 (indicating clarity) to 3.00 (representing a high density of crystals), with increments of 0.25, as described by Gahl et al. [11] Additionally, AS-OCT was performed to measure the depth of corneal crystal deposition (Fig. 4). IVCM was performed in one patient (Fig. 5), while others were uncooperative for examination. The IVCM score was employed to assess the distribution of crystal deposits spanning a 400 × 400 µm surface area within different layers of the central cornea, encompassing the epithelium, Bowman’s layer, anterior stroma, deep stroma, and endothelium. The IVCM score was classified as 0 if there were no crystals, 1 if covering 25%, 2 if 25–50%, 3 if 50–75%, and 4 over 75% of the determined area [4]. Furthermore, two consenting patients underwent genetic testing to determine the genetic variation involved in the disease.
Genetic testing
Whole-exome sequencing (WES) was performed using the Illumina Nova Platform (Redcliffe Labs, Hyderabad, Telangana, India). Thirty MB regions of the human exome were analysed for variants, including genes related to the patient’s phenotype. The genome was sequenced to mean > 80–100X coverage and a minimum of ~ 90% bases for at least 20X coverage. Filtered sequence output reads were aligned with the NCBI reference sequence (GRCh37.75). Variants compared to reference were called using the Genome Analysis Tool Kit (GATK). Variants were annotated and categorized using Golden Helix VarSeq and Varsome analysis workflow implementing ACMG (American College of Medical Genetics and Genomics) guidelines.
Results
Case 1
Patient 1 was a 16-year-old girl presenting with photophobia and diminished vision in both eyes with raised intraocular pressure documented at a different hospital. She was born out of a non-consanguineous marriage and had a history of neonatal jaundice, for which phototherapy was given for 4 days. She had also developed bony abnormalities secondary to rickets (Fig. 1a) and experienced multiple episodes of tetany in the first decade of life. She had a short stature with a broad nasal bridge and cubitus valgus in the upper limbs and genu valgus in the lower limbs (Fig. 1b, c). She had complaints of polyuria shortly after she was a year old and had gone through multiple consultations but received only symptomatic treatment. At 10 years of age, she was diagnosed with infantile nephropathic cystinosis with Fanconi syndrome. Her best-corrected visual acuity was 20/60 (0.5 logMAR) in the right eye and 20/50 (0.4 logMar) in the left eye. The intraocular pressure measured by Goldmann applanation tonometry was 20 mmHg in both eyes. Both eyes had diffused corneal crystalline deposits in the anterior and mid-stroma. Slit lamp photographs in the optical section showed CCC score (CCCS) of 3.0 (Figs. 2a and 3). AS-OCT imaging showed cornea crystals in the anterior and mid-stroma (Fig. 4a). IVCM showed the presence of needle-shaped crystals in the stroma with a score of 2 (Fig. 5). Retinal OCT and fundus examination in both eyes were within normal limits, with no evidence of pigmentary retinopathy (Fig. 6).

In patient 1. a Bony abnormalities due to hypophosphatemic rickets in cystinosis. b Cubitus valgus in upper limbs. c Short stature with genu valgus (knock-knee) in the lower limbs

Slit lamp images of patients 1, 2, 3, 4 corresponding a, b, c, d, respectively, showing diffuse cystine corneal crystals

Slit lamp photographs (a & b) in the optical section showing corneal cystine crystals (CCC) in patient 1

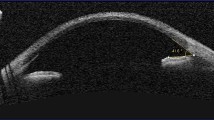
Anterior segment optical coherence tomography (AS-OCT) showing corneal crystals in the anterior and mid-stroma (a, b, c, d) (yellow arrows) in patients 1, 2, 3, and 4, respectively

In vivo confocal microscopy (IVCM) images showing cystine crystals (of score − 2) in the epithelium-stromal section in left (OS) and right (OD) eye of patient 1

Posterior segment imaging in patient 1 observed within normal limits. a Unremarkable optical coherence tomography. b and c Unremarkable coloured fundus of the right and left eye respectively. Imaging of the posterior segment was tedious to capture given the intense blepharospasm
The patient underwent genetic testing, which revealed the previously reported homozygous pathogenic variant c.18_21delGACT (p.Thr7PhefsX7) in the CTNS gene. She was asked to continue cysteamine eye drops thrice daily and advised to follow up closely with a paediatric nephrologist.
Case 2
Patient 2 was a 10-year-old male child. He had undergone screening at 1 year of age for ocular manifestations of cystinosis, which were absent during that period. Later on, he presented with symptoms of photophobia and diminished vision. Slit lamp examination revealed diffuse CCC deposition in both eyes with a CCCS of 3.0 (Fig. 2b). The AS-OCT showed diffused stromal crystalline deposits in both eyes (Fig. 4b). His diagnosis status was cystinosis with Fanconi syndrome, characterized by growth retardation and hypothyroidism. The best-corrected visual acuity was 20/125 (0.8 logMar) in the right eye and 20/25 (0.1 logMAR) in the left eye. The intraocular pressure was 15 mmHg in the right and 19 mmHg in the left eye. Fundus appearance was within normal limits. He was recommended to undergo genetic testing but was lost to follow-up. He was advised to take cysteamine drops in both eyes and consult a paediatric nephrologist for systemic management.
Case 3
Patient 3 was an 8-year-old female child who presented to our OPD with her mother with symptoms of photophobia, redness, and watering in both eyes for 2 months. She had previously received a diagnosis of microsporidial keratitis at another medical facility. She had begun treatment for this before being referred to our centre, where she was subsequently diagnosed with cystinosis. She had a history of failure to thrive as diagnosed by her paediatrician but no sign or symptoms suggestive of renal disorder or hypophosphatemia rickets. The best-corrected visual acuity was 20/30 (0.2 logMAR) in the right eye and 20/40 (0.3 logMAR) in the left eye. The intraocular pressure was digitally normal in both eyes. The CCCS in both eyes were 2.0 (Fig. 2c). Both eyes showed diffuse crystalline deposits in the stroma confirmed on anterior segment OCT (Fig. 4c). The fundus examination was unremarkable in both eyes. She was recommended to undergo genetic testing but was lost to follow-up. She was advised to take cysteamine drops in both eyes and consult a paediatric nephrologist for systemic management.
Case 4
Patient 4, a 4-year-old girl, visited our OPD accompanied by her mother, presenting with photophobia in both eyes. She was born out of a third-degree consanguineous marriage. She had a history of delayed achievement of developmental milestones and had been experiencing polyuria and polydipsia since the age of 1. She had consulted elsewhere and was diagnosed with granular dystrophy. Upon consultation with a nephrologist, it was discovered that she had renal tubular acidosis. Consequently, she was directed to our centre for an assessment to ascertain the presence of ocular cystinosis. On ophthalmic examination at our centre, her visual acuity using the Teller Acuity chart was 20/60 (0.5 logMAR) in the right eye and 20/80 (0.6 logMAR) in the left eye. Both eyes showed the presence of CCC on slit lamp examination with CCCS 2.5 (Fig. 2d). Both eyes had diffused stromal corneal crystalline deposits validated on AS-OCT (Fig. 4d). Fundus examination revealed no abnormalities. Upon genetic testing, she was identified with a homozygous pathogenic variant c.18_21delGACT in the CTNS gene, mirroring the findings in patient 1. She was asked to follow up with her renal function test reports and was advised to start cysteamine eye drops.
Discussion
The extreme rarity of cystinosis is evident when considering its occurrence in different regions. In North America, the incidence of infantile nephropathic cystinosis is 1 in 100,000 to 200,000. The incidence reported in various other countries, such as France, Germany, Denmark, Sweden, and Australia, ranges from 1 in 115,000 to 1 in 260,000 live births. In Poland, the incidence of cystinosis is just one person in 2.92 million. Interestingly, no confirmed cases of cystinosis in Finland have been reported. There is insufficient information concerning the prevalence of cystinosis in regions spanning the Middle East, North Africa, and China [10, 11, 12]. Reports of cystinosis from India are scarce considering its larger population, and only a few studies have demonstrated the related genetic information. Table 2 summarizes the status of cystinosis reports from the Indian perspective. Through our present case series, we further expanded the clinical and genetic aspects of cystinosis within the Indian context.
Infantile nephropathic cystinosis is the most critical form, usually presenting with features of renal Fanconi syndrome, such as failure to thrive, persistent polyuria, polydipsia, and acidosis. It can also affect the endocrine, skeletal, muscular, and central nervous systems [29]. Hypophosphatemic rickets can occur due to renal tubular damage, as was observed in one of our patients who presented with genu valgum. Thyroid dysfunction develops in 50–70% of untreated children with cystinosis and usually renders by 10 years of age, as evidenced in two patients in our series [30]. Juvenile nephropathic cystinosis usually presents during late adolescence and is a milder variant with minimal proximal tubular dysfunction and proteinuria. Non-nephropathic or ocular cystinosis presents with pristine renal function and symptoms of photophobia. All the patients in our series were preadolescent with signs and symptoms that lie within the spectrum of renal Fanconi syndrome. Therefore, we characterized all four patients as having infantile nephropathic cystinosis. Moreover, owing to the sporadic nature and rarity of the disease, the diagnosis of cystinosis is often delayed leading to ‘diagnostic odyssey’ as was seen in 3/4 of our patients. For instance, one patient underwent multiple hospital consultations for several years before receiving appropriate management. Another patient was misdiagnosed with microsporidial keratitis before being diagnosed at our institute, while the patient 4 was considered to be suffering from granular dystrophy in a prior diagnosis.
The CTNS gene on chromosome 17p13.3 encodes the cystinosin protein, a vital component of the lysosomal membrane responsible for transporting cystine out of lysosomes. It is characterized by its seven-transmembrane domain structure and a pair of extensively preserved proline-glutamine motifs and becomes an integral component of the lysosomal membrane [31]. Since 1998, when a mutation in the CTNS gene was first established as a causative factor for cystinosis, more than 140 variations have been reported [32]. The most common genetic alteration, a 57-kb deletion affecting the gene’s promoter and initial exons, is predominantly found in individuals of North European descent due to a potential founder effect. However, this genetic variation is less prevalent among populations from the Middle East, Asia, and Africa [33]. Two out of four of our patients underwent genetic testing, revealing a homozygous pathogenic variant c.18_21delGACT (p.Thr7PhefsX7) in the CTNS gene. This identified variation appears relatively a common cause of cystinosis as it has been reported previously in patients from Polish, Spanish, Turkish, American, German, and Swiss-based populations [34, 35, 36]. The variation is also indexed in the ClinVir database as a pathogenic variation. It causes the replacement of threonine (Thr) at position 7 by phenylalanine (Phe) and a frameshift that results in a premature stop codon seven amino acids downstream. The variation is anticipated to result in either a truncation of the encoded protein or its absence, attributed to nonsense-mediated decay. The presence of this variation in two Indian cystinosis cases, and various other ethnic populations, indicates its role as founder pathogenic variation and, therefore, can be targeted for genetic screening in cystinosis-related cases. The other two patients were recommended for genetic evaluation but were unfortunately lost to follow-up.
Corneal and conjunctival crystal deposition is pathognomonic of cystinosis and was initially described by Burki et al. [37]. Corneal crystals present as needle-shaped refractile opacities deposited in all layers of the cornea. They first occur during infancy and are evident by 16 months of age, usually causing symptoms of photophobia in the first few years of life. Deposition begins in the anterior periphery and progresses posteriorly and centripetally. By the age of 7, the peripheral stroma is packed with crystals; by 20 years of age, the entire corneal stroma is usually involved [38]. Intense photophobia was observed in all our four patients as a result of these cystine crystals. The cases in our study showed relatively reduced visual acuity. While the corneal crystals do not typically lead to a decrease in visual acuity, we suspect that due to the individual’s age and the significant blepharospasm triggered by photophobia, assessing visual acuity may be unreliable. On the other hand, regular fundus examination might help to monitor retinal changes if they present subclinical retinal or optic nerve changes. Pigmentary retinopathy has been reported in children with cystinosis after age of 6; however, none of our patients presented with the same.
Noninvasive ophthalmic examination tools are crucial for diagnosing ocular manifestations related to cystinosis, especially in young children [39]. Among these tools, the slit lamp, commonly available in clinical settings, is vital for detecting corneal crystals. These crystals, appearing as spindle-shaped or fusiform hyper-reflective bodies, can be assessed using optical sections or parallelepiped beams to gauge their depth. However, due to their small size and variable distribution, they may not always be readily visible during routine examinations. Furthermore, the current CCCS system is based on subjective analysis with the references which may have limitations in scoring the progression in advanced cases.
AS-OCT provides another noninvasive option for rapid and semiautomated analysis of corneal crystals, in cornea. In AS-OCT images, cystine crystals appear as hyperreflective punctate deposits in the anterior stroma. This method, simpler to perform, provides the depth of corneal crystal and can also serve to qualitatively evaluate the progression of corneal crystal deposition. However, the instrument setup is costly, and it may not capture the high-resolution distribution of corneal crystals in the other regions such as the ciliary body.
IVCM is considered the gold standard for measuring high-resolution cystine deposition in all layers of the cornea. It provides a wide-scale comprehensive assessment of corneal pathology which can be useful in accessing the outcome of given cystinosis management. However, it is a contact-based procedure that necessitates placing the objective lens on the corneal surface throughout imaging. This can be challenging in young patients due to photophobia and blepharospasm. Due to its very high cost, the instrument accessibility is limited.
There is no cure for cystinosis, and its primary management involves symptomatic-based treatment. Cystine-depleting therapy for cystinosis was first described in 1976 and continues to be the mainstay cystinosis therapy using cysteamine [40]. Cysteamine bitartrate (Cystagon, Mylan Pharma, UK) is currently the most widely used oral formulation. Cysteamine’s precise mechanism of action, though, is unclear; it is known for its capacity to transform accumulated lysosomal cystine into a combination of other forms, such as cysteine and cysteine-cysteamine mixed disulphide [41]. This transformation effectively diminishes cystine buildup and facilitates bypassing the need for a cystine transporter to move cystine. Cysteamine can be available in both an immediate release and delayed release formulation, and both slow down the progression of the disease. While systemic cysteamine therapy can be used to halt the progression of systemic features of cystinosis and posterior segment ocular afflictions such as pigmentary retinopathy, it does not help prevent corneal crystal accumulation due to the avascular nature of the cornea. Topical cysteamine therapy has proven effective in impeding corneal and conjunctival crystal accumulation [42]. The regimen is usually a 0.55% cysteamine hydrochloride solution with 0.01% benzalkonium chloride, used 6–12 times daily. However, this requires refrigeration since, at room temperature, cysteamine oxidizes to cystamine, its disulphide form, rendering the solution ineffective. To circumvent the problem of repeated administration and refrigeration requirements, a viscous gel-like formulation of cysteamine hydrochloride was devised, which could be stored at room temperature for 7 days and required administration four times daily [43]. In our study, only one patient had previously initiated topical cysteamine therapy prior to their visit, while we advised the other three patients to commence the same treatment. A regular longer follow-up is necessary to monitor the response and tolerance to therapy, and a multidisciplinary approach in conjunction with a paediatric nephrologist is warranted. Therefore, all the patients in our study will be followed up to assess the impact of cysteamine therapy, and the outcomes will be reported in future updates.
A study conducted by Bertholet-Thomas et al. comparatively analysed the management of nephropathic cystinosis in developing nations (DiN) versus developed nations (DeN). The analysis of data from 213 patients across 41 centres in 30 nations revealed substantial disparities in crucial aspects of cystinosis care [44]. Significant differences were observed in the timing of treatment initiation, genetic testing practices, progression to end-stage renal disease (ESRD), and outcomes of renal transplantation, with a disadvantageous trend in DiN. In addition, accessibility to cysteamine therapy, pivotal for treatment, encounters obstacles such as the absence of cysteamine in stock, prohibitive costs, lack of licensing, or considerations of its unacceptability within the context of national priorities. These challenges highlight the necessity for concerted efforts to overcome barriers and improve the global management of cystinosis, ensuring equitable access to essential treatments across diverse socioeconomic settings.
Various case reports from different countries have shed light on the crucial aspects in diagnosing cystinosis. A report from China highlights that initial symptoms of nephrotic cystinosis may resemble those of Fanconi syndrome, and ocular manifestations might not be evident in early infancy. Therefore, infants exhibiting Fanconi syndrome symptoms with normal eye exams should be monitored for signs of corneal cystine deposition, with early CSTN gene testing enabling a prompt diagnosis of cystinosis in such cases [45]. A report from Tunisia emphasizes early diagnosis along with cysteamine therapy to prevent progression to ESRD, which could necessitate haemodialysis and kidney transplant. Additionally, post-renal transplantation, continuing cysteamine treatment, is advocated to delay extrarenal complications [46]. In Egypt, where leukocyte cystine tests are not readily accessible, programmes like EGORD (Egyptian Group of Orphan Diseases) target high suspicion index of cystinosis, characterized by symptoms such as fluid and electrolyte depletion, aminoaciduria, glycosuria, phosphaturia, renal tubular acidosis, rickets, and growth impairment. Such cases undergo follow-up assessments for cystinosis diagnosis and subsequent treatment with cysteamine therapy [47]. A multicentric report from Brazil underscores the necessity of a comprehensive diagnostic approach involving various healthcare professionals such as social assistants, psychologists, nurses, nutritionists, dentists, and multiple medical specialists for effective cystinosis diagnosis and management [48]. Collaboration among international experts and the sharing of best practices are essential for improving the timely diagnosis and management of cystinosis on a global scale.
Emerging regenerative medicine strategies, encompassing recombinant adeno-associated virus (r-AAV)-based gene therapy and stem cell therapy, are advancing through preclinical and clinical phases to treat cystinosis. In recent developments, the Food and Drug Administration granted clinical trial approval for autologous cell therapy involving CTNS gene-modified haematopoietic stem and progenitor cells (HSPCs) following promising outcomes in preclinical studies [49]. This autologous haematopoietic stem cell gene therapy offers a hopeful novel approach for mitigating cystinosis-related complications. Cystinosis case reports and access to the registry of cystinosis patients are crucial for ensuring that individuals can reap the advantages of upcoming therapeutic interventions.
In conclusion, this case series highlights significant aspects of cystinosis and sheds light on the often overlooked and misdiagnosed condition of cystinosis in India emphasizing the importance of early diagnosis and best practised management. Genetic testing is pivotal in expediting diagnosis, enabling early intervention, and, thus, better survival conditions for patients with cystinosis.
Availability of data and materials
The datasets analysed during the current study are available from the corresponding author upon reasonable request.
References
Nesterova G, Gahl WA. Cystinosis: the evolution of a treatable disease. Pediatr Nephrol. 2013;28(1):51–9.
Deshpande AA, Ravichandran R, Bachhawat AK. Molecular analysis of the CTNS gene in Indians with nephropathic cystinosis. Indian J Pediatr. 2017;84(3):240–1.
Wilmer MJ, Schoeber JP, van den Heuvel LP, Levtchenko EN. Cystinosis: practical tools for diagnosis and treatment. Pediatr Nephrol Berl Ger. 2011;26(2):205–15.
Kowalczyk M, Toro MD, Rejdak R, Załuska W, Gagliano C, Sikora P. Ophthalmic evaluation of diagnosed cases of eye cystinosis: a tertiary care center’s experience. Diagn Basel Switz. 2020;10(11):911.
bvAriceta G, Camacho JA, Fernández-Obispo M, Fernández-Polo A, Gamez J, García-Villoria J, Lara Monteczuma E, Leyes P, Martín-Begué N, Oppenheimer F, Perelló M, Morell GP, Torra R, Santandreu AV, Güell A, Grupo T-CiS.bcn. Cystinosis in adult and adolescent patients: recommendations for the comprehensive care of cystinosis. Nefrologia. 2015;35(3):304–21.
Pinxten AM, Hua MT, Simpson J, Hohenfellner K, Levtchenko E, Casteels I. Clinical practice: a proposed standardized ophthalmological assessment for patients with cystinosis. Ophthalmol Ther. 2017;6(1):93–104.
Csorba A, Maka E, Maneschg OA, Szabó A, Szentmáry N, Csidey M, et al. Examination of corneal deposits in nephropathic cystinosis using in vivo confocal microscopy and anterior segment optical coherence tomography: an age-dependent cross-sectional study. BMC Ophthalmol. 2020;20(1):73.
Bondue T, Kouraich A, Berlingerio SP, Veys K, Marie S, Alsaad KO, et al. The pitfall of white blood cell cystine measurement to diagnose juvenile cystinosis. Int J Mol Sci. 2023;24(2):1253.
Levtchenko E, Servais A, Hulton SA, Ariceta G, Emma F, Game DS, Lange K, Lapatto R, Liang H, Sberro-Soussan R, Topaloglu R, Das AM, Webb NJA, Wanner C. Expert guidance on the multidisciplinary management of cystinosis in adolescent and adult patients. Clin Kidney J. 2022;15(9):1675–84.
Elmonem MA, Veys KR, Soliman NA, van Dyck M, van den Heuvel LP, Levtchenko E. Cystinosis: a review. Orphanet J Rare Dis. 2016;22(11):47.
Gahl WA, Kuehl EM, Iwata F, Lindblad A, Kaiser-Kupfer MI. Corneal crystals in nephropathic cystinosis: natural history and treatment with cysteamine eyedrops. Mol Genet Metab. 2000;71(1–2):100–20.
Vashist N, Deshpande AA, Kanakaraj A, Ravichandran R, Bachhawat AK. Cystinosis: status of research and treatment in India and the world. J Biosci. 2023;48:50.
Kumar PA, Subramanyam G. Early onset of chronic renal failure in infantile nephropathic cystinosis. Indian Pediatr. 2004;41(11):1172–3.
Tang S, Danda S, Zoleikhaeian M, Simon M, Huang T. An Indian boy with nephropathic cystinosis: a case report and molecular analysis of CTNS mutation. Genet Test Mol Biomark. 2009;13(4):435–8.
Sharma A, Gupta R, Sethi SK, Bagga A, Dinda AK. Giant cell transformation of podocytes: a unique histological feature associated with cystinosis. Indian J Nephrol. 2011;21(2):123–5.
Dhooria GS, Bains HS. Nephrotic range proteinuria as a presenting feature of classical nephropathic cystinosis. Indian J Pediatr. 2014;81(7):712–4.
Kiran BV, Barman H, Iyengar A. Clinical profile and outcome of renal tubular disorders in children: a single center experience. Indian J Nephrol. 2014;24(6):362–6.
Kanthila J, Dsa S, Bhat KG. Nephropathic cystinosis presenting as renal fanconi syndrome without glycosuria. J Clin Diagn Res JCDR. 2015;9(3):SD05-06.
Mittal D, Bagga A, Tandon R, Sharma MC, Bhatnagar V. Hirschsprung’s disease with infantile nephropathic cystinosis. J Indian Assoc Pediatr Surg. 2015;20(3):153–4.
More V, Shanbag P. Infantile nephropathic cystinosis with incomplete fanconi syndrome, hypothyroidism, hydrouretero-nephrosis, and megacystis. Saudi J Kidney Dis Transplant Off Publ Saudi Cent Organ Transplant Saudi Arab. 2016;27(3):598–601.
Priyanka, Bhatt GC, Kumar A, Takkar B. Crystalline keratopathy in nephropathic cystinosis. Sudan J Paediatr. 2019;19(2):169–70.
Naik MP, Sethi HS, Dabas S. Ocular cystinosis: rarity redefined. Indian J Ophthalmol. 2019;67(7):1158–9.
Matthai SM, Jacob S, Bindra MS, David VG, Varughese S. Nephropathic cystinosis presenting with uveitis: report of a “Can’t See, Can’t Pee” situation. Indian J Pathol Microbiol. 2019;62(3):457–60.
Das G, Nanda PM, Kaur A, Kumar R. Bartter syndrome and hypothyroidism masquerading cystinosis in a 3-year-old girl: rare manifestation of a rare disease. BMJ Case Rep. 2021;14(7):e242954.
Deepthi B, Krishnamurthy S, Karunakar P, Barathidasan G, Rajavelu TN. Atypical manifestations of infantile-onset nephropathic cystinosis: a diagnostic challenge. CEN Case Rep. 2022;11(3):347–50.
Namratha Vaghdevi C, Jahnavi B, Sindhu D, P AL, R R, Shah M, et al. Cystinosis: a 6.5-year-follow-up study. Indian J Pediatr. 2022;89(8):824.
Gurnani B, Kaur K, Gupta I. Sparkling eye-a rare presentation in cystinosis. Asia-Pac J Ophthalmol Phila Pa. 2023;12(2):269.
Singh A, Patel R, Abhinay A, Prasad R, Mishra D, Mishra OP. Nephrogenic diabetes insipidus in a 10-mo-old infant: complication of nephropathic cystinosis. Indian J Pediatr. 2023;90(5):521.
Atmis B, K Bayazit A, Cevizli D, Kor D, Fidan HB, Bisgin A, Kilavuz S, Unal I, Erdogan KE, Melek E, Gonlusen G, Anarat A, Onenli Mungan N. More than tubular dysfunction: cystinosis and kidney outcomes. J Nephrol. 2022;35(3):831–40.
Bako D, Kılavuz S, YasinKöksoy A, UzanTatli Z, Beydogan E. A different approach to cystinosis: ultrasound, doppler, and shear wave elastography findings of thyroid gland. Orphanet J Rare Dis. 2023;18(1):173.
Guo X, Schmiege P, Assafa TE, Wang R, Xu Y, Donnelly L, et al. Structure and mechanism of human cystine exporter cystinosin. Cell. 2022;185(20):3739-3752.e18.
David D, Princiero Berlingerio S, Elmonem MA, Oliveira Arcolino F, Soliman N, van den Heuvel B, et al. Molecular basis of cystinosis: geographic distribution, functional consequences of mutations in the CTNS gene, and potential for repair. Nephron. 2019;141(2):133–46.
Najafi M, Tamandani DMK, Azarfar A, Bakey Z, Behjati F, Antony D, et al. A 57 kB genomic deletion causing CTNS loss of function contributes to the CTNS mutational spectrum in the Middle East. Front Pediatr. 2019;7:89.
Macías-Vidal J, Rodés M, Hernández-Pérez JM, Vilaseca MA, Coll MJ. Analysis of the CTNS gene in 32 cystinosis patients from Spain. Clin Genet. 2009;76(5):486–9.
Shotelersuk V, Larson D, Anikster Y, McDowell G, Lemons R, Bernardini I, et al. CTNS mutations in an American-based population of cystinosis patients. Am J Hum Genet. 1998;63(5):1352–62.
Kiehntopf M, Schickel J, von der Gönne B, Koch HG, Superti-Furga A, Steinmann B, et al. Analysis of the CTNS gene in patients of German and Swiss origin with nephropathic cystinosis. Hum Mutat. 2002;20(3):237.
Burki E. Ueber die Cystinkrankheit im Kleinkindesalter unter besonderer Berücksichtigung des Augenbefundes [About the cystinosis in infancy with special reference to eye findings]. Ophthalmologica. 1941;101:331–42.
Tsilou E, Zhou M, Gahl W, Sieving PC, Chan CC. Ophthalmic manifestations and histopathology of infantile nephropathic cystinosis: report of a case and review of the literature. Surv Ophthalmol. 2007;52(1):97–105.
Biswas S, Gaviria M, Malheiro L, Marques JP, Giordano V, Liang H. Latest clinical approaches in the ocular management of cystinosis: a review of current practice and opinion from the ophthalmology cystinosis forum. Ophthalmol Ther. 2018;7(2):307–22.
Cherqui S. Cysteamine therapy: a treatment for cystinosis, not a cure. Kidney Int. 2012;81(2):127–9.
Gahl WA, Tietze F, Butler JD, Schulman JD. Cysteamine depletes cystinotic leucocyte granular fractions of cystine by the mechanism of disulphide interchange. Biochem J. 1985;228(3):545–50.
Kaur S, Sarma P, Kaur H, Prajapat M, Shekhar N, Bhattacharyya J, Kaur H, Kumar S, Medhi B, Ram J, Das D, Avti P, Prakash A, Singh R, Bhattacharyya A. Efficacy and safety of topical cysteamine in corneal cystinosis: a systematic review and meta-analysis. Am J Ophthalmol. 2021;223:275–85.
Liang H, Labbé A, Le Mouhaër J, Plisson C, Baudouin C. A new viscous cysteamine eye drops treatment for ophthalmic cystinosis: an open-label randomized comparative phase III pivotal study. Invest Ophthalmol Vis Sci. 2017;58(4):2275–83.
Bertholet-Thomas A, Berthiller J, Tasic V, Kassai B, Otukesh H, Greco M, et al. Worldwide view of nephropathic cystinosis: results from a survey from 30 countries. BMC Nephrol. 2017;18(1):210.
Ling C, Liu X, Chen Z, Jiang Y, Fan J, Meng Q, Fu Q, Yu J. Corneal cystine crystals in cystinosis. Arch Dis Child. 2017;102(12):1185.
Jellouli M, Ferjani M, Abidi K, Zarrouk C, Abdelmoula J, Gargah T. Infantile cystinosis: from dialysis to renal transplantation. Saudi J Kidney Dis Transpl. 2017;28(5):1180–3.
Soliman NA, El-Baroudy R, Rizk A, Bazaraa H, Younan A. Nephropathic cystinosis in children: an overlooked disease. Saudi J Kidney Dis Transpl. 2009;20(3):436–42.
Vaisbich MH, Koch VH. Report of a Brazilian multicenter study on nephropathic cystinosis. Nephron Clin Pract. 2010;114(1):c12–8.
Cherqui S. Cystinose - De la découverte du gène aux premiers essais de thérapie génique [Cystinosis: from the gene identification to the first gene therapy clinical trial]. Med Sci (Paris). 2023;39(3):253–61.
Acknowledgements
The author would like to acknowledge the patients and their family for their consent and contributions.
Funding
The financial support is provided by Hyderabad Eye Research Foundation and Hyderabad Eye Institute, Hyderabad, India.
Author information
Authors and Affiliations
Contributions
AH played key roles in clinical diagnosis, data analysis, and manuscript composition. AV made significant contributions to genetic analysis and manuscript preparation. DSA was instrumental in imaging and data collection. DPE provided valuable expertise in overall case analysis, diagnosis, and manuscript quality. MR contributed to the design, clinical diagnosis, data analysis, management, and manuscript writing.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval for this study was waived by the Ethics Committee at L V Prasad Eye Institute, Hyderabad (Ethics Ref. No. LEC-BHR-R-05–24-1234) under the retrospective nature of the study. Informed written consent to participate in the study was provided by all participants or their parent or legal guardian in the case of children under 16 years old.
Consent for publication
Written informed consent was obtained from all participants or their parent or legal guardian in the case of children under 16 years old for the publication of this case report and any sharing of images or data. A copy of the written consent is available on request by the editor in chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Heroor, A., Verma, A., Achanta, D.S. et al. Unveiling cystinosis in India. J Rare Dis 3, 25 (2024). https://doi.org/10.1007/s44162-024-00046-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44162-024-00046-x