
Abstract
Porphyrias are a cluster of inherited metabolic diseases. Acute intermittent porphyria (AIP) is inherited autosomal dominantly that presents with multi-systemic symptoms and acute repetitive attacks in any age of lifespan. Spinal muscular atrophy (SMA) is a motor neuron disease that is autosomal recessively inherited and seen with a relatively higher incidence in Turkey. In this case report, we discuss a 27-year-old male with gait problems and fatigue. Here, we report a familial heterozygous mutation in hydroxymethylbilane synthase (HMBS) gene together with homozygous deletion in the survival motor neuron 1 (SMN1) gene in a Turkish patient.
Similar content being viewed by others
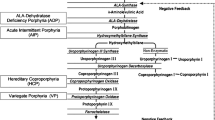


Porphyrias are a group of metabolic diseases due to mutations causing different enzyme defects and resulting in the accumulation of metabolic intermediates causing gastrointestinal, skin, and neurological manifestations [1]. There are eight types of porphyria associated with each enzyme on the heme biosynthesis pathway.
Acute intermittent porphyria (AIP) is the most commonly encountered form of hepatic porphyria caused by the deficiency of the hydroxymethylbilane synthase (HMBS) enzyme. As the disease is inherited autosomal dominantly, heterozygote patients may experience acute attacks when exacerbating factors are introduced, despite most remaining symptom-free [2].
Spinal muscular atrophy (SMA) is an autosomal recessively inherited disease that occurs as a result of degeneration in lower motor neurons with a high childhood incidence of 1/6000 live births in Turkey. SMA disease arises from homozygous mutations of survival motor neuron (SMN) which present as two similar copies: SMN1 gene deletions constitute most of the cases whereas mutations in the SMN2 gene are not known to cause spinal muscle atrophy [3]. Type 3 SMA (Kugelberg-Welander) is a milder form of the disease with childhood or adolescent onset; thus, milder limitations than SMA1 or SMA2 are observed in the gait of patients [4]. Here, we present a phenomenal case of a 27-year-old man who harbours mutations in both HMBS and SMN1 genes.
Case report
A now 27-year-old man has first attended the paediatrics department with sudden falls and a tendency to tiptoe-walk starting at the age of 4. He is a 4 pack-year smoker and does not consume alcohol at present. He has no known prescribed medications. He has no other known comorbid diseases and has not undergone any procedures other than two muscle biopsies.
He was born term, vaginally without complications and his independent walking age was 11 months. An otherwise completely healthy 4-year-old boy sought information from the paediatric neurology department with complaints of fatigue and weakness in all extremities, predominantly on the lower extremities. His serum creatine kinase (CK) level ranged between 178 and 869 (N: 39–308, IU/L) throughout the years. Initially diagnosed at the age of 4 with myopathy, diagnosis of SMA was only possible after having EMG at age 5 which showed slowly progressive anterior horn disease. Homozygous deletion in the exons 7–8 of the SMN1 gene, with 4 copies of SMN2, confirmed the SMA type 3 diagnosis. At age 27, he still is ambulatory with difficulty in walking and limited walking distance but has a full-time home-based profession. Although he does not need a wheelchair for daily chores, he usually needs it for long distances. In the last year, his weakness worsened even more, especially prominent on his left side. On his final neurological examination, cranial area and neck muscles were normal, axial muscles were moderately weak, and muscle strength in extremities was around 4/5 Medical Research Council (MRC) in the upper and 2–3/5 MRC in the lower proximal muscles. Bilateral but more prominent left, Achilles contracture was observed. The right calf was hypertrophic. Deep tendon reflexes were absent. Superficial and deep sensory examinations were normal.
His family history is negative for parental consanguinity or SMA but consists of three cousins suffering from AIP. We started following mentioned cousins in our institute after one of the cousins experienced an acute attack. Due to having porphyria in cousins, a family pedigree was crafted (Fig. 1) and urine porphobilinogen test and genetic analysis were requested from the family members to detect porphyria. Our patient’s genetic result revealed a heterozygous mutation in the HMBS gene, c913.2A > G (IVS13-2A > G) suggesting AIP. Similarly, his mother who migrated from the Balkans also had heterozygous mutations in HMBS, hemochromatosis (HFE), and 5-aminolevulinate synthase 2 (ALAS-2) genes. Both family members tested negative for urine porphobilinogen. Furthermore, in our patient’s lab results over the years, ALT (NR: 0–40U/L) and AST (NR: 0–38U/L) levels were within normal range except for a 2-U/L increase in ALT in 2015 of 52U/L (NR: 0–50 U/L). Additionally in 2015, the patient’s direct bilirubin was found slightly elevated as 0.25 mg/dL (NR: 0–0.20 mg/dL), while in 2014, his direct bilirubin level was observed at 0.40 mg/dL (NR: 0–0.2 mg/dL). In his lab results in 2013, 2015, and 2016, decreased creatinine levels of 0.40, 0.41, and 0.38 mg/dL (NR: 0.70–1.30 mg/dL) were noted respectively. Contributing to these results, a urine myoglobin level of 85 ng/mL (NR: 16–76 ng/mL) was observed in 2011.

Family pedigree of the patient illustrating the HMBS gene status
Upon questioning, our patient did not claim any porphyria-suggesting symptoms such as constipation, abdominal pain, skin lesions, and abnormal urine colour. His mother affirmed she had unbearable abdominal pain during menstruation once that she attributed to menstrual cramps. Besides, she also denied any porphyria-like symptoms.
Discussion
AIP is a rare disease with a prevalence of 1 to 8 per 100,000 in European countries. Together with SMA, the possibility of having both mutated genes in the same individual varies between 1 per 75 to 600 million making our case a unique report [5].
Both diseases have challenges starting from the diagnosis. Porphyria presents with nonspecific symptoms making differential diagnosis more challenging, although it can easily be diagnosed with urine porphobilinogen. The diagnosis of SMA, especially SMA 3, may also be challenging as the clinical picture is very similar to a limb-girdle type myopathy and elevated CK levels. Therefore, children with gait problems bring dystrophy to mind initially. Diagnosis is instituted with EMG and genetic analysis. However, screening tests and genetic analysis have been started to be implemented recently [6]. Similarly, in our patient, the SMA diagnosis was made 2 years after the myopathy diagnosis and genetic analysis for porphyria was only requested owing to the family history.
Increased production of 5-aminolaevulinic acid synthase 1 (ALAS-1) enzyme in AIP can lead to impairment in mitochondrial bioenergetic pathways of the liver, which can lead to liver damage [7]. Our patient’s laboratory results exhibit a normal functioning liver in relation to liver function parameters such as ALT and AST. However, in 2015, the patient’s ALT and direct bilirubin levels were slightly increased. While a 2-U/L increase in ALT (NR: 0–50 U/L) and a 0.05-mg/dL increase in direct bilirubin (NR: 0–0.20 mg/dL) do not indicate a hepatic involvement of AIP, the laboratory values may be linked to a mild hepatic disease or hepatic function disruption due to the use of a specific drug at the time. On the other hand, increasing the level of AST/ALT can be associated with a high level of CK enzyme.
Our patient did not present with any porphyria-like symptoms so far which might be related to his heterozygous mutation [8]. Porphyria-suggesting laboratory results such as urine porphobilinogen, ALAS-1, and urine protoporphyrin were negative. However, it should be emphasized that positivity is only captured during attacks. Although our patient has not had an attack of porphyria until now, he might encounter an attack in the future.
Consanguineous marriages are known to increase the risk of autosomal recessive genetic diseases. Our patient comes from a family with no known previous SMA patients. Upon questioning, we did not find any kinship between his parents. We could not screen his grandparents, however we believe a consanguineous marriage may be the initial reason.
It is known that SMA is a progressive disease affecting predominantly proximal muscles [9]. In the neurological examination performed, predominant proximal weakness and atrophy were detected in our patient. Proximal muscular atrophy had also been reported in several cases of porphyria without any aberrant symptoms such as abdominal pain, mainly due to neurovisceral neuropathy [10]. Therefore, this information and given the faster progression of the disease in the past year, could it be possible that the existing HMBS mutation acts as a modifier in the progression of SMA in our patient? Another explanation is that our patient has slowly progressive SMA and has not been exposed to stressors such as porphyrinogenic drugs to initiate a porphyria attack yet.
Porphyria education is a fundamental element to overcome this enigma. Necessary awareness seminars amongst physicians explaining the symptoms should be executed and they should be briefed about treatment options, that a simple glucose infusion might cease attacks. Prevention of the occurrence of attacks by eliminating porphyrinogenic drugs, behavioural adjustments, and stress avoidance should be advised to patients. Although there were no porphyria-like attacks in our case, knowing beforehand that there is a disease that can be exacerbated by triggering factors such as smoking might facilitate the diagnostic process in the future if there are complaints and can prevent complications.
To our knowledge, this is the first case ever reported of an individual possessing mutations for both SMA and porphyria. An individual carrying mutated genes for both diseases is similar to the odds of two pins colliding in mid-air. Further gene analyses are needed in patients that are known to suffer from a congenitally inherited disease to uncover a possible secondary disease.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
References
Ramanujam VS, Anderson KE. Porphyria diagnostics-part 1: a brief overview of the porphyrias. Curr Protoc Hum Genet. 2015;86:17.20.1–17.20.26. Published 2015 Jul https://doi.org/10.1002/0471142905.hg1720s86
Chen B, Solis-Villa C, Hakenberg J, et al. Acute intermittent porphyria: predicted pathogenicity of HMBS variants indicates extremely low penetrance of the autosomal dominant disease. Hum Mutat. 2016;37(11):1215–22. https://doi.org/10.1002/humu.23067.
Sel S. K, Kasap H, Koç F, Güzel A. İ. Spinal Müsküler Atrofi ve Moleküler Genetiği. aktd. 2012;21(1):1–26.
Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120–33. https://doi.org/10.1016/S0140-6736(08)60921-6.
Nordmann Y, Puy H, Da Silva V, et al. Acute intermittent porphyria: prevalence of mutations in the porphobilinogen deaminase gene in blood donors in France. J Intern Med. 1997;242(3):213–7. https://doi.org/10.1046/j.1365-2796.1997.00189.x.
Prior TW. Spinal muscular atrophy: a time for screening. Curr Opin Pediatr. 2010;22(6):696–702. https://doi.org/10.1097/MOP.0b013e32833f3046.
Homedan C, Laafi J, Schmitt C, et al. Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model. Int J Biochem Cell Biol. 2014;51:93–101. https://doi.org/10.1016/j.biocel.2014.03.032.
Yasuda M, Chen B, Desnick RJ. Recent advances on porphyria genetics: inheritance, penetrance & molecular heterogeneity, including new modifying/causative genes. Mol Genet Metab. 2019;128(3):320–31. https://doi.org/10.1016/j.ymgme.2018.11.012.
Liewluck T, Saperstein DS. Progressive muscular atrophy. Neurol Clin. 2015;33(4):761–73. https://doi.org/10.1016/j.ncl.2015.07.005.
Albertyn CH, Sonderup M, Bryer A, Corrigall A, Meissner P, Heckmann JM. Acute intermittent porphyria presenting as progressive muscular atrophy in a young black man. S Afr Med J. 2014;104(4):283–5. https://doi.org/10.7196/samj.7785.
Acknowledgements
Contents of this case report were partially published at ICPP (International Congress on Porphyrins and Porphyrias) between 4 and 7 September 2022 in Sofia, Bulgaria.
Funding
No funding was received for conducting this study.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by SC, İA, EBA, and GY. The first draft of the manuscript was written by SC, İA, EBA, and PO, and GS supervised the process. All authors commented on previous versions of the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The patient has given written consent for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Çavdaroğlu, S., Altun, İ., Atasay, E.B. et al. Acute intermittent porphyria and spinal muscular atrophy: two rare diseases seen in one patient. J Rare Dis 2, 4 (2023). https://doi.org/10.1007/s44162-023-00007-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44162-023-00007-w