
Abstract
The conversion of biomass to chemicals/fuels has emerged as a valuable solution that offers both environmental and economic benefits, with the transformation of carbohydrate into formic acid garnering escalating scholar interest. However, the relative limited efficiency of catalyzed-oxidation or expensive cost of H2O2 and alkali in wet hydrothermal oxidation impose limitations on industrialization. This paper proposed a new idea for formic acid production by O2-H2O2 co-oxidation of carbohydrate. A two-step reaction method was developed, where the initial step is engineered to regulate the carbon chain cleavage of carbohydrates to augment the production of active intermediate. Oxygen was employed in the subsequent step as effective oxidant through free radical mechanism, resulting in a formic acid yield of 82.6%. Theoretical calculation, intermediates detection and real time EPR confirmed the reaction mechanism. Finally, the universality of the reaction was verified by using disaccharides and polysaccharides such as cellulose as substrates.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Non-renewable fossil fuels constitute a significant portion of the global energy composition and are progressively being exhausted [1,2,3]. In response to the energy crisis, the exploration of biomass has garnered the attention of numerous researchers [4]. Biomass, characterized by its abundance, cost-effectiveness, and environmentally friendly nature, fundamentally owns the chemical energy encapsulated within the chemical compounds that make up the organism, and the synthesis and utilization of biomass energy is a process of zero carbon emission [5, 6]. To truly harvest this energy source for the resolution of energy crisis, economical and sustainable conversion methodologies are required, necessitating careful consideration of reaction methodologies and catalyst design [7].
Rich functional groups are contained in biomass, including hydroxyl, carboxyl, amino, sulfonic acid and other functional groups, which facilitate the production of a diverse array of chemicals [8, 9]. Among these, organic acids serve as crucial raw materials for high-energy–density and value-added chemical products in the industrial sector. A variety of organic acids are produced from biomass, including formic acid, lactic acid, levulinic acid and glycolic acid [10,11,12]. Formic acid (FA), the simplest aliphatic carboxylic acid, is characterized by its relative non-toxicity, non-corrosiveness, biodegradability, safety, and ease of storage and transportation, making it environmentally friendly [13]. It is widely used in fine chemical industry, such as drug synthesis, pesticide production, chemical products, etc. In addition to that, formic acid is a key energy carrier, as formic acid has a higher hydrogen content (4.4wt %) and volume capacity (53.7 g/L) [14, 15] compared to gaseous hydrogen. At the same time, formic acid is a carbon monoxide carrier that can be decomposed in a controlled manner [15]. Consequently, formic acid contributes to the construction of new framework for the renewable energy economy [16].
The demand of formic acid for these various applications has greatly promoted the development of the approaches for its production. Compared with industrial production of formic acid from fossil energy, production of formic acid from biomass has environmental and economic benefits [17]. With 75% of biomass on Earth being carbohydrates, the production of formic acid from carbohydrates attracted significant research attention. As shown in Table 1, previous studies are mainly divided into two categories: catalytic oxidation and hydrothermal oxidation. In catalytic oxidation reactions with oxygen (O2), heteropoly acids and vanadyl groups-based catalysts are normally required [18,19,20,21,22,23,24]. However, the limited reaction rate and conversion rate as well as the high cost of catalysts pose application challenges [25,26,27]. High-temperature water is an environmentally benign solvent and exhibits exceptional performance in biomass conversion due to its unique properties, thereby representing a potential reaction media in this field [28]. In hydrothermal oxidation of carbohydrate, hydrogen peroxide (H2O2) is typically employed as the oxidant in conjunction with a strong homogeneous alkali in high concentrations, which yields high reaction efficiency owing to the mechanism of free radical reactions [29]. However, it is expensive to use a large amount of H2O2, and the overuse of homogeneous alkali is detrimental to reactors and environment [26, 30,31,32,33]. Therefore, to develop an efficient approach for FA production from carbohydrate, employing the advantages of hydrothermal reactions, while replacing or reducing H2O2 with cheaper oxidant such as O2 becomes a pursuit.
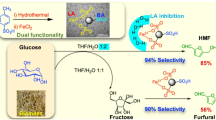
Indeed, under hydrothermal conditions, the density, dielectric constant, and viscosity of water are reduced [34], thereby enhancing the solubility of the gaseous phase and non-polar organic matter. This could disrupt the phase interface between O2 and organic matter, rendering O2 an efficient oxidant [35, 36]. Concurrently, our group’s prior studies have determined that the key to converting carbohydrates into high-yield formic acid lies in the ability to cleave carbon bonds and accumulate two-carbon aldoses. Certain transition metal oxides can selectively regulate the bond-breaking mode of monosaccharides such as glucose [30, 31, 37]. In light of these considerations, we propose a strategy that employs both O2 and H2O2 as oxidants to facilitate the free radical reaction via transition metal oxides as the catalyst. In detail, a two-step reaction method with O2-H2O2 co-oxidation via a 10% Co3O4/C catalyst was designed. When a small quantity of H2O2 and dominant O2 are used in tandem, the hydroxyl radical produced by H2O2 stimulates the activity of O2, yielding a substantial number of superoxide anions and hydroxyl radicals [38, 39]. Simultaneously, in the presence of hydroxyl radical, Co3O4/C catalyst regulates the bond-breaking mode of glucose, culminating in the accumulation of glycolaldehyde as the primary product. These two factors finally lead to the high yield of formic acid as 82.6%, and other polysaccharides could also be converted as substrates. Thus, the purpose of ensuring the yield of formic acid with economic O2 as oxidant via recoverable catalyst was achieved simultaneously with the effect of 1 + 1 > 2 by capitalizing on the separate advantages of catalytic and hydrothermal oxidation (as shown in Fig. 1).

The illustration of O2-H2O2 co-oxidation of carbohydrate to achieve highly efficient FA production
2 Experimental section
2.1 Experimental procedures
The experiment of O2-H2O2 co-oxidation of glucose to produce formic acid was carried out in SUS 316 six-strand-reactors (see Figure S1a) [40]. The procedure involved two main steps. In a typical run, in the first step, Co3O4/C catalyst, glucose, hydrogen peroxide (H2O2), sodium hydroxide (NaOH), deionized water and agitator were added to the reactor successively. After sealing, the reactor was placed in a water bath at 25 ℃ for a certain amount of time. In the second step, desired amount of oxygen (O2) was purged to the reactor. After the heating system of six-strand-reactor reached designed temperature, the reactor was quickly transferred to the heating system. After the preset reaction time, the reactor was cooled rapidly. Subsequently, gaseous, and liquid products were gathered for analysis. The liquid products were filtered through a 0.22 µm aqueous phase filter (hydrophilic PTFE film) and preserved in an analytical bottle, which were analyzed by high performance liquid chromatography (HPLC) and total organic carbons (TOC). The solid sample post-reaction was subjected to repeated washing with deionized water and ethanol, followed by vacuum drying, and then collected for further analysis. Details of the analysis can be found in the Supplementary Information.
2.2 Co3O4/C catalyst synthesis and recycling tests
The Co3O4/C catalyst was synthesized by the following steps. Firstly, a certain amount of carbon carrier was added to 50 mL water and continuously stirred at 500 r/min at room temperature for 1 h, then cobaltous nitrate hexahydrate was added to the mixture and stirred at the same speed for 6 h to form the precursor. A solution of 0.5 mol·L−1 NaOH was then dropped into the suspension until the pH reaches 10.0. Finally, the suspension was washed several times with a 50 mL deionized water suction filter to make its pH neutral, and the obtained solids were then placed in a freeze-drying machine to dry overnight to obtain the Co3O4/C catalyst [9].
To elucidate the microstructure of the catalyst, it was characterized by inductively coupled plasma atomic emission spectroscopy (ICP-AES), nitrogen adsorption/desorption tests, transmission electron microscopy (TEM), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS). Details of the analysis can be found in the Supplementary Information.
When the used Co3O4/C was tested for recyclability, the typical regeneration procedures are as follows. Initially, the retrieved Co3O4/C was subjected to a washing process with saturated sodium carbonate and water, each conducted three times in succession. Following an overnight lyophilization process, the regenerated Co3O4/C was obtained.
2.3 Brief description of EPR free radical test and VASP theory calculation
The in-situ examinations of free radicals were conducted in a reactor designed with capabilities for ventilation, pressure maintenance, and real-time sampling. The samples, procured in real time throughout the reaction process, were instantaneously dispatched to the Electron Paramagnetic Resonance spectrometer (EPR) for the detection of free radical production (see Figure S1b).
Spin-polarized electronic structure calculations were performed using the plane-wave basis set approach as implemented in the Vienna ab initio simulation package (VASP). The projector augmented wave (PAW) method was used to represent the ion–core electron interactions. The valence electrons were represented with a plane wave basis set with an energy cutoff of 450 eV. Electronic exchange and correlation were described with the Perdew-Burke-Ernzerhof (PBE) functional. The DFT-D3 method was used to treat van der Waals interactions. A 15 Å vacuum space was used to avoid interactions between surface slabs. The convergence criteria for the self-consistent electronic structure and geometry were set to 10−5 eV and 0.05 eV/Å, respectively. The energy released by binding different substrates to Co3O4 crystals indicated the closeness of adsorption.
2.4 Determination of reaction efficiency
According to the chemical equations:
The theoretical formic acid yield is 6 times of the substrate glucose addition. The yield of product is defined as the ratio of the actual product quantity obtained by the experiment to the theoretical product quantity. The substrate glucose will be partially or completely converted into various products, and the glucose conversion rate is defined as the ratio of the amount of glucose involved in the reaction to the amount of glucose added before the reaction. A 100% hydrogen peroxide refers to the quantity of hydrogen peroxide required to completely oxidize the glucose substrate into formic acid, which is six times the molar quantity of glucose. The formula mentioned above are as follows:
3 Results and discussion
3.1 O2-H2O2 co-oxidation of glucose via two-step reaction method and the screening of active catalyst
We start the investigation of carbohydrate oxidation to FA with glucose as the substrate with the addition of 20% H2O2 or 2 MPa O2 in the temperature range of 100–220 ℃. As shown in Figure S2, if only 2 MPa O2 was added, formic acid yield increased with the increase of temperature, but the overall yield was low (Figure S2a). When only 20% H2O2 was added, the highest formic acid yield was obtained at 180 ℃ and decreased when the temperature was higher (Figure S2a). If O2 and H2O2 were added at the same time, O2-H2O2 had obvious synergistic effect when the reaction temperature was between 160 and 200 ℃, which resulted in FA yield over 30% (Figure S2a).
To enhance FA yield from glucose oxidation, we put forward the two-step reaction strategy with catalysts addition (Table S1). As aforementioned, the key to convert carbohydrates into high-yield formic acid is to break carbon bonds and accumulate two-carbon aldoses [41]. Previous studies have used transition metal oxides as catalysts to regulate the bond breaking of carbohydrate, and it has been found that Vanadium pentoxide was an active catalyst to activate O2 for the oxidation of chitin monomers [42]. Thus, H2O2, glucose, several metal oxides and slight homogeneous alkali were added to the reactor for the formation of two-carbon aldoses in the first step, after which O2 was added at high temperature for FA production. MnO, Fe3O4, Co3O4, NiO, CuO, ZnO and V2O5 were tested as catalysts for the first step. As shown in Figure S2b, Co3O4 has the best catalytic activity and brings 65.3% formic acid yield, which is 1.4 times of the controlling experiment without the catalyst. On the other hand, the effects of other transition metal oxides were significantly different. For example, NiO, in contrast to Co3O4, played a certain inhibitory role in the reaction, which may because that Ni can promote carbon–carbon coupling, as the yield of acetic acid yield was improved than that of the blank group [43]. Similar to the effect of NiO, V2O5 is more likely to convert glucose into acetic acid, which was also reported in previous studies [42].
For further improving the catalyst performance, active carbon C was used as the carrier to synthesize Co3O4/C [44, 45]. By exploring metal loading capacity, the optimal catalyst was determined to be 10% Co3O4/C (Figure S2c). The microstructure of 10% Co3O4/C was elucidated by various analyses. ICP test determined that the actual content of the active component cobalt tetroxide was 9.29%, which was near the designed value of 10%. N2 adsorption–desorption test examined the specific surface area and pore size of the synthesized catalyst, which were slightly smaller than that of the carbon carrier, suggesting that Co3O4 entered the pore of the active carbon (Figure S3a). XRD results show that the crystal structure of Co3O4 was not changed by carbon loading (Figure S3b). XPS test was carried out to determine the valence states of Co and oxygen elements, which remained the same when loaded on carbon support (Fig. 2) [46, 47]. TEM and mapping images of element distribution showed that the catalyst was spherical with particle size ranging from 30–50 nm, and the active component Co3O4 was evenly dispersed on the carrier carbon (Figure S3c).

XPS spectrum of 10% Co3O4/C
The optimization of FA yield was obtained by exploring the effects of different reaction conditions on formic acid production from O2-H2O2 co-oxidation of glucose. The experiment was divided into two steps to discuss. In the first step, the quantity of glucose, reaction time, the amount of H2O2, homogeneous alkali and 10% Co3O4/C were altered (Figure S4). In the second step, the factors as O2 flux, reaction temperature, time and water filling were optimized (Figure S5). After systematically changing the reaction parameters, the optimal conditions were as follow: In the first step, 100 mmol/L glucose, 80 mg 10% Co3O4/C catalyst, 0.5 M NaOH, 20% H2O2 solution, 25 ℃, 1 h were adopted; In the second step, 1.5 MPa O2, 150 ℃, 30 min and 50% water filling were applied, and formic acid yield reached 82.6%. Further analysis of gaseous products indicated that only unreacted O2 remained, while no other carbon containing products such as CO2, CO was detected. In liquid phase, apart from formic acid, acetic acid, lactic acid as byproducts were detected, along with little remained arabinose and erythrose. The corresponding quantity proportions to glucose are depicted in Figure S6. As a comparison, the formic acid yield with the optimal conditions while in one-step reaction was evaluated, that is glucose, 20% H2O2, 80 mg 10% Co3O4/C catalyst, 1.5 MPa O2 were added together at 150 ℃. Only 30.1% formic acid yield was obtained in the one-step experiment, further underscoring the enhancing effect of the two-step reaction strategy.
Notably, the maximum formic acid yield of O2-H2O2 co-oxidation via heterogeneous catalyst Co3O4/C was comparable to that in previous studies under strong alkaline condition with a large amount of H2O2 oxidation or using vanadium catalysts, and comparing to research with O2 as the sole oxidant, the FA yield reached over 80%, which is an obvious increasement even with much less reaction time (Table 1).
The stability of Co3O4/C catalyst was investigated by recycling experiments. After the initial catalytic reaction, Co3O4/C catalyst was collected, rinsed with deionized water for three times, vacuum dried, and applied for the same experimental, denoted as 1st#. The yield of formic acid decreased much with Co3O4/C 1st#, indicating that the catalyst was deactivated after the reaction. It was speculated that the products of glucose oxidation, mainly carboxylic acid, covered the active sites on the catalyst surface. To remove the acid, the catalyst was rinsed with saturated sodium carbonate solution three times before rinsing with deionized water [31]. Co3O4/C obtained from this alkali regeneration process was used for several cycles and recorded as 1st, 2nd, 3rd, respectively. The formic acid yield was almost unchanged after three times experiment, indicating that the alkaline regeneration steps were effective for maintaining the catalytic ability of Co3O4/C catalyst (Figure S7).
The Co3O4/C catalyst before and after the reaction was characterized by N2 adsorption–desorption test and thermogravimetric analysis. Almost the same BET surface area was determined for Co3O4/C catalyst after the reaction (Table S2), and the thermogravimetric test results showed that the content of Co3O4 in Co3O4/C were 9.3% and 9.2% before and after the reaction, respectively. Thus, the composition and content of the active substance of Co3O4/C catalyst were almost unchanged in reaction. In addition, ICP test was carried out to detect the dissolved Co ion in the solution. The content of Co ion in solution was at ppm level, indicating that the Co3O4/C catalyst was hardly dissolved (Table S3). Co3O4/C catalyst before and after reaction was further determined by XRD and TEM (Figure S8 and S9). The crystal structure of Co3O4 barely changed before and after the reaction. Furthermore, Co3O4 was uniformly distributed on C carrier during the cycle, and the crystal structure maintained well. Thus, no agglomeration deactivation was observed in Co3O4 particles during the recycle experiments.
3.2 Investigation of reaction pathway and catalytic mechanism in the first step reaction
To see whether glucose scission to glycolaldehyde was achieved with the designed reaction strategy in the first step, detailed products distribution and reaction pathway were investigated. For the first step, time-sharing experiments with time gradient of 10 min were performed at room temperature, and the products were quantitatively analyzed by HPLC. As shown in Fig. 3, glucose was continuously consumed, and the contents of 5-carbon arabinose and 4-carbon erythrose first increased and then decreased, indicating them as important intermediates. The content of glycolaldehyde and the target product formic acid gradually increased and the amount of glycolaldehyde increased even more, which is the shining point of the first step reaction at room temperature, since glycolaldehyde has great potential to form formic acid. Indeed, when other products that are usually formed in glucose degradation, including glyceraldehyde, dihydroxyacetone, glycolic acid and ethylene glycol, were used as substrates in the second step reaction, the yield of formic acid decreased tremendously compared to that of glycolaldehyde (Figure S10). Almost no 3-carbon aldose was produced during the whole process, which was also noteworthy as they are easily converted to lactic and acetic acid, decreasing the selectivity of product formic acid [48].

a The high-performance liquid chromatography (HPLC) chromatograms of liquid samples in time-gradient experiments of the first step. b The product distributions in time-gradient experiments of the first step. The product distributions in time-gradient experiments of arabinose (c) and erythrose (d) for the first step. The product distributions in time-gradient experiments without oxidant H2O2 (e) or catalyst Co3O4/C (f) for the first step (Reaction conditions: 100 mmol·L.−1 glucose or other substrates, 0.5 M NaOH, 20% H2O2/80 mg Co3O4/C, 25 ℃)
As arabinose and erythrose are important intermediates, they were further used as substrates in the first step of the time-sharing reaction. Arabinose was converted to glycolaldehyde and formaldehyde within 30 min (Fig. 3c), and within 20 min, more than 90% of erythrose was converted to glycolaldehyde (Fig. 3d), and the formic acid yield of 86.2% and 89.8% were obtained by the second step oxygen-supply reaction directly. On the other hand, further increasing reaction time to over 40 min in the first step resulted in the unselective oxidation of glycolaldehyde into glycolic acid, and even carbon chain grown products as erythrose and lactic acid were detected, which was formed by the dehydration of glycolaldehyde and subsequent inadequate oxidation. Thus, the selective formation of formic acid relies not only on the accumulation of glycolaldehyde in the first step but also the timely and adequate oxidation of glycolaldehyde in the second step.
To explore the respective role of oxidant H2O2 and catalyst Co3O4/C in the first step reaction, a time-sharing experiment with only H2O2 or Co3O4/C was conducted at room temperature. When there is no oxidant H2O2 in the first step, fructose rises first and then falls, and lactic acid is the main product (Fig. 3e). Since only Co3O4/C and NaOH were added in this step, these results showed that the base and Co3O4/C catalyst could only promote the alkaline isomerization and reverse aldol condensation of glucose. When catalyst Co3O4/C was not added, the reaction products are complex, and there are various bond-breaking ways of glucose (Fig. 3f). Therefore, the reliable 5-carbon aldose multi-site bond breaking to produce glycolaldehyde and formaldehyde, and the 4-carbon aldose bond breaking to produce two glycolaldehyde require the participation of both catalysts Co3O4/C and oxidant H2O2.
We then used DFT theoretical calculation to study why Co3O4/C afforded the selective glycolaldehyde production from glucose. The energy released by binding different substrates to Co3O4 crystals indicates the closeness of adsorption [49]. Under the condition of the same carbon chain length, the aldehyde group binds Co3O4 more closely than the carboxyl group and the hydroxyl group, and the 2-carbon glycolaldehyde binds Co3O4 more closely than the 3-carbon glyceraldehyde (Fig. 4), which directly support the finding that Co3O4 was beneficial for 2-carbon glycolaldehyde formation from glucose.

Changes in adsorption energy of different compounds bind to Co3O4
Operando diffuse reflectance infrared Fourier transform spectroscopy (Operando–DRIFTS) was next applied to provide insights into the catalytic mechanism of glucose transformation over Co3O4/C by analyzing key species in time–dependent measurements (Figure S11). The signals detected in the 1800–1550 cm–1 region were assigned to C = O stretching vibrations and were attributed to the carbonyl groups of glucose–derived products, which include arabinose and erythrose [50]. These findings demonstrated that glucose was effectively activated on the surface of the Co3O4/C catalyst. A peak of increasing intensity at 1384 cm–1 was attributed to symmetrical stretched vibrations of ‒COO in glycolaldehyde, demonstrating that glycolaldehyde was successfully formed from glucose over Co3O4/C, which was probably through retro-aldol condensation. In the meantime, other products such as fructose, lactate, glyceraldehyde, dihydroxyacetone easily obtained from dehydration, decarboxylation, and isomerization of glucose were not observed [30, 31]. With increased reaction time, a slight peak at 1317 cm–1 corresponded to symmetrical stretching vibration of ‒COO in glycolate (HOCH2COO‒) was observed [51, 52], which was possibly caused by glycolaldehyde oxidation.
Based on these results, we infer the bond-breaking pathway of glucose in the first step (Fig. 5a). With the oxidant H2O2, glucose molecules break α bond to form arabinose, and break β bond to form erythrose and glycolaldehyde. With catalyst Co3O4/C, erythrose continues to break to two molecules of glycolaldehyde, while arabinose is more inclined to break the two-point bond to directly produce two molecules of glycolaldehyde and one molecule of formaldehyde via the mechanism of retro-aldol condensation. Thus, the first step reaction ends with the accumulation of a large amount of glycolaldehyde. Noteworthy, with the combination of oxidant H2O2 and catalyst Co3O4/C, glucose isomerization, dehydration, decarboxylation and other side reactions rarely occur.

a Glucose selective bond breaking to glycolaldehyde in the presence of H2O2 and Co3O4/C. b Whole reaction mechanism of O2-H2O2 co-oxidation of glucose into formic acid catalyzed by Co3O4/C
3.3 Investigation of reaction pathway and catalytic mechanism in the second step reaction
The oxidation mechanism of second step high-temperature reaction was explored by analyzing the changes of free radicals after O2 introduced with EPR instrument. The free radical content in the system was captured and detected at 10 min, 20 min and 30 min, respectively, as showed in Fig. 6. From the beginning of the reaction to 30 min, the content of OH· and O2−· free radicals increased continuously and reached a peak value at 30 min (Fig. 6a, b). On the other hand, when O2 was not added in the second step, only a small number of OH· free radicals were detected in the system, and almost no O2−· free radicals were produced (Fig. 6c, d). Furthermore, when H2O2 was not added, only a small amount of O2−·and OH· free radicals were detected (Fig. 6e, f), indicating that the addition of H2O2 was a prerequisite for the generation of a large number of OH· and O2−·. Thus, the coexistence of oxygen and H2O2 was vital for the abundant formation of O2−·and OH· free radicals. Further, to explore the effect of free radicals for the formic acid production, free radical quenching experiments were carried out. As shown in Table 2, OH· quencher (methanol) and O2-· quencher (p-benzoquinone) were added to the second high temperature system, and formic acid yield was significantly reduced, demonstrating that both O2− and OH· are crucial oxidants for formic acid formation.

In-situ EPR detection of free radical production in the second-step reaction a OH· radical production, b O2−· radical production, c OH· radical production without O2 addition; d O2−· radical production without O2 addition; e OH· radical production without H2O2; f O2.−· radical production without H2O2. (Reaction condition: First step: 100 mmol/L glucose, 80 mg 10% Co3O4/C catalyst, 0.5 M NaOH, 25 ℃, 1 h. Second step: 150 ℃, 30 min, 50% water filling)
Since OH· was vital for the abundant formation of O2−·free radicals in the second high-temperature step, we further investigated the effect of the concentration of the remaining H2O2 on the second step. The experiments of H2O2 with different concentrations were carried out. We used glycolaldehyde as the substrate as it was the main product after the first step reaction. With the introduction of O2, the formic acid yield of 90% was reached when H2O2 exceeds 10% (Table S4). Furthermore, the mixed substrate of glucose, arabinose, erythrose and glycolaldehyde were also applied to mimic the real solution after the first step reaction, and the yield of formic acid was 79.1%, which was similar to that obtained by the previous two-step method (See Table S5). Therefore, with this active hydrothermal atmosphere, relatively small amount of H2O2 was capable to activate O2 sufficiently.
Based on the results, the reaction mechanism of formic acid formation in the second step was proposed (Fig. 5b). First, the remaining H2O2 as the heuristic agent produces a small amount of OH· free radical (H2O2 → 2 OH·). Subsequently, OH· free radical activates O2 to produce O2-·.(2 OH· + O2 + OH− → 2O2−· + 2 H2O). Thirdly, a large number of OH· and O2-· free radicals are produced by radical chain reaction (OH− + O2 → OH· + O2−·). Afterwards, they oxidize the substrate glycolaldehyde adsorbed on the active catalyst Co3O4/C to form the target product formic acid. Finally, we combined the first step of room temperature reaction with the second step of high temperature reaction to illustrate the whole process of the reaction. Glycolaldehyde is more easily adsorbed at the active site of the catalyst and reacts with the O2-· and OH· free radicals to form formaldehyde, which is further oxidized to formic acid (Fig. 5a).
3.4 Conversion of multiple carbohydrates with the proposed reaction strategy
With promising formic acid production obtained from O2-H2O2 co-oxidation of glucose, whether the system is identically effective in converting complicated carbohydrate was explored. Firstly, we expanded the reaction substrates to other monosaccharides, including galactose and fructose. The yield of formic acid from glucose and galactose was similar, indicating that the difference of the chiral structure of aldose had little effect on the yield of formic acid. The formic acid yield of ketose, however, is lower than that of aldose (see Table S6), which is because there is no aldehyde group at the end of the carbon chain, making it difficult to generate glycolaldehyde via the designed reaction approach. At the same time, the keto structure makes it easier to break the bond to form three-carbon glyceraldehyde by reverse aldol condensation, which will further undergo dehydration and isomerization, resulting in the poor selectivity of formic acid [53].
The substrate is further extended to disaccharides. By appropriately increasing the concentration of homogeneous base to 1 M and increasing the first step reaction time at room temperature to promote the break of glycoside and monosaccharide bond, the formic acid yield of cellobiose, maltose, lactose and sucrose was 24.6%, 16.5%, 23.6%, 3.2%, respectively (see Table S6). Cellobiose, maltose and lactose are different in the monomer composition and the connection mode of glucoside bond, which slightly affects the formic acid yield, while sucrose is non-reductive disaccharide, and its formic acid yield is significantly less than that of reducing disaccharide.
With polysaccharide as the substrate, by extending the time of the first step to 8 h, 9.4% and 8.1% formic acid yield was obtained from starch and inulin, and xylan showed promising formic production with 26.3% yield. However, because cellulose is water-insoluble, almost no formic acid is obtained, thus it is pretreated by ball milling, and improved formic acid production was realized [54]. To further increase the formic acid yield of xylan and cellulose, the dosage of H2O2 and oxygen were increased to 50% or 2 MPa, respectively. Under the improved experimental conditions, the formic acid yields of xylan and cellulose were 42.1% and 28.1%, respectively (Figure S12), which was quite an improvement comparing to other researches where formic acid yield were normally less than 20% under similar conditions [55, 56].
4 Conclusions
We propose a method of low cost and high yield method to produce formic acid from carbohydrate biomass. A straight-forward two-step reaction mode was designed with O2-H2O2 co-oxidation. By designing transition metal oxides catalyst 10% Co3O4/C with stable catalytic performance and optimizing the reaction conditions, the formic acid yield was up to 82.6%. Oxidant H2O2 and catalyst Co3O4/C selectively modulate the bond-breaking mode of glucose in the first room temperature reaction and accumulate a large amount of primary product glycolaldehyde. In the second high temperature reaction, OH· radical generated by H2O2 activates O2 to form O2−·, which are the direct oxidants of glycolaldehyde to formic acid. VASP theory calculation shows that glycolaldehyde is more easily combined with Co3O4/C catalyst and oxidized by free radicals to formic acid. In addition, this co-oxidation strategy was broadened to extended substrates as various disaccharides and polysaccharides. This study reveals the potential of O2 as a direct oxidant for efficient formic acid production from carbohydrate under mild hydrothermal conditions and gives hint for catalysts design of regulating glucose carbon chain scission for enhanced oxidation, and thus hopefully to provide insights for industrial utilization of carbohydrate biomass.
Availability of data and materials
All the data are presented in the main paper or additional supporting files.
References
Wise M, Calvin K, Thomson A, Clarke L, Bond-Lamberty B, Sands R, Smith SJ, Janetos A, Edmonds J (2009) Implications of limiting CO2 concentrations for land use and energy. Science 324:1183–1186
Hu YH, Jin F (2019) Special column: solar energy conversion, Front. Energy 13:205–206
Yan N, Chen X (2015) Sustainability: Don’t waste seafood waste. Nature 524:155–157
Meier D, van de Beld B, Bridgwater AV, Elliott DC, Oasmaa A, Preto F (2013) State-of-the-art of fast pyrolysis in IEA bioenergy member countries. Renew Sust Energ Rev 20:619–641
Wang M, Ma J, Liu H, Luo N, Zhao Z, Wang F (2018) Sustainable Productions of Organic Acids and Their Derivatives from Biomass via Selective Oxidative Cleavage of C-C Bond. ACS Catal 8:2129–2165
Li L, Ge Y (2017) System-level cost evaluation for economic viability of cellulosic biofuel manufacturing. Appl Energy 203:711–722
Naik SN, Goud VV, Rout PK, Dalai AK (2010) Production of first and second generation biofuels: A comprehensive review. Renew Sustain Energy Rev 14:578–597
Sharma A, Pareek V, Zhang D (2015) Biomass pyrolysis—A review of modelling, process parameters and catalytic studies. Renew Sust Energ Rev 50:1081–1096
Li J, Zhu P, Zhong H, Yang Y, Cheng J, Wang Y, Jin F (2021) Hydrothermal Reduction of NaHCO3 into Formate with Protein-Based Biomass over Pd/γ-Al2O3 Nanocatalysts. ACS Sustain Chem Eng 9:4791–4800
Adam YS, Fang Y, Huo Z, Zeng X, Jing Z, Jin F (2013) Production of carboxylic acids from glucose with metal oxides under hydrothermal conditions. Res Chem Intermed 41:3201–3211
Zhang Q, Xu S, Cao Y, Ruan R, Clark JH, Hu C, Tsang DCW (2022) Sustainable production of gluconic acid and glucuronic acid via microwave-assisted glucose oxidation over low-cost Cu-biochar catalysts. Green Chem 24:6657–6670
Chen Y, Yang Y, Liu X, Shi X, Wang C, Zhong H, Jin F (2023) Sustainable production of formic acid and acetic acid from biomass. Molecular Catalysis 545:113199
Wei G-H, Lu T, Liu H-Y, Bai J-X, Wang Q, Li G-Y, Liang Y-H (2023) Exploring the continuous cleavage-oxidation mechanism of the catalytic oxidation of cellulose to formic acid: A combined experimental and theoretical study. Fuel 341:127667
Bielinski EA, Lagaditis PO, Zhang Y, Mercado BQ, Würtele C, Bernskoetter WH, Hazari N, Schneider S (2014) Lewis Acid-Assisted Formic Acid Dehydrogenation Using a Pincer-Supported Iron Catalyst. J Am Chem Soc 136:10234–10237
Grasemann M, Laurenczy G (2012) Formic acid as a hydrogen source – recent developments and future trends. Energy Environ Sci 5:8171
Supronowicz W, Ignatyev IA, Lolli G, Wolf A, Zhao L, Mleczko L (2015) Formic acid: a future bridge between the power and chemical industries. Green Chem 17:2904–2911
Jiang J, Wieckowski A (2012) Prospective direct formate fuel cell. Electrochemistry Commun 18:41–43
Gromov NV, Medvedeva TB, Lukoyanov IA, Panchenko VN, Timofeeva MN, Taran OP, Parmon VN (2022) Formic Acid Production via One-Pot Hydrolysis-Oxidation of Starch over Quaternary Ammonium Salts of Vanadium-Containing Keggin-Type Heteropoly Acids. Catalysts 12:1252
Li J, Ding DJ, Deng L, Guo QX, Fu Y (2012) Catalytic Air Oxidation of Biomass-Derived Carbohydrates to Formic Acid. Chemsuschem 5:1313–1318
Gromov NV, Taran OP, Delidovich IV, Pestunov AV, Rodikova YA, Yatsenko DA, Zhizhina EG, Parmon VN (2016) Hydrolytic oxidation of cellulose to formic acid in the presence of Mo-V-P heteropoly acid catalysts. Catal Today 278:74–81
Zhang J, Sun M, Liu X, Han Y (2014) Catalytic oxidative conversion of cellulosic biomass to formic acid and acetic acid with exceptionally high yields. Catal Today 233:77–82
Voß D, Pickel H, Albert J (2020) Improving the fractionated catalytic oxidation of lignocellulosic biomass to formic acid and cellulose by using design of experiments. Chem Ing Tech 92:1343–1343
Tang Z, Deng W, Wang Y, Zhu E, Wan X, Zhang Q, Wang Y (2014) Transformation of Cellulose and its Derived Carbohydrates into Formic and Lactic Acids Catalyzed by Vanadyl Cations. Chemsuschem 7:1557–1567
Niu M, Hou Y, Wu W, Ren S, Yang R (2018) Successive C1–C2 bond cleavage: the mechanism of vanadium(v)-catalyzed aerobic oxidation of d-glucose to formic acid in aqueous solution. Phys Chem Chem Phys 20:17942–17951
Khenkin AM, Leitus G, Neumann R (2010) Electron transfer-oxygen transfer oxygenation of sulfides catalyzed by the H5PV2Mo10O40 polyoxometalate. J Am Chem Soc 132:11446–11448
Li J, Smith RL, Xu S, Li D, Yang J, Zhang K, Shen F (2022) Manganese oxide as an alternative to vanadium-based catalysts for effective conversion of glucose to formic acid in water. Green Chem 24:315–324
Sahoo PK, Zhang T, Das S (2021) Oxidative Transformation of Biomass into Formic Acid. Eur J Org Chem 2021:1331–1343
F. Jin, J. Yun, G. Li, A. Kishita, K. Tohji, H (2008) Enomoto, Hydrothermal conversion of carbohydrate biomass into formic acid at mild temperatures, Green Chem 10
Yun J, Yao G, Jin F, Zhong H, Kishita A, Tohji K, Enomoto H, Wang L (2016) Low-temperature and highly efficient conversion of saccharides into formic acid under hydrothermal conditions. AIChE J 62:3657–3663
Wang C, Chen X, Qi M, Wu J, Gözaydın G, Yan N, Zhong H, Jin F (2019) Room temperature, near-quantitative conversion of glucose into formic acid. Green Chem 21:6089–6096
Wu L, Yang Y, Cheng J, Shi X, Zhong H, Jin F (2022) Highly Efficient Conversion of Carbohydrates into Formic Acid with a Heterogeneous MgO Catalyst at Near-Ambient Temperatures. ACS Sustain Chem Eng 10:15423–15436
Jiang Z, Zhang Z, Song J, Meng Q, Zhou H, He Z, Han B (2015) Metal-Oxide-Catalyzed Efficient Conversion of Cellulose to Oxalic Acid in Alkaline Solution under Low Oxygen Pressure. ACS Sustainable Chemistry & Engineering 4:305–311
Wang G, Meng Y, Zhou J, Zhang L (2018) Selective hydrothermal degradation of cellulose to formic acid in alkaline solutions. Cellulose 25:5659–5668
Akiya N, Savage PE (2002) Roles of Water for Chemical Reactions in High-Temperature Water. Chem Rev 102:2725–2750
Nakhshiniev B, Biddinika MK, Gonzales HB, Sumida H, Yoshikawa K (2014) Evaluation of hydrothermal treatment in enhancing rice straw compost stability and maturity. Bioresour Technol 151:306–313
Jin F, Enomoto H (2007) Application of hydrothermal reaction to conversion of plant-origin biomasses into acetic and lactic acids. J Mater Sci 43:2463–2471
Liu X, Zhong H, Wang C, He D, Jin F (2022) CO2 reduction into formic acid under hydrothermal conditions: A mini review. Energy Sci Enginer 10:1601–1613
Les A, Adamowicz L (1990) Theoretical-Study of Simple Electron-Transfer Reactions Involving Oxo Radicals and Anions. Chem Phys Lett 175:187–191
Guo J, Xie T, Yang S, Xie Q, Liu Q, Qin J (2021) Free-Radical and Non-Free-Radical Based Reaction Pathways of Iodide Oxidation by Hydrogen Peroxide in Acid Solution–Ab Initio Calculations. Russ J Phys Chem A 95:S15–S22
Jin F-M, Kishita A, Moriya T, Enomoto H (2001) Kinetics of oxidation of food wastes with H2O2 in supercritical water. J Supercrit Fluids 19:251–262
Huo Z, Fang Y, Yao G, Zeng X, Ren D, Jin F (2015) Improved two-step hydrothermal process for acetic acid production from carbohydrate biomass. J Energy Chem 24:207–212
Chen X, Yang H, Zhong Z, Yan N (2017) Base-catalysed, one-step mechanochemical conversion of chitin and shrimp shells into low molecular weight chitosan. Green Chem 19:2783–2792
Zhong H, Yao G, Cui X, Yan P, Wang X, Jin F (2019) Selective conversion of carbon dioxide into methane with a 98% yield on an in situ formed Ni nanoparticle catalyst in water. Chem Eng J 357:421–427
Munnik P, de Jongh PE, de Jong KP (2015) Recent Developments in the Synthesis of Supported Catalysts. Chem Rev 115:6687–6718
Wang X, Yang Y, Zhong H, Wang T, Cheng J, Jin F (2021) Molecular H2O promoted catalytic bicarbonate reduction with methanol into formate over Pd0.5Cu0.5/C under mild hydrothermal conditions. Green Chem 23:430–439
Chen Z, Kronawitter CX, Koel BE (2015) Facet-dependent activity and stability of Co3O4 nanocrystals towards the oxygen evolution reaction. Phys Chem Chem Phys 17:29387–29393
Ma L, Seo CY, Chen X, Sun K, Schwank JW (2018) Indium-doped Co3O4 nanorods for catalytic oxidation of CO and C3H6 towards diesel exhaust. Appl Catal B 222:44–58
Yan W, Jin B, Cheng J, Shi X, Zhong H, Jin F (2022) Ru/ZrO2 as a Facile and Efficient Heterogeneous Catalyst for the Catalytic Hydrogenation of Bicarbonate Using Biodiesel-Waste Glycerol as a Hydrogen Donor. ACS Sustain Chem Eng 10:5374–5383
Yang W, Li X, Jiang Z, Li C, Zhao J, Wang H, Liao Q (2020) Structure-dependent catalysis of Co3O4 crystals in persulfate activation via nonradical pathway. Appl Surf Sci 525:146482
Jobson E, Baiker A, Wokaun A (1989) Interaction of ethylene glycol with alumina supported copper — an FTIR study. Ber Bunsen-Ges Phys Chem Chem Phys 93:64–70
Wang L, Meng H, Shen PK, Bianchini C, Vizza F, Wei Z (2011) In situ FTIR spectroelectrochemical study on the mechanism of ethylene glycol electrocatalytic oxidation at a Pd electrode. Phys Chem Chem Phys 13:2667–2673
Yang Y, Zhong H, Cheng J, Hu YH, Smith RL, Jin F (2023) Carbohydrates via hot water as catalyst for CO2 reduction reaction. Next Energy 1:100037
Xu H, Ye X, Shi X, Zhong H, He D, Jin B, Jin F (2022) ZnO as a simple and facile catalyst for acid-base coordination transformation of biomass-based monosaccharides into lactic acid. Molecular Catalysis 522:112241
Chen X, Gao Y, Wang L, Chen H, Yan N (2015) Effect of Treatment Methods on Chitin Structure and Its Transformation into Nitrogen-Containing Chemicals. ChemPlusChem 80:1565–1572
Demesa AG, Laari A, Turunen I, Sillanpää M (2015) Alkaline Partial Wet Oxidation of Lignin for the Production of Carboxylic Acids, Catal. Sci Technol 38:2270–2278
Gromov NV, Medvedeva TB, Lukoyanov IA, Panchenko VN, Prikhod’ko SA, Parmon VN, Timofeeva MN (2023) Hydrolysis-oxidation of cellulose to formic acid in the presence of micellar vanadium-containing molybdophosphoric heteropoly acids. Results in Engineering 17:100913
Funding
The authors appreciatively acknowledge the financial support of the National Natural Science Foundation of China (No. 21978170 & 22108171), the Natural Science Foundation of Shanghai (No. 23ZR1435200), and Shanghai Key Laboratory of Hydrogen Science & Center of Hydrogen Science, Shanghai Jiao Tong University, China.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. YJC: Conceptualization, Methodology, Investigation, Writing original draft. YY: Conceptualization, Methodology, Validation, Supervision, Writing-review and editing, Project administration. XL: Resources, Methodology. FMJ: Conceptualization, Resources, Methodology, Validation, Supervision.
Corresponding authors
Ethics declarations
Competing interests
Fangming Jin is an Associate Editor of Carbon Neutrality and was not involved in the editorial review, nor the decision to publish this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Y., Yang, Y., Liu, X. et al. O2-H2O2 high-efficient co-oxidation of carbohydrate biomass to formic acid via Co3O4/C nanocatalyst. Carb Neutrality 3, 23 (2024). https://doi.org/10.1007/s43979-024-00099-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s43979-024-00099-3