
Abstract
Diadumene lineata is a colorful sea anemone with orange stripe tissue of the body column and plain tentacles with red lines. We subjected Diadumene lineata to expression cloning and obtained genes encoding orange (OFP: DiLiFP561) and red fluorescent proteins (RFPs: DiLiFP570 and DiLiFP571). These proteins formed obligatory tetramers. All three proteins showed bright fluorescence with the brightness of 58.3 mM−1·cm−1 (DiLiFP561), 43.9 mM−1·cm−1 (DiLiFP570), and 31.2 mM−1·cm−1 (DiLiFP571), which were equivalent to that of commonly used red fluorescent proteins. Amplitude-weighted average fluorescence lifetimes of DiLiFP561, DiLiFP570 and DiLiFP571 were determined as 3.7, 3.6 and 3.0 ns. We determined a crystal structure of DiLiFP570 at 1.63 Å resolution. The crystal structure of DiLiFP570 revealed that the chromophore has an extended π-conjugated structure similar to that of DsRed. Most of the amino acid residues surrounding the chromophore were common between DiLiFP570 and DiLiFP561, except M159 of DiLiFP570 (Lysine in DiLiFP561), which is located close to the chromophore hydroxyl group. Interestingly, a similar K-to-M substitution has been reported in a red-shifted variant of DsRed (mRFP1). It is a striking observation that the naturally evolved color-change variants are consistent with the mutation induced via protein engineering processes. The newly cloned proteins are promising as orange and red fluorescent markers for imaging with long fluorescence lifetime.
Graphical abstract

Similar content being viewed by others

1 Introduction
Fluorescent proteins are widely used in the field of biology for exploring various phenomena in cells and in organisms [1,2,3,4,5,6,7]. Among them, red florescent proteins are preferable for deep tissue imaging due to less light scattering at longer wavelengths [8, 9]. Various red fluorescent proteins have been discovered mainly from Cnidarians [10]. Diadumene lineata (syn. Haliplanella lineata, Diadumene luciae) is a small sea anemone originally inhabiting the coast of Japan [11]. The sea anemone is called ‘orange-striped green sea anemone’ since it has conspicuous orange stripes in the column [12]. Careful observation shows that it has red stripes on its tentacles. These stripes and tentacles emit bright fluorescence under blue light. This observation motivated us to acquire both orange and red fluorescent proteins from the same animal species. We performed expression cloning with Diadumene lineata cDNA employing a frame-insensitive expression vector, pRSET-TriEX [13], and obtained a gene encoding an orange fluorescent protein. We further acquired two genes encoding novel red fluorescent proteins by PCR cloning using the sequence information of the orange fluorescent protein. Here we report photophysical properties and oligomeric states of the new fluorescent proteins, as well as the crystal structure of one of the red fluorescent proteins.
2 Materials and methods
2.1 Expression cloning
Total RNA was extracted from a single orange stripe tissue of the body column of Diadumene lineata with TRIzol reagent according to the manufacturer’s protocol [13]. The extracted total RNA samples were converted to cDNA using SuperScript Double-Stranded cDNA Synthesis Kit (11917-010, Invitrogen, Carlsbad, CA, USA). The double-stranded cDNA was phosphorated and inserted at the SmaI sight of pRSET-TriEX by blunt-end ligation to construct a plasmid for expression cloning. Transformation of Escherichia coli (JM109(DE3)) was performed using this plasmid. A colony that emitted fluorescence was selected and a plasmid encoding the fluorescent protein was prepared and sequenced. The entire coding sequence (CDS) was amplified by PCR and cloned to the BamHI/EcoRI sites of pRSET B for protein expression, spectral analysis, and oligomeric state analysis.
2.2 PCR cloning
Total RNA was prepared from a single tentacle. cDNA was prepared from the total RNA with the same protocol as described above [13]. The cDNA was used as PCR template using the same primers as for amplifying the CDS region of the orange fluorescent protein. The pRSET vector containing the cDNA was used for the transformation of E. coli competent cells. Two red colonies, slightly differently colored, were selected and plasmids were prepared for sequencing and protein expression.
2.3 Photophysical properties
Photophysical characterization (absorption, emission, lifetime measurements) of DiLiFPs was performed in phosphate buffer (10 mM, pH 7.0) supplemented with 150 mM NaCl at 21 °C. Absorption spectra were recorded with a Cary-4000 spectrometer (Agilent Technologies). Fluorescence spectra were recorded with Fluorolog 3 spectrofluorometer (Horiba Jobin Yvon) with 2 nm excitation/emission bandpass and corrected for the instrumental response characteristics. The theoretical extinction coefficient of DiLiFPs at 280 nm was calculated from the amino acid sequence (W, Y and C) [14] and used to determine the total protein concentration. In this calculation, we assumed that there is no intramolecular cysteine S–S bonding. The concentrations of matured chromophore were determined by the alkaline denaturation method [15,16,17]. Based on this matured chromophore concentration, the chromophore extinction coefficient was calculated. The fraction of properly folded fluorescent protein is calculated from the matured chromophore concentration over the total protein concentration. The fluorescence quantum yield was determined by a comparative method using rhodamine 101 in ethanol (Φf = 0.915) as a standard [18].
Fluorescence lifetime was measured using a setup reported previously [19], a single photon counting experiment, the FT200 Picoquant spectrometer coupled with 200 fs excitation laser. Excitation pulse (530 nm, 200 fs, 4 MHz) was provided by a femtosecond Ti:sapphire laser (Coherent Chameleon Ultra II) coupled to an intracavity frequency-doubled OPO (APE) and a pulse picker (4 MHz). Briefly, here the emission from protein solution (absorbance < 0.1 at 530 nm, Helma spectroscopic cell 1 × 1 cm) was collected at 90° through a polarizer set at the magic angle and a monochromator (bandpass 4 nm). The single-photon events were collected by a cooled microchannel plate photomultiplier tube R3809U (Hamamatsu) and recorded by a PicoHarp 300 TCSPC system (PicoQuant, 4 ps bin time). The instrumental response function was recorded at the laser excitation wavelength using colloidal silica (Ludox), and its full width at half maximum was ~ 40 ps. All decays were collected until reaching 104 counts. The decays were recorded at three different emission wavelengths covering the steady-state emission spectra (DiLiFP561: 575–600–625 nm/DiLiFP570 & DiLiFP571: 600–625–650 nm) and globally analyzed by a sum of exponentials convolved by the measured IRF using the FluoFit software (Picoquant). The quality of the fit was judged by the weighted residuals and their autocorrelation function.
To determine pKa, buffers supplemented with 150 mM NaCl were prepared for pH3, 4, 5, 6, 7, 8, 9, 10, 11 and 12 (pH 3, 4, 5: citrate buffer, pH 6, 7, 8, 9: Tris buffer, pH 10, 11, 12: citrate glycine phosphate buffer). The concentrations of DiLiFPs were adjusted to be 10 µg/mL. The fluorescence spectra excited at 530 nm were measured with a fluorometer (FP-8200, JASCO) at 25 °C.
Photostabilities of DiLiFPs were analyzed by determining photobleaching quantum yield based on the protocol reported by De Keersmaecker et al. [20]. Details are available in the supporting materials.
Maturation of DiLiFPs was measured by monitoring the increase of fluorescence intensity in a protein solution rapidly prepared (<1 h) on ice from bacteria in which the protein expression was induced for 1 h. Details are available in the supporting information.
2.4 Expression of fluorescent proteins for spectral and oligomeric states analyses
Fluorescent proteins were prepared as reported previously [13]. Briefly, E. coli competent cells (JM109(DE3)) were transformed with pRSET-DiLiFP561, pRSET-DiLiFP570 and pRSET-DiLiFP571 used for protein expression. The expressed proteins were extracted from E. coli cells and purified by immobilized metal affinity chromatography using Ni–NTA agarose resin (QIAGEN) according to the manufacturer’s protocol. Desalting columns (PD-10, GE Healthcare Life Sciences) were used for removing imidazole and changing buffer to 10 mM phosphate-buffered saline (pH 7.4).
2.5 Pseudo-native polyacrylamide gel electrophoresis
Pseudo-native polyacrylamide gel electrophoresis (PN-PAGE) analysis [21] was performed on 15% polyacrylamide gel (15% acrylamide, 0.09% bis-acrylamide, 0.375 M Tris HCl, pH 8.8, and 0.1% SDS) with 5% polyacrylamide stacking gel (5% acrylamide, 0.13% bis-acrylamide, 0.125 M Tris–HCl, pH 6.8, and 0.1% SDS). Samples (7.5 µg of fluorescent protein into each well) were loaded on the gel in a buffer containing 0.0625 M Tris HCl, pH 6.8, 1% SDS, 10% glycerol without boiling. A protein unstained marker (XL-Ladder Broad, APRO Life Science Institute, Inc., Naruto, Japan) was used as the molecular weight standards. The transmission and the fluorescence images of the gel were taken with a digital camera (PowerShot G11, Canon). After taking the fluorescence image, the gel was stained with a Coomassie brilliant blue stain (CBB-G250, nacalai tesque, Kyoto, Japan) to detect all protein bands. The gel was illuminated with a light table for the transmission image or Transilluminator (TFML-26, UVP) equipped with Visi-Blue Plate (with Orange Cover) for the fluorescence image [13].
2.6 DiLiFP570 expression for crystallization
For structure determination, we subcloned the DiLiFP570 gene into a pET28b vector, whose N-terminal hexahistidine tag can be removed by thrombin digestion. The DiLiFP570 gene was PCR-amplified. In order to use the NdeI site on the vector for subcloning, intrinsic NdeI sites in the fluorescent proteins were removed by introducing a silent mutation: the codon coding 61st histidine ‘CAT’ was changed to ‘CAC’. The DiLiFP570 gene was cloned into the pET28b vector.
DiLiFP570 (pET28b) was expressed in BL21(DE3) using LB medium at 28 °C. After overnight expression, cells were harvested and disrupted by 15 min sonication. Cell suspension after sonication was centrifuged and the supernatant was applied onto a Ni–NTA column. An N-terminus hexahistidine tag of the purified DiLiFP570 sample was cleaved by thrombin and was removed by a subsequent Ni–NTA column chromatography step.
2.7 Crystallization of DiLiFP570
Crystallization of DiLiFP570 was done by the hanging-drop vapor diffusion method. The crystallization condition was 3.0 M ammonium sulphate, 0.1 M imidazole at pH 6.0 at 20 °C with a protein concentration of 2.5 mg/mL. X-ray diffraction data were collected at a beamline BL-5A of KEK photon factory, Tsukuba, Japan. The collected data were processed by XDS [22]. The initial phase was determined by molecular replacement using MOLREP [23]. The closest homologous protein structure was a GFP-like non-fluorescent chromoprotein (PDBID: 3VK1) with a sequence homology of 67%, which was used as a search model. Structural refinement was performed by PhenixRefine [24] and Refmac5 [25]. A structural model was built using Coot [26]. The constructed structural model was deposited to protein data bank through PDBj (pdbj.org) with ID 7X2B. Pictures of the protein structure were created with Pymol (www.pymol.org).
3 Results and discussion
3.1 Cloning of fluorescent protein genes
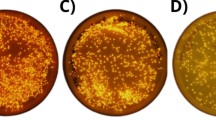
We collected a sea anemone, Diadumene lineata, from the Japan Sea coast. It showed orange stripes in the column and red stripes along the tentacles under blue light (Fig. 1a). The expression cloning of fluorescent proteins was performed from the body that has an orange stripe and found a bright fluorescent colony on the plate (Fig. 1b). From that colony, a gene encoding orange fluorescent protein was obtained. We named this fluorescent protein ‘DiLiFP561’, based on the absorption maximum in the visible range shown in the following section. BLAST [27] search revealed that two very similar fluorescent protein sequences (plum-1 and plum-2) have been reported recently by Sarper et al. [28]. The amino acid sequence of DiLiFP561 is 99.09% and 97.73% identical with plum-2 and plum-1, respectively.

a A small sea anemone Diadumene lineata under blue LED (470 nm) illumination. Stripes at the side of the body contain orange fluorescent protein and red fluorescent protein is expressed in the tentacles. b Expression cloning of fluorescent protein from Diadumene lineata with the pRSET-TriEX vector. The fluorescent colony is indicated by the red circle. c PCR cloning of red fluorescent proteins
By using the N-terminal and C-terminal DNA sequence of DiLiFP561, we performed PCR cloning on the tentacle and found red fluorescent colonies on the plate (Fig. 1c). We obtained genes encoding red fluorescent proteins from two bright clones and named ‘DiLiFP570’ and ‘DiLiFP571’. When compared to DiLiFP561, sequence alignments for DiLiFP570 and DiLiFP571 showed 15 and 12 amino acid substitutions out of 220, respectively, which correspond to identity percentages of 93.18% and 94.55% (Fig. 2). We registered the sequence information of DiLiFP561, DiLiFP570, and DiLiFP571 to DDBJ with the accession number of LC682408, LC682409, and LC682410, respectively.

Alignment of the amino acid sequences of DiLiFP561 with DiLiFP570 and DiLiFP571. Identical amino acids are indicated with asterisks. Non-conserved residues are highlighted with green and indicated with dots or colons, or are highlighted with yellow color and indicated with blanks
3.2 Oligomeric states of DiLiFPs
Oligomeric states of the proteins were analyzed by pseudo-native SDS PAGE (Fig. 3) [13, 21]. The theoretical molecular weight of DiLiFP monomers calculated from their sequence was ~ 25 kDa. All DiLiFPs migrated almost the same distances as tetrameric fluorescent protein (Kaede [7]) with molecular weight ~ 100 kDa (Fig. 3b). A small fraction of DiLiFPs was seen at the same distance as monomeric fluorescent protein (eGFP) (Fig. 3a), however, it was non-fluorescent. From this result, we concluded that DiLiFPs formed obligate tetramers.

Tetramer formation of DiLiFPs in solution revealed by pseudo-native gel-electrophoresis. Proteins were visualized by CBB staining (a) or blue LED illumination (b). eGFP, ScSuFP and Kaede were used as a control of monomer, obligatory dimer, and obligatory tetramer, respectively. Molecular weight standards are shown on the left of the gel. Lanes: 1—eGFP (monomer); 2—ScSuFP (dimer); 3—Kaede (tetramer); 4—DiLiFP561; 5—DiLiFP570; 6—DiLiFP571
3.3 Photophysical characterization of purified fluorescent proteins
Absorption and fluorescence spectra of the purified DiLiFPs were measured at pH7.0 (Table 1, Fig. 4). The absorption bands at 280 nm were assigned to amino acid residues. The visible absorption maxima originated from the chromophores of DiLiFP561, DiLiFP570, and DiLiFP571 were located at 561, 570, and 571 nm, respectively. A shoulder is also observed at higher energy revealing a vibronic structure. The extinction coefficients at these maxima were 1.08 × 105, 9.54 × 104, and 8.44 × 104 M−1·cm−1, respectively, by using the matured chromophore concentration [15,16,17]. The fractions of properly folded DiLiFP561, DiLiFP570, and DiLiFP571 were 55.6%, 50.6%, and 47.8%, respectively.

Absorption and fluorescence emission (excitation at 530 nm) spectra of DiLiFP561 DiLiFP570, and DiLiFP571 at pH 7.0
The emission spectrum of DiLiFP561 was the mirror image of the absorption spectra with a vibrational structure (Fig. 4). The fluorescence spectrum has its maximum at 578 nm, and the fluorescence quantum yield was calculated as 0.54. In contrast, emission spectra for DiLiFP570 and DiLiFP571 were characterized by a broad band with only one maximum and relatively large Stokes shift. The maximum wavelength of the fluorescence spectrum of DiLiFP570 and DiLiFP571 were 600 and 605 nm, respectively, and fluorescence quantum yields were 0.46 and 0.37. The broadband emission of the red fluorescent proteins could be a sign of higher flexibility, due to a larger rearrangement in the excited state concomitant with the existence of a non-radiative pathway.
Absorption and fluorescence spectra of DiLiFP561 were similar to those of plum in Ref. [28]. This result was expected, since the amino acid sequence of DiLiFP561 was 99.09% and 97.73% identical with plum-2 and plum-1, respectively. In contrast, the absorption and fluorescence spectra of DiLiFP570 and DiLiFP571 were red-shifted compared to those of plum-2 and plum-1.
The fluorescence decays were measured at three different wavelengths: the emission maximum, 25 and 50 nm longer than the maximum. Two-phase exponential regression analyses were performed for all three proteins. Fast pre-exponential component (0.3 ns) was also involved for DiLiFP570 and DiLiFP571. The pre-exponential component appeared as a decay component at 600 nm, and a rise component at 625 and 650 nm. The amplitude-weighted average fluorescence lifetimes of DiLiFP561, DiLiFP570 and DiLiFP571 were calculated to 3.7, 3.6 and 3.0 ns, respectively, at their emission maximum wavelength (Table 1, Fig. 5). No significant difference was observed on the average fluorescence lifetime detected at the longer wavelength (Table S2, Supporting information). So far, only five orange and red fluorescent proteins have been reported with longer lifetimes than DiLiFP561 and DiLiFP570: mKO (4.1 ns), KO (4.2 ns) [29], mCherry-XL (3.9 ns) [30], mCyRFP1 (3.7 ns) [31] and mScarlet (3.9 ns) [32]. The shorter lifetime of DiLiFP571 than others was in line with the value of fluorescence quantum yield. DiLiFP570 had 95% contribution from the longest component, whereas the decay curve of DiLiFP571 had about 75% contribution from this component. This can be linked to the existence of more than two states in the ground state [33] or that formed only after excitation. The short 0.3 ns component observed for DiLiFP570, and DiLiFP571 is changing sign (decay/rise) and could be assigned to excited state proton transfer or Förster resonance energy transfer between a neutral and anionic species, as observed for red Kaede species [34, 35].

Fluorescence decay at emission maximum wavelength of DiLiFPs in phosphate buffer (10 mM, pH 7.0) supplemented with 150 mM NaCl at 21℃. A 200-fs pulsed laser at 530 nm at 4 MHz was used for excitation. TCSPC bin time was 4 ps. The full width at half maximum of IRF was ~ 40 ps
To evaluate the pH dependency of DiLiFPs, emission spectra at various pH were acquired (Fig. 6). A blue shift of the emission spectra at acidic pH was observed, only for DiLiFP570 and DiLiFP571. Fluorescence of DiLiFPs became dimmer at lower pH (Fig. 6D). The apparent pKa values, which were estimated from pH values that showed a half maximum of the fluorescence intensity, were 4.3, 4.5 and 4.6, for DiLiFP561, DiLiFP570, DiLiFP571, respectively.

pH dependency of DiLiFP fluorescence excited at 530 nm. Fluorescence emission spectra of a DiLiFP561, b DiLiFP570, c DiLiFP571. d pH titration curve using fluorescence intensities at 578 nm (DiLiFP561), 600 nm (DiLiFP570), or 605 nm (DiLiFP571)
The brightness of fluorescent protein is defined as the product of the molecular absorption coefficient and the fluorescence quantum yield. The brightness at pH 7 of DiLiFP561 and DiLiFP570 was 58.3 and 43.9 mM−1·cm−1, respectively. The brightness of DiLiFP571 was 31.2 mM−1·cm−1, which was lower than other DiLiFPs. Among the long-lifetime orange and red fluorescent proteins, so far reported only mScarlet shows higher brightness (70.0 mM−1·cm−1) than DiLiFP561 and DiLiFP570.
Photostability of DiLiFPs was evaluated by observing photobleaching using proteins immobilized on a cover glass observed with a fluorescence microscope [36]. The bleaching curves were fitted to a two-component exponential model. The bleaching quantum yields of DiLiFPs were determined as 1.45 × 10–5 (45.6%) and 1.39 × 10–6 (54.4%) for DiLiFP561, 7.03 × 10–6 (26.7%) and 8.46 × 10–7 (73.3%) for DiLiFP570 and 5.30 × 10–6 (31.7%) and 9.36 × 10–7 (68.3%) for DiLiFP571 (Fig. S3, supporting information). The bleaching quantum yields were the same order of magnitude as that of EGFP (8.8 × 10–6) [37] and LSSmOrange (1.3 × 10–6) [20]. We concluded that DiLiFPs were enough stable to use for imaging applications (details are in supporting information).
Final evaluation we performed for imaging application was the maturation. DiLiFPs for this experiment were purified rapidly (less than 1 h) on ice from bacteria in which the expression of the proteins was induced for 1 h. The fluorescence intensities of DiLiFP570 and DiLiFP571 were saturated in less than 24 h and the level was kept for 96 h (Fig. S1, Supporting information). Their fluorescence intensities at 3 h were more than half of the plateau level. The maturation of DiLiFP561 showed a similar trend, although the fluorescence intensity at 24 h was ~ 80% of that at 96 h (Fig. S1d, Supporting information). Therefore, the time required for half maturation of all three DiLiFPs were shorter than 3 h. From these results, we concluded that the maturation of DiLiFPs is fast enough for an imaging application.
3.4 Crystal structure of DiLiFP570
The crystal of DiLiFP570 diffracted to a resolution of 1.63 Å. The crystal belonged to I222 space group, and one protein molecule was contained in the asymmetric unit (Table 2).
The DsRed-like tetramer can be generated by the symmetry operation (Fig. 7a). The protein molecule contains an artifact sequence after thrombin cleavage at its N-terminus (Gly–Ser–His) and these residues were disordered in the structure. Except for these disordered terminal residues, electron density map for the rest of the entire protein molecule was clear. Met62–Tyr63–Gly64 forms the DsRed-like chromophore in the structure with the extended two double bonds, which was confirmed by the positioning of N and Cα atoms of Met62 on the same plane of chromophore aromatics (Fig. 7b).

Crystal structure of DiLiFP570. a The DsRed-like tetramer. The tetramer was generated by crystal symmetry operations. Each monomer is shown in different colors (magenta, red, blue, and green) with a ribbon representation. b Chromophore structure of DiLiFP570. c Left: different amino-acid residues between DiLiFP570 and DiLiFP561 mapped onto the DiLiFP570 structure with yellow stick representations. Non-conserved residues from Fig. 1 are selected. Right: expanded view around chromophore. Met159 (corresponding residue for DsRed is Lys163) is highlighted in yellow and labeled
The photophysical properties of fluorescent proteins are influenced by the stabilization of β-strand and hydrogen bond [38, 39]. To reveal the molecular mechanism of the fluorescent color difference between DiLiFP561 and DiLiFP570, we mapped the residues which differ between the two FPs (Fig. 7c; non-conserved residues are shown with yellow stick representation). We found that most of these residues are located at relatively distant positions from the chromophore and are exposed to the solvent. Among them, only Met159 was located in the vicinity of the chromophore (Fig. 7c). For DiLiFP561, residue 159 is Lys, a positively charged residue, while there is no charge in DiLiFP570 at this position. The residue corresponding to Lys159 is Lys163 in DsRed [40]. The primary amine of the Lys163 side chain is considered as a critical group for maintaining the negative charge of the chromophore phenol oxygen via a salt bridge at all times. Similar Lys/Met substitution was observed in a monomeric DsRed variant, mRFP1 generated by the directed evolution method [41]. Red shifts of the emission and excitation peak are also reported in mRFP1; the emission and excitation peak wavelengths of mRFP1 are 24 and 26 nm longer than those of DsRed, while DiLiFP570/ DiLiFP571 show 9/10 nm red shift in their excitation and 22/27 nm in their emission peak wavelengths compared with DiLiFP561. The redshifts of fluorescence for DiLiFP570/DiLiFP571 are also consistent with the difference between DsRed and mRFP1. We think that the absence of the positive charge and associated interactions in DiLiFP570/DiLiFP571 could be reasons for the redshifts of fluorescence. The positive charge of Lys159 localizes electron density towards hydroxyl oxygen in the chromophore. We consider that the effect of this localization is eliminated with methionine substitution at position 159, and the delocalization stabilizes the π-conjugation resulting in a red shift of fluorescence.
4 Conclusion
Considering that all DiLiFPs shown in this study originate from natural organisms, it is interesting that the naturally obtained color-change mutation is consistent with the mutation which was found in the protein engineering processes during the DsRed modifications. To our knowledge, this type of observation is the first in the fluorescence protein research field. Hence, cloning of fluorescent proteins from uninvestigated species will still provide interesting insights for developing fluorescent proteins.
DiLiFP561 and DiLiFP570 were orange and red fluorescent proteins with long fluorescence lifetime and relatively pH stable. The newly cloned orange and red fluorescent proteins are promising as fluorescent markers for imaging that requires a long lifetime and/or under mild acidic conditions.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Mishin, A. S., Subach, F., Yampolsky, I., King, W., Lukyanov, K. A., & Verkhusha, V. (2008). The first mutant of the Aequorea victoria green fluorescent protein that forms a red chromophore. Biochemistry, 47, 4666–4673.
Miyawaki, A., Shcherbakova, D. M., & Verkhusha, V. (2012). Red fluorescent proteins: Chromophore formation and cellular applications. Current Opinion in Structural Biology, 22, 679–688.
Fradkov, A. F., Chen, Y., Ding, L., Barsova, E., Matz, F., & Lukyanov, S. A. (2000). Novel fluorescent protein from Discosoma coral and its mutants possesses a unique far-red fluorescence. FEBS Letters, 479, 127–130.
Lambert, G. G., Depernet, H., Gotthard, G., Schultz, D. T., Navizet, I., Lambert, T., Adams, S. R., Torreblanca-Zanca, A., Chu, M., Bindels, D. S., Levesque, V., Nero Moffatt, J., Salih, A., Royant, A., & Shaner, N. C. (2020). Aequorea’s secrets revealed: New fluorescent proteins with unique properties for bioimaging and biosensing. PLoS Biology, 18, e3000936.
Adam, V., Lelimousin, M., Boehme, S., Desfonds, G., Nienhaus, K., Field, M. J., Wiedenmann, J., McSweeney, S., Nienhaus, G. U., & Bourgeois, D. (2008). Structural characterization of IrisFP, an optical highlighter undergoing multiple photo-induced transformations. Proceedings of the National Academy of Sciences, 105, 18343–18348.
Cormack, B. P., Valdivia, R. H., & Falkow, S. (1996). FACS-optimized mutants of the green fluorescent protein (GFP). Gene, 173, 33–38.
Ando, R., Hama, H., Yamamoto-Hino, M., Mizuno, H., & Miyawaki, A. (2002). An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proceedings of the National Academy of Sciences, 99, 12651–12656.
Deliolanis, N. C., Kasmieh, R., Wurdinger, T., Tannous, B. A., Shah, K., & Ntziachristos, V. (2008). Performance of the red-shifted fluorescent proteins in deep-tissue molecular imaging applications. Journal of Biomedial Optics, 13, 044008.
Hojman, P., Eriksen, J., & Gehl, J. (2009). In vivo imaging of far-red fluorescent proteins after DNA electrotransfer to muscle tissue. Biological Procedures Online, 11, 253–262.
Rodriguez, E. A., Campbell, R. E., Lin, J. Y., Lin, M. Z., Miyawaki, A., Palmer, A. E., Shu, X., Zhang, J., & Tsien, R. Y. (2017). The growing and glowing toolbox of fluorescent and photoactive proteins. Trends in Biochemical Sciences, 42, 111–129.
Newcomer, K., Flenniken, M. M., & Carlton, J. T. (2019). Home and away and home again: Discovery of a native reproductive strategy of the globally invading sea anemone Diadumene lineata (Verrill, 1869) in a satellite population. Biological Invasions, 21, 1491–1497.
Uchida, T. (1932). Occurrence in Japan of Diadumene luciae, a remarkable actinian of rapid dispersal. Journal of the Faculty of Science Hokkaido Imperial University Series VI Zoology, 2, 69–82.
Horiuchi, Y., Laskaratou, D., Sliwa, M., Ruckebusch, C., Hatori, K., Mizuno, H., & Hotta, J. I. (2018). Frame-insensitive expression cloning of fluorescent protein from Scolionema suvaense. International Journal of Molecular Sciences, 19, 371.
Pace, C. N., Vajdos, F., Fee, L., Grimsley, G., & Gray, T. (1995). How to measure and predict the molar absorption coefficient of a protein. Protein Science, 4, 2411–2423.
Ward, W. W., Cody, C. W., Hart, R. C., & Cormier, M. J. (1980). Spectrophotometric identity of the energy transfer chromophores in Renilla and Aequorea green-fluorescent proteins. Photochemistry and Photobiology, 31, 611–615.
Ward, W. W. (1981). Properties of the coelenterate green-fluorescent proteins. Bioluminescence and chemiluminescence (pp. 235–242). Amsterdam: Elsevier.
Gross, L. A., Baird, G. S., Hoffman, R. C., Baldridge, K. K., & Tsien, R. Y. (2000). The structure of the chromophore within DsRed, a red fluorescent protein from coral. Proceedings of the National Academy of Sciences, 97, 11990–11995.
Würth, C., Grabolle, M., Pauli, J., Spieles, M., & Resch-Genger, U. (2013). Relative and absolute determination of fluorescence quantum yields of transparent samples. Nature Protocols, 8, 1535–1550.
Ghose, A., Rebarz, M., Maltsev, O., Hintermann, L., Ruckebusch, C., Fron, E., Hofkens, J., Mély, Y., Naumov, P., Sliwa, M., & Didier, P. (2015). Emission properties of oxyluciferin and its derivatives in water: Revealing the nature of the emissive species in firefly bioluminescence. Journal of Physical Chemistry B, 119, 2638–2649.
de Keersmaecker, H., Fron, E., Rocha, S., Kogure, T., Miyawaki, A., Hofkens, J., & Mizuno, H. (2016). Photoconvertible behavior of LSSmOrange applicable for single emission band optical highlighting. Biophysical Journal, 111, 1014–1025.
Yanushevich, Y. G., Staroverov, D. B., Savitsky, A. P., Fradkov, A. F., Gurskaya, N. G., Bulina, M. E., Lukyanov, K. A., & Lukyanov, S. A. (2002). A strategy for the generation of non-aggregating mutants of Anthozoa fluorescent proteins. FEBS Letters, 511, 11–14.
Kabsch, W. (2010). XDS. Acta Crystallographica. Section D, Biological Crystallography, 66, 125–132.
Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E. B., Leslie, A. G. W., McCoy, A., McNicholas, S. J., Murshudov, G. N., Pannu, N. S., Potterton, E. A., Powell, H. R., Read, R. J., Vagin, A., & Wilson, K. S. (2011). Overview of the CCP4 suite and current developments. Acta Crystallographica. Section D, Biological Crystallography, 67, 235–242.
Liebschner, D., Afonine, P., Baker, M. L., Bunkoczi, G., Chen, V. B., Croll, T. I., Hintze, B., Hung, L. W., Jain, S., McCoy, A. J., Moriarty, N. W., Oeffner, R. D., Poon, B. K., Prisant, M. G., Read, R. J., Richardson, J. S., Richardson, D. C., Sammito, M. D., Sobolev, O., … Adams, P. D. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallographica D Structural Biology, 75, 861–877.
Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F., & Vagin, A. A. (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallographica. Section D, Biological Crystallography, 67, 355–367.
Emsley, P., Lohkamp, B., Scott, W. G., & Cowtan, K. (2010). Features and development of Coot. Acta Crystallographica. Section D, Biological Crystallography, 66, 486–501.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215, 403–410.
Sarper, S. E., Hirai, T., Matsuyama, T., Kuratani, S., & Fujimoto, K. (2021). Polymorphism in the symmetries of gastric pouch arrangements in the sea anemone D. lineata. Zoological Letters. https://doi.org/10.1186/s40851-021-00180-0
Karasawa, S., Araki, T., Nagai, T., Mizuno, H., & Miyawaki, A. (2004). Cyan-emitting and orange-emitting fluorescent proteins as a donor/acceptor pair for fluorescence resonance energy transfer. Biochemical Journal, 381, 307–312.
Mukherjee, S., Manna, P., Hung, S. T., Vietmeyer, F., Friis, P., Palmer, A. E., & Jimenez, R. (2022). Directed evolution of a bright variant of mCherry: Suppression of nonradiative decay by fluorescence lifetime selections. Journal of Physical Chemistry B, 126, 4659–4668.
Laviv, T., Kim, B. B., Chu, J., Lam, A. J., Lin, M. Z., & Yasuda, R. (2016). Simultaneous dual-color fluorescence lifetime imaging with novel red-shifted fluorescent proteins. Nature Methods, 13, 989–992.
Bindels, D. S., Haarbosch, L., van Weeren, L., Postma, M., Wiese, K. E., Mastop, M., Aumonier, S., Gotthard, G., Royant, A., Hink, M. A., & Gadella, T. W. J. (2016). MScarlet: A bright monomeric red fluorescent protein for cellular imaging. Nature Methods, 14, 53–56.
Ghosh, A., Isbaner, S., Veiga-Gutiérrez, M., Gregor, I., Enderlein, J., & Karedla, N. (2017). Quantifying microsecond transition times using fluorescence lifetime correlation spectroscopy. Journal of Physical Chemistry Letters, 8, 6022–6028.
Hosoi, H., Mizuno, H., Miyawaki, A., & Tahara, T. (2006). Competition between energy and proton transfer in ultrafast excited-state dynamics of an oligomeric fluorescent protein red Kaede. Journal of Physical Chemistry B, 110, 22853–22860.
Fron, E., Sliwa, M., Adam, V., Michiels, J., Rocha, S., Dedecker, P., Hofkens, J., & Mizuno, H. (2014). Excited state dynamics of the photoconvertible fluorescent protein Kaede revealed by ultrafast spectroscopy. Photochemical and Photobiological Sciences, 13, 867–874.
Adam, V., Mizuno, H., Grichine, A., Hotta, J. I., Yamagata, Y., Moeyaert, B., Nienhaus, G. U., Miyawaki, A., Bourgeois, D., & Hofkens, J. (2010). Data storage based on photochromic and photoconvertible fluorescent proteins. Journal of Biotechnology, 149, 289–298.
Peterman, E. J. G., Brasselet, S., & Moerner, W. E. (1999). The fluorescence dynamics of single molecules of green fluorescent protein. Journal of Physical Chemistry A, 103, 10553–10560.
Goedhart, J., von Stetten, D., Noirclerc-Savoye, M., Lelimousin, M., Joosen, L., Hink, M. A., van Weeren, L., Gadella, T. W. J., & Royant, A. (2012). Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nature Communications, 3, 751.
Golub, M., Guillon, V., Gotthard, G., Zeller, D., Martinez, N., Seydel, T., Koza, M. M., Lafaye, C., Clavel, D., von Stetten, D., Royant, A., & Peters, J. (2019). Dynamics of a family of cyan fluorescent proteins probed by incoherent neutron scattering. Journal of Royal Society Interface, 16, 1–10.
Yarbrough, D., Wachter, R. M., Kallio, K., Matz, M., & Remington, S. J. (2001). Refined crystal structure of DsRed, a red fluorescent protein from coral, at 2.0-Å resolution. Proceedings of the National Academy of Sciences, 98, 462–467.
Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A., & Tsien, R. Y. (2002). A monomeric red fluorescent protein. Proceedings of the National Academy of Sciences, 99, 7877–7882.
Acknowledgements
We thank the beamline stuff of KEK- PF, Tsukuba, Japan, for the X-ray diffraction data collection. This work was supported in part by YU-COE program of Yamagata University, the Category 1 research grant from KU Leuven (C14/16/053) and PRESTO of JST (JPMJPR10QA). DL acknowledges KU Leuven Facultaire Luik Onderzoeksfonds (FLOF) for financial support. Chevreul Institute (FR 2638), Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation, Hauts-de-France Region and FEDER are acknowledged for supporting and funding partially this work. We thank the reviewers for their insightful and helpful comments.
Author information
Authors and Affiliations
Contributions
YH, KM, KH, MS, HM and J-IH conceived and supervised the study; KM, KH, MS, HM and J-IH designed experiments; YH, KM, DL, KH, MS, J-IH performed experiments; YH, KM, DL, MS, HM, J-IH analyzed data; YH, KM, DL, KH, MS, HM, and J-IH wrote the manuscript. KM, KH, MS, HM and J-IH made the manuscript revision.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Horiuchi, Y., Makabe, K., Laskaratou, D. et al. Cloning and structural basis of fluorescent protein color variants from identical species of sea anemone, Diadumene lineata. Photochem Photobiol Sci 22, 1591–1601 (2023). https://doi.org/10.1007/s43630-023-00399-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43630-023-00399-0