
Abstract
This study investigated whether liquiritin can alleviate cerebral ischemia–reperfusion injury by regulating Nurr1 to mediate mitochondrial homeostasis. SH-SY5Y cells were subjected to glucose deprivation and reperfusion to establish a cerebral ischemia–reperfusion injury model in vitro. Cell viability and apoptosis were then determined using a cell counting kit and flow cytometry analysis. The degree of mitochondrial swelling was evaluated using a cell mitochondria isolation kit. Reactive superoxide generation, mitochondrial membrane potential, adenosine triphosphate (ATP) content, and mitochondrial ultrastructure were analyzed using dihydroethidium, JC-1 (5,5′,6,6′-tetrachloro1,1′,3,3′-tetramethylbenzimidazolylcarbocyanine iodide), luciferase-based ATP bioluminescent assays, and transmission electron microscopy, respectively. Quantitative reverse transcription PCR and western blot assays were conducted to detect levels of mitochondrial fission-related factors. Glucose deprivation and reperfusion exposure significantly reduced the viability and induced apoptosis of SH-SY5Y cells, indicating that glucose deprivation and reperfusion exposure successfully induced cerebral ischemia–reperfusion injury. Glucose deprivation and reperfusion exposure also increased the degree of mitochondrial swelling, promoted an increase in superoxide, and decreased mitochondrial membrane potential and ATP enzyme levels. Cerebral ischemia–reperfusion injury also significantly increases Drp1 and Fis1 protein expression, reduces mitofusin-2 and optic atrophy 1 levels, increases nuclear receptor-related 1 and inverted formin-2 expression, and decreases yes-associated protein expression. Electron microscopy further revealed sparse mitochondria and broken cristae. However, these findings were reversed by liquiritin in a dose-dependent manner and were further abolished after carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone treatment. Our study suggests that the protective effects of liquiritin on cerebral ischemia–reperfusion injury are linked to nuclear receptor-related 1 upregulation, followed by the regulation of yes-associated protein-inverted formin-2-mitochondrial fission pathways. Liquiritin may represent a novel therapeutic agent for treating cerebral ischemia–reperfusion injury.
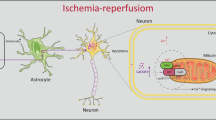
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cerebrovascular disease is a common and frequently occurring disease that seriously endangers human health. The incidence rate of ischemic cerebrovascular disease accounts for 75% of all cerebrovascular diseases (Papadakis and Buchan 2009; Zhang 2019). Mechanisms underlying brain tissue damage in patients with ischemic cerebrovascular disease include ischemia and ischemia–reperfusion (I/R) injury. Cerebral I/R injury is frequently considered the primary cause of cerebral damage and is a vital clinical problem in cerebral injury treatment (Li et al. 2020a). Multiple studies have revealed that glucose/oxygen deprivation can be applied to generate a cerebral I/R injury model in vitro and have illustrated the mechanism of cerebral I/R injury. For instance, Wang et al. (2022) revealed that the overexpression of microRNA-149-5p attenuates cerebral I/R injury by targeting Notch2. Huang et al. (2019) have suggested that microRNA-34b protects against focal cerebral I/R injury by targeting Keap1. Therefore, reducing I/R injury is crucial in the treatment of ischemic cerebrovascular disease.
The maintenance of mitochondrial homeostasis plays an important role in cell growth, and damaged mitochondria may lead to oxidative stress and ATP depletion (Sun et al. 2022). A large body of evidence has confirmed that mitochondrial fission is an important regulator of mitochondrial homeostasis and that mitochondria have become potential targets for regulating the progression of cerebral IR injury (Guo et al. 2022). Excessive mitochondrial division impairs ATP production and promotes apoptosis (Ju et al. 2007). Mitochondrial fission has been found to be associated with cardiovascular disease (Jin et al. 2021), cancer metastasis and invasion (Bandopadhyay et al. 2022), as well as ischemia/reperfusion injury (Anzell et al. 2021). However, the involvement of mitochondrial fission in cerebral I/R injuries requires further investigation.
An increasing number of reports have confirmed that many factors have revealed protective effects on cerebral I/R injuries—such as vitexin compound B-1 (Hu et al. 2023), interleukin-11 (Zhang et al. 2019), and curcumin (Huang et al. 2021). Liquiritin (1), a flavonoid found in the roots of liquorice, has been shown to exert protective effects against polycystic ovary syndrome (Cui et al. 2022), Alzheimer’s disease (Huang et al. 2015), and bone cancer (Ni et al. 2020). Liu et al. (2020) have also shown that liquiritin protects against LPS-induced acute lung injury. However, whether liquiritin protects against cerebral I/R injury remains unclear.

Therefore, our study aimed to (i) explore the roles of liquiritin in glucose deprivation and reperfusion (OGD)-induced SH-SY5Y cells, (ii) illustrate the relationship between mitochondrial fission and cerebral I/R injury, and (iii) analyze whether liquiritin regulates cerebral I/R injury by regulating Nurr1 via the YAP-INF2-mitochondrial fission pathway, explaining the latent mechanism. Our findings may offer a theoretical basis for the diagnosis and treatment of cerebral I/R injury.
Materials and Methods
OGD/R Model Establishment
The SH-SY5Y cells were acquired from American Type Culture Collection (ATCC) and maintained in a Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% fetal bovine serum (FBS, Procell) and 1% penicillin/streptomycin (Procell) in an incubator containing 5% CO2 at 37 °C. An OGD/R model for SH-SY5Y cells was established. Briefly, SH-SY5Y cells were transferred to the cultural condition with 1% O2, 94% N2, 5% CO2, and glucose-free RPMI-1640 medium at 37 °C for 4 h. The cells were then placed in a normal medium containing 5% CO2 at 37 °C for 48 h. The SH-SY5Y cells were induced by different concentrations (20, 40, and 80 µM) of liquiritin (Lot No. CB-1004, purity > 98%, ChengDu Conbon Biotech Co., LTD, Chengdu, China) (Mou et al. 2021) or carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) in order to conduct follow-up experiments.
CCK-8 Assay
The CCK-8 assay was used to evaluate cell proliferation. SH-SY5Y cells were plated into 96-well plates and cultured for 24 h, and CCK-8 solution was added to each well. Absorbance (450 nm) was assessed via an ELISA reader (Thermo Fisher Scientific), according to the manufacturer’s protocol.
Flow Cytometry Analysis
Flow cytometry was used to analyze apoptosis. After OGD/R treatment, the SH-SY5Y cells were collected by centrifugation at 1000 × g at 4 °C for 5 min. The cells were then washed with PBS. Apoptosis was detected using the Annexin-V/propidium iodide Apoptosis Detection Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Finally, apoptotic cells were quantified using a flow cytometer (BD Biosciences, USA) and analyzed using FlowJo.
Western Blot Assay
After OGD/R treatment, total proteins were extracted from SH-SY5Y cells using a RIPA lysis buffer (Beyotime) and measured using a bicinchoninic acid protein assay kit (Invitrogen, USA) following the manufacturer’s protocols. Average protein concentrations were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene fluoride membrane. After blocking with 5% skim milk in phosphate buffer solution-Tween-20 (PBST) for 2 h, the membranes were cultured in primary antibodies against Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (ab181602, 1:10000, Abcam, Cambridge, MA, USA), Fis1 (10956-1-AP, 1:1000, Wuhan Sanying Biotechnology, Wuhan, China), Drp1 (ab184247, 1:3000, Abcam, Cambridge, MA, USA), Mfn2 (#9482, 1:1000, Cell signalling Technology, Danvers, MA, USA), Opa1 (ab157457, 1:500, Abcam, Cambridge, MA, USA), nuclear receptor-related 1 (Nurr1, 10975-2-AP, 1:2000, Wuan Sanying Biotechnology, Wuhan, China), yes-associated protein (YAP, ab245286, 1:500, Abcam, Cambridge, MA, USA), or inverted formin-2 (INF2, 20466-1-AP, 1:1000, Wuan Sanying Biotechnology, Wuhan, China) overnight at 4 °C. After washing thrice with TBST, the membranes were cultivated with secondary antibodies for 2 h. Protein signals were visualized using enhanced chemiluminescence detection system reagents (Pierce, Rockford, IL, USA) and quantified using ImageJ software.
qRT-PCR Analysis
The levels of GAPDH, Fis1, Drp1, Mfn2, Opa1, Nurr1, and YAP-INF2 were measured using qRT-PCR. RNA was isolated from cells using the TRIzol reagent (ELK Biotechnology) according to the manufacturer’s protocol. Total RNA was reverse transcribed to cDNA using the PrimeScript RT Reagent Kit (TaKaRa, China), and qRT-PCR analysis was conducted using the SYBR PrimeScript RT-PCR Kit (TaKaRa) with an ABI 7500 Real-Time PCR System (Applied Biosystems). Target gene expressions were calculated using the 2−ΔΔCT method (Livak and Schmittgen 2001). Primer sequences for PCR were listed in Table S1.
Mitochondrial Isolation
To isolate enriched mitochondria, SH-SY5Y cells were washed with cold PBS and incubated in a lysis buffer for 30 min. The cells were then centrifuged at 2000 × g at 4 °C to remove insoluble matter. The supernatant was treated according to the instructions of the Cell Mitochondria Isolation Kit (C3601, Beyotime). The absorbance at 520 nm was measured for 10 min in accordance with the manufacturer’s instructions. To measure mitochondrial swelling, the decrease in absorbance was used as the standard, indicating an inversely proportional relationship between absorbance and mitochondrial swelling.
ATP Production Assay
To measure the ATP levels, SH-SY5Y cells were washed with cold PBS and incubated in a lysis buffer for 30 min. The cells were then centrifuged at 2000 × g at 4 °C. ATP levels were measured using a firefly luciferase-based ATP assay kit (Nanjing Jiancheng Bioengineering Institute). A hybrid luminometer multimode microplate reader (BioTek, VT, USA) was used to analyze the results.
Mitochondrial Potential Assay
To measure mitochondrial potential, the cells were centrifuged at 300 × g at 4 °C. After removing the supernatant, the sample was washed three times with PBS, with fresh culture medium being subsequently added. A JC-1 kit (Beyotime, China) was used to measure the mitochondrial potential. Flow cytometry was performed to analyze the results. After OGD/R treatment, the SH-SY5Y cells were seeded in 96-well plates.
Mitochondrial Superoxide Assay
To measure mitochondrial superoxide, the cells were centrifuged at 300 × g at 4 °C. The growth medium was replaced with dihydroethidium (DHE) in serum-free RPMI-1640 medium, and the cells were cultured for 30 min. Flow cytometry was used to analyze the DHE fluorescence.
Mitochondrial Ultrastructure Assay
To observe mitochondrial ultrastructure, the cells were centrifuged at 300 × g at 4 °C. The culture medium was removed and the electron microscope fixative was added and fixed overnight at 4 °C. After dehydration, infiltration, embedding polymerization, sectioning, and staining, the cells were observed using transmission electron microscopy.
Statistical Analysis
All experiments were performed in triplicate. All results were presented as the mean ± standard deviation (SD) and analyzed via SPSS v.20.0 (IBM Corp., Armonk, NY, USA). Differences among groups were estimated using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test and Student’s t-test. *p < 0.05 and **p < 0.01 indicated statistically significant differences.
Results and Discussion
FCCP Reversed Liquiritin Protection
SH-SY5Y cells were cultured in an oxygen–glucose deprivation medium for 4 h and reoxygenated for another 18 h so as to induce cerebral I/R injury in vitro. Cell viability and apoptosis were determined using CCK-8 and flow cytometry analyses, respectively, to evaluate cerebral I/R injury. As shown in Fig. 1A, SH-SY5Y cell viability was remarkably reduced in the OGD/R group compared with that in the control group. Furthermore, compared with the OGD/R group, liquiritin relieved the degree of damage caused by OGD/R-induced cerebral I/R injury in a dose-dependent manner, as confirmed by increased cell viability. In addition, we analyzed apoptosis in the control, OGD/R-stimulated, and OGD/R + liquiritin groups. As shown in Fig. 1B, the percentage of apoptotic SH-SY5Y cells was higher in the OGD/R group than in the control group. We also found that cell apoptosis was remarkably increased in the OGD/R + liquiritin group in a dose-dependent manner compared to that in the OGD/R group (Fig. 1B and C). However, these findings were reversed by FCCP. In summary, our results revealed that OGD/R successfully stimulated a cerebral I/R injury model in vitro, and that FCCP reversed the effects of liquiritin on OGD/R-induced cerebral I/R injury.

Establishment of OGD/R-induced cerebral I/R injury model in vitro. SH-SY5Y cells were cultured in an oxygen–glucose deprivation medium and reoxygenated to stimulate cerebral I/R injury in vitro. A Cell viability was determined using a CCK-8 assay. B and C Flow cytometry was performed to detect apoptotic cells. **p < 0.01 vs Control; ##p < 0.01 vs OGD/R; &&p < 0.01 vs OGD/R + liquiritin-80
Reversion on OGD/R-Activated Mitochondrial Fission
Previous investigations have found that reperfusion-associated mitochondrial injury is caused by enhanced mitochondrial fragmentation, and that mitochondrial fission has been proven to be an inducer of mitochondrial apoptosis in heart reperfusions (Guo et al. 2021a). To determine whether liquiritin triggers mitochondrial damage through mitochondrial fragmentation, we measured the degree of mitochondrial membrane swelling in each group. As shown in Fig. 2, when compared with the control group, the degree of mitochondrial membrane swelling increased after OGD/R injury, indicating that OGD/R stress caused mitochondrial fragmentation. Moreover, liquiritin maintained the mitochondrial network and weakened some mitochondrial fragments in a dose-dependent manner. However, we observed the opposite results in the FCCP-treated group. Our findings confirmed that liquiritin blocked OGD/R-induced mitochondrial fission in a dose-dependent manner.

The effects of liquiritin (1) and FCCP on OGD/R-activated mitochondrial membrane swelling. The OGD/R model was then treated with liquiritin and the cells were divided into six groups: Control, OGD/R, OGD/R + 20 µM liquiritin, OGD/R + 40 µM liquiritin, OGD/R + 80 µM liquiritin, and OGD/R + 80 µM liquiritin + FCCP groups. The degree of mitochondrial membrane swelling was quantified by analyzing the value OD520. **p < 0.01 vs Control; ##p < 0.01 vs OGD/R; &&p < 0.01 vs OGD/R + liquiritin-80
Reversion on OGD/R-Induced Mitochondrial Dihydroethidium
We also used the fluorescent probe DHE to measure superoxide anion production in the cells. As shown in Fig. 3A, compared to the control group, OGD significantly increased intracellular superoxide anion-free radicals. All liquiritin treatments effectively inhibited OGD-induced production of superoxide anion-free radicals. When mitochondria are damaged, JC-1 aggregates decrease, and the accumulation of JC-1 monomers increases. As shown in Fig. 3B and C, compared to the control group, OGD caused significant damage to the mitochondria, and the mitochondrial membrane potential was significantly reduced. However, liquiritin improved OGD-induced mitochondrial damage in a dose-dependent manner, as confirmed by increased mitochondrial membrane potential.

Effects of liquiritin (1) and FCCP on OGD/R-activated mitochondrial DHE, mitochondrial membrane potential, and ATP. The OGD/R model was then treated with liquiritin and the cells were divided into six groups: Control, OGD/R, OGD/R + 20 µM liquiritin, OGD/R + 40 µM liquiritin, OGD/R + 80 µM liquiritin, and OGD/R + 80 µM liquiritin + FCCP groups. A–C DHE and JC-1 fluorescence were analyzed by flow cytometry. D The ATP levels were determined using an ATP assay. **p < 0.01 vs Control; #, ##p < 0.05, 0.01 vs OGD/R; &&p < 0.01 vs OGD/R + liquiritin-80
Mitochondria are also the center of cellular ATP production (Wang et al. 2020). Thus, we measured ATP production in the mitochondria and found that OGD treatment reduced ATP production (Fig. 3D). However, liquiritin reversed the ATP production in a dose-dependent manner in OGD treatment cells. These effects were partially reversed by FCCP, indicating that liquiritin relieves cerebral I/R injury by regulating mitochondrial DHE, mitochondrial membrane potential, and ATP.
Reversion on Mitochondrial Fission-Related Proteins
Previous studies have suggested that Mfn2 is essential for maintaining mitochondrial morphology, and especially mitochondrial fusion (Huo et al. 2022). Therefore, we used qRT-PCR and western blotting to analyze the levels of mitochondrial division-associated genes. As presented in Fig. 4A–E, compared to the control group, the protein levels and mRNA expression of mitochondrial division-related genes, such as Fis1 and Drp1, were increased in the OGD/R treatment group. In contrast, the expression levels of the mitochondrial fusion proteins Mfn2 and Opa1 decreased due to OGD/R injury. Interestingly, liquiritin reversed the balance between fission- and fusion-related factors in a dose-dependent manner, suggesting that brain I/R injuries lead to excessive mitochondrial division and disruption of mitochondrial fusion. However, FCCP treatment increased Drp1 and Fis1 expression, reduced Mfn2 and Opa1 levels, and ameliorated the imbalance between mitochondrial fission and fusion. Taken together, these data confirm that liquiritin alleviates cerebral I/R injury through the mitochondrial fission pathway.

The effects of FCCP and liquiritin (1) on mitochondrial fission-related proteins and Nurr1-regulated YAP-INF2 pathways. The OGD/R model was then treated with liquiritin and the cells were divided into six groups: Control, OGD/R, OGD/R + 20 µM liquiritin, OGD/R + 40 µM liquiritin, OGD/R + 80 µM liquiritin, and OGD/R + 80 µM liquiritin + FCCP group. A Protein expression of Fis1, Drp1, Mfn2, Opa1, Nurr1, YAP, and INF2 was determined by western blotting. B–H mRNA levels of Fis1, Drp1, Mfn2, Opa1, Nurr1, YAP, and INF2 in SH-SY5Y cells from each treatment group were assessed using qRT-PCR. **p < 0.01 vs Control; #, ##p < 0.05, 0.01 vs OGD/R; &&p < 0.01 vs OGD/R + liquiritin-80
Reversion by Regulating Nurr1
It has been reported that increased Nurr1 is detrimental to cell viability (Al-Nusaif et al. 2022). To understand the roles of Nurr1 in cerebral I/R injury, the expression of Nurr1 in SH-SY5Y cells was detected using qRT-PCR and western blotting. Compared to the control group, the protein levels of Nurr1 were significantly increased after OGD/R treatment (Fig. 4A). This finding was further supported by the determination of Nurr1 mRNA expression. Based on these results, we concluded that IRI may be associated with an increase in Nurr1 (Fig. 4F).
Furthermore, it was found that Nurr1 is the upstream activator of INF2 under I/R conditions and that INF2 plays a decisive role in initiating Nurr1-mediated mitochondrial fission (Zhang and Yu 2018). Previous studies identified the YAP pathway as an inhibitor of reperfusion-mediated mitochondrial fission (Xue et al. 2022). Based on this information, we investigated whether Nurr1 regulates INF2 and mitochondrial fission through the YAP signaling pathway. qRT-PCR and western blot analyses showed that OGD/R treatment decreased the expression of YAP1 and increased the expression of INF2 when compared to the control group (Fig. 4A, G, H). Liquiritin reversed INF2 and YAP1 expression in a dose-dependent manner, which was eliminated by FCCP. These findings suggest that the YAP pathway is involved in Nurr1-mediated mitochondrial division and neuronal mitochondrial apoptosis.
Reversion Effect on Mitochondrial Ultrastructure
Finally, we observed the ultrastructure of the mitochondria using transmission electron microscopy, with the results (Fig. 5) showing that mitochondrial fission increased, that the mean mitochondrial size decreased, and that the number of mitochondria increased after treatment with OGD/R (Fig. 5). The OGD/R-induced increase in mitochondrial fission was reversed by liquiritin treatment. However, we observed the opposite results in the FCCP-treated group, indicating that liquiritin has a protective effect on the mitochondria in cerebral ischemic cells.

Effects of FCCP and liquiritin (1) on mitochondrial ultrastructure. The OGD/R model was then treated with liquiritin and the cells were divided into six groups: Control, OGD/R, OGD/R + 20 µM liquiritin, OGD/R + 40 µM liquiritin, OGD/R + 80 µM liquiritin, and OGD/R + 80 µM liquiritin + FCCP group. Transmission electron microscopy revealed the mitochondrial morphology in each group of cells
Cerebral I/R injury is a major health problem in the treatment of cerebrovascular disease. Therefore, it is important to clarify the mechanisms underlying cerebral I/R injury for early prevention and therapy (Guo et al. 2021b). OGD/R exposure is often applied for inducing cells to cause I/R injury (Zhang et al. 2021). In our study, a cerebral I/R injury model was established using OGD/R, and cell damage was assessed by detecting cell viability and apoptosis. Our data revealed that OGD/R inhibited SH-SY5Y cell growth and induced apoptosis, indicating the successful establishment of an in vitro model of I/R injury.
Liquiritin, a natural inhibitor of aldo–keto reductase family one member C1 (AKR1C1), is the main antitussive component of flavonoids derived from liquorice and interferes with progesterone metabolism (Zeng et al. 2019). Wang et al. (2023) indicated that liquiritin reduces ferroptosis in doxorubicin-induced cardiotoxicity by targeting the SLC7A11/glutathione peroxidase 4 (GPX4) pathway. Moreover, Yang et al. (2021) demonstrated that liquiritin reduced LPS-induced inflammation in HaCaT cells via the regulation of microRNA-31/MyD88. Sun et al. (2010) also revealed the neuroprotective effects of liquiritin against focal cerebral I/R injuries in mice via its antioxidant and antiapoptotic properties. However, the specific mechanism of action of liquiritin in the OGD/R-induced injury model is not fully understood. Therefore, we analyzed the protective effects of liquiritin in OGD/R-stimulated injury models using CCK-8 and flow cytometry assays to assess the effects of liquiritin on SH-SY5Y cells after OGD/R exposure. Our findings indicated that liquiritin relieved cerebral I/R injury in OGD/R-induced cells in a dose-dependent manner.
The pathogenesis of cerebral I/R injury is linked to a series of complex processes, including cellular oxidation (Zhao et al. 2021), inflammatory responses (Zheng et al. 2020), and mitochondrial fission (Guo et al. 2022). Mitochondrial fission is a physiological response that occurs during many biological processes, including cell proliferation, migration, and metabolism (Li et al. 2019). In addition, the functions and upstream regulatory mechanisms of mitochondrial fission in cerebral I/R injury remain unclear. Thus, we evaluated the effects of FCCP on SH-SY5Y cell viability and apoptosis and found that FCCP reversed the effects of liquiritin on OGD/R-induced cerebral I/R injury, suggesting the involvement of mitochondrial fission in cerebral I/R injury. Mitochondrial homeostasis plays a vital role in cell viability and stress responses. Disordered mitophagy mediates neuronal oxidative stress, thereby causing cerebral I/R injury. Li et al. (2020b) suggested that liquiritin protects PC12 cells from corticosterone-induced neurotoxicity via the regulation of metabolic disorders, attenuation of the ERK1/2-NF-kappaB pathway, activation of the Nrf2-Keap1 pathway, and inhibition of the mitochondrial apoptosis pathway. In the present study, we observed concomitant mitochondrial damage following cerebral I/R injury, as confirmed by increased mitochondrial superoxide anion production, decreased mitochondrial membrane potential, and increased mitochondrial swelling. Our findings are similar to those of a previous study that showed that preserving mitochondrial homeostasis sends pro-survival signals to the reperfused brain. Mitochondria not only require ATP for movement, but also to maintain cell integrity and viability (Tang et al. 2022). Lu et al. (2018) demonstrated that artesunate suppresses oxidative and inflammatory processes by activating Nrf2 and ROS-dependent p38 MAPK, and protects against cerebral I/R injury. Moreover, Chen et al. (2008) suggested the effect of electroacupuncture on mitochondrial membrane potential and apoptosis in the cerebral cortex of rats with focal cerebral I/R injury. Yu and Gao (2017) suggested that propofol affects mitochondrial ATP content and ATPase activity in the hippocampus of rats with cerebral I/R injury. Our data suggested that FCCP reversed the effects of liquiritin on OGD/R-induced mitochondrial DHE, mitochondrial membrane potential, and ATP.
In addition, we used qRT-PCR and western blotting to analyze the expression levels of genes involved in mitochondrial fission, including Fis1, Drp1, Mfn2, and Opa1. Our results showed that liquiritin reversed the balance between fission- and fusion-related factors in a dose-dependent manner, whereas FCCP corrected the imbalance between mitochondrial fission and fusion, as confirmed by the increased Fis1 and Drp1 expression and decreased Mfn2 and Opa1 levels. Previous studies have shown that mitochondrial damage is caused by the upregulation of Nurr1. Nurr1 is an orphan nuclear receptor that regulates mitochondrial function via multiple mechanisms (Zhao et al. 2019). Wei et al. (2016) revealed that Nurr1 was found to trigger osteoarthritis by translocating to the mitochondrial membrane and inducing mitochondrial apoptosis. Our study provides further evidence that mitochondrial damage is caused by Nurr1 dysregulation. INF2 is a well-established factor involved in mitochondrial fragmentation (Zhao et al. 2018). Our results showed that Nurr1 controlled mitochondrial fission by increasing INF2 expression and inhibiting the YAP pathway. Meanwhile, a large body of evidence has confirmed the role of YAP in initiating mitochondrial fission and identified the YAP/INF2 axis as being responsible for mitochondrial fission during cerebral I/R injuries (Zhang and Yu 2018). In accordance with these findings, the results of qRT-PCR and western blot analysis in this study showed that liquiritin reversed INF2 and YAP1 expression in a dose-dependent manner, and that these effects were eliminated by FCCP. At the same time, compared with the control group, the OGD/R model group showed mitochondrial intraventricular edema and a significant reduction in the number of cristae, and that the cristae in the liquiritin treatment group were arranged in an orderly and stratified manner. However, the opposite result was observed in the FCCP-treated group, indicating that liquiritin has a protective effect on the mitochondria in cerebral ischemic cells.
There are also some limitations of this study. Firstly, this study did not conduct in vivo experiments. Other signaling pathways may be involved in the protective effect of liquiritin on cerebra I/R injury, which requires further exploration. We will conduct additional in-depth research in the future.
Conclusion
A novel understanding of the potential mechanism of liquiritin regulating Nurr1 to reduce cerebral I/R injury via the YAP-INF2-mitochondrial fission pathway was elucidated. These findings provide potential new targets to recognize the occurrence and development of cerebral I/R injury and may provide new methods for the treatment of ischemic stroke.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Anzell AR, Fogo GM, Gurm Z, Raghunayakula S, Wider JM, Maheras KJ, Emaus KJ, Bryson TD, Wang M, Neumar RW, Przyklenk K, Sanderson T (2021) Mitochondrial fission and mitophagy are independent mechanisms regulating ischemia/reperfusion injury in primary neurons. Cell Death Dis 12:475. https://doi.org/10.1038/s41419-021-03752-2
Al-Nusaif M, Yang Y, Li S, Cheng C, Le W (2022) The role of NURR1 in metabolic abnormalities of Parkinson’s disease. Mol Neurodegener 17:46. https://doi.org/10.1186/s13024-022-00544-w
Bandopadhyay S, Prasad P, Ray U, Das GD, Roy SS (2022) SIRT6 promotes mitochondrial fission and subsequent cellular invasion in ovarian cancer. FEBS Open Bio 12:1657–1676. https://doi.org/10.1002/2211-5463.13452
Cui X, Zhou S, Lin Y (2022) Protective effects of liquiritin on polycystic ovary syndrome through modulating ovarian granulosa cell proliferation and apoptosis via miR-206/PI3K/AKT pathway. Cytotechnology 74:385–393. https://doi.org/10.1007/s10616-022-00531-5
Chen XY, Zhang QL, Bai B (2008) Effect of electroacupuncture on mitochondrial membrane potential and apoptosis in the cerebral cortex in rats with focal cerebral ischemia/reperfusion injury. Zhen Ci Yan Jiu 33:107–110
Guo Z, Zhao M, Jia G, Ma R, Li M (2021a) LncRNA PART1 alleviated myocardial ischemia/reperfusion injury via suppressing miR-503-5p/BIRC5 mediated mitochondrial apoptosis. Int J Cardiol 338:176–184. https://doi.org/10.1016/j.ijcard.2021.05.044
Guo X, Shen X, Yong Z (2021b) MiR-101 protects against the cerebral I/R injury through regulating JAK2/STAT3 signaling pathway. Neuropsychiatr Dis Treat 17:2791–2802. https://doi.org/10.2147/NDT.S292471
Guo J, Zhang L, Bu Y, Li W, Hu J, Li J (2022) Ras-related protein Rab-20 inhibition alleviates cerebral ischemia/reperfusion injury by inhibiting mitochondrial fission and dysfunction. Front Mol Neurosci 15:986710. https://doi.org/10.3389/fnmol.2022.986710
Hu ZY, Yang ZB, Zhang R, Luo XJ, Peng J (2023) The protective effect of vitexin compound B-1 on rat cerebral I/R injury through a mechanism involving modulation of miR-92b/NOX4 pathway. CNS Neurol Disord Drug Targets 22:137–147. https://doi.org/10.2174/1871527321666220324115848
Huang X, Wang Y, Ren K (2015) Protective effects of liquiritin on the brain of rats with Alzheimer’s disease. West Indian Med J 64:468–472. https://doi.org/10.7727/wimj.2016.058
Huang R, Ma J, Niu B, Li J, Chang J, Zhang Y, Liu P, Luan X (2019) MiR-34b protects against focal cerebral ischemia-reperfusion (I/R) injury in rat by targeting Keap1. J Stroke Cerebrovasc Dis 28:1–9. https://doi.org/10.1016/j.jstrokecerebrovasdis.2018.08.023
Huang L, Li X, Liu Y, Liang X, Ye H, Yang C, Hua L, Zhang X (2021) Curcumin alleviates cerebral ischemia-reperfusion injury by inhibiting NLRP1-dependent neuronal pyroptosis. Curr Neurovasc Res 18:189–196. https://doi.org/10.2174/1567202618666210607150140
Huo Y, Sun W, Shi T, Gao S, Zhuang M (2022) The MFN1 and MFN2 mitofusins promote clustering between mitochondria and peroxisomes. Commun Biol 5:423. https://doi.org/10.1038/s42003-022-03377-x
Ju WK, Liu Q, Kim KY, Crowston JG, Lindsey JD, Agarwal N, Ellisman MH, Perkins GA, Weinreb RN (2007) Elevated hydrostatic pressure triggers mitochondrial fission and decreases cellular ATP in differentiated RGC-5 cells. Invest Ophthalmol vis Sci 48:2145–2151. https://doi.org/10.1167/iovs.06-0573
Jin JY, Wei XX, Zhi XL, Wang XH, Meng D (2021) Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol Sin 42:655–664. https://doi.org/10.1038/s41401-020-00518-y
Li P, Liu Y, Liu W, Li G, Tang Q, Zhang Q, Leng F, Sheng F, Hu C, Lai W, Liu Y, Zhou M, Huang J, Zhou H, Zhang R, Zhao Y (2019) IR-783 inhibits breast cancer cell proliferation and migration by inducing mitochondrial fission. Int J Oncol 55:415–424. https://doi.org/10.3892/ijo.2019.4821
Li X, Cheng S, Hu H, Zhang X, Xu J, Wang R, Zhang P (2020a) Progranulin protects against cerebral ischemia-reperfusion (I/R) injury by inhibiting necroptosis and oxidative stress. Biochem Biophys Res Commun 521:569–576. https://doi.org/10.1016/j.bbrc.2019.09.111
Li X, Qin X, Tian J, Gao X, Wu X, Du G, Zhou Y (2020b) Liquiritin protects PC12 cells from corticosterone-induced neurotoxicity via regulation of metabolic disorders, attenuation ERK1/2-NF-kappaB pathway, activation Nrf2-Keap1 pathway, and inhibition mitochondrial apoptosis pathway. Food Chem Toxicol 146:111801. https://doi.org/10.1016/j.fct.2020.111801
Liu Z, Wang P, Lu S, Guo R, Gao W, Tong H, Yin Y, Han X, Liu T, Chen X, Zhu MX, Yang Z (2020) Liquiritin, a novel inhibitor of TRPV1 and TRPA1, protects against LPS-induced acute lung injury. Cell Calcium 88:102198. https://doi.org/10.1016/j.ceca.2020.102198
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Lu H, Wang B, Cui N, Zhang Y (2018) Artesunate suppresses oxidative and inflammatory processes by activating Nrf2 and ROS-dependent p38 MAPK and protects against cerebral ischemia-reperfusion injury. Mol Med Rep 17:6639–6646. https://doi.org/10.3892/mmr.2018.8666
Mou SQ, Zhou ZY, Feng H, Zhang N, Lin Z, Aiyasiding X, Li WJ, Ding W, Liao HH, Bian ZY, Tang QZ (2021) Liquiritin attenuates lipopolysaccharides-induced cardiomyocyte injury via an AMP-activated protein kinase-dependent signaling pathway. Front Pharmacol 12:648688. https://doi.org/10.3389/fphar.2021.648688
Ni H, Xu M, Xie K, Fei Y, Deng H, He Q, Wang T, Liu S, Zhu J, Xu L, Yao M (2020) Liquiritin alleviates pain through inhibiting CXCL1/CXCR2 signaling pathway in bone cancer pain rat. Front Pharmacol 11:436. https://doi.org/10.3389/fphar.2020.00436
Papadakis M, Buchan A (2009) Approaches to neuroprotective and reperfusion injury therapy. Handb Clin Neurol 94:1205–1223. https://doi.org/10.1016/S0072-9752(08)94059-8
Sun YX, Tang Y, Wu AL, Liu T, Dai XL, Zheng QS, Wang ZB (2010) Neuroprotective effect of liquiritin against focal cerebral ischemia/reperfusion in mice via its antioxidant and antiapoptosis properties. J Asian Nat Prod Res 12:1051–1060. https://doi.org/10.1080/10286020.2010.535520
Sun S, Yu W, Xu H, Li C, Zou R, Wu NN, Wang L, Ge J, Ren J, Zhang Y (2022) TBC1D15-Drp1 interaction-mediated mitochondrial homeostasis confers cardioprotection against myocardial ischemia/reperfusion injury. Metabolism 134:155239. https://doi.org/10.1016/j.metabol.2022.155239
Tang W, Li Y, He S, Jiang T, Wang N, Du M, Cheng B, Gao W, Li Y, Wang Q (2022) Caveolin-1 alleviates diabetes-associated cognitive dysfunction through modulating neuronal ferroptosis-mediated mitochondrial homeostasis. Antioxid Redox Signal 37:867–886. https://doi.org/10.1089/ars.2021.0233
Wang Z, Peng Q, Hou Y, Gao X, Zhong S, Fang Y, Liu C, Liu X (2020) Resistance assessment for SYP-14288 in Phytophthora capsici and changes in mitochondria electric potential-associated respiration and ATP production confers resistance. Pest Manag Sci 76:2525–2536. https://doi.org/10.1002/ps.5795
Wang X, Xu Q, Wang S (2022) Overexpression of miR-149-5p attenuates cerebral ischemia/reperfusion (I/R) injury by targeting Notch2. Neuromol Med 24:279–289. https://doi.org/10.1007/s12017-021-08685-9
Wang M, Liu M, Tang L, Shen L, Xiao J, Li R (2023) Liquiritin reduces ferroptosis in doxorubicin-induced cardiotoxicity through targeting SLC7A11/GPX4 pathway. Naunyn Schmiedebergs Arch Pharmacol 2023. https://doi.org/10.1007/s00210-023-02515-4
Wei X, Gao H, Zou J, Liu X, Chen D, Liao J, Xu Y, Ma L, Tang B, Zhang Z, Cai X, Jin K, Xia Y, Wang Q (2016) Contra-directional coupling of Nur77 and Nurr1 in neurodegeneration: a novel mechanism for memantine-induced anti-inflammation and anti-mitochondrial impairment. Mol Neurobiol 53:5876–5892. https://doi.org/10.1007/s12035-015-9477-7
Xue LX, Chen SF, Xue SX, Zhang XZ, Lian YJ (2022) P2RY2 alleviates cerebral ischemia-reperfusion injury by inhibiting YAP phosphorylation and reducing mitochondrial fission. Neuroscience 480:155–166. https://doi.org/10.1016/j.neuroscience.2021.11.013
Yang X, Dang X, Zhang X, Zhao S (2021) Liquiritin reduces lipopolysaccharide-aroused HaCaT cell inflammation damage via regulation of microRNA-31/MyD88. Int Immunopharmacol 101:108283. https://doi.org/10.1016/j.intimp.2021.108283
Yu DJ, Gao HY (2017) Effect of propofol on mitochondrial ATP content and ATPase activity in hippocampus of rats with cerebral ischemia-reperfusion injury. Saudi J Biol Sci 24:246–250. https://doi.org/10.1016/j.sjbs.2016.09.007
Zeng C, Zhu D, You J, Dong X, Yang B, Zhu H, He Q (2019) Liquiritin, as a natural inhibitor of AKR1C1, could interfere with the progesterone metabolism. Front Physiol 10:833. https://doi.org/10.3389/fphys.2019.00833
Zhang J (2019) Advances in surgical treatment of ischemic cerebrovascular disease. Zhejiang Da Xue Xue Bao Yi Xue Ban 48(3):233–240. https://doi.org/10.3785/j.issn.1008-9292.2019.06.01
Zhang Z, Yu J (2018) Nurr1 exacerbates cerebral ischemia-reperfusion injury via modulating YAP-INF2-mitochondrial fission pathways. Int J Biochem Cell Biol 104:149–160. https://doi.org/10.1016/j.biocel.2018.09.014
Zhang B, Zhang HX, Shi ST, Bai YL, Zhe X, Zhang SJ, Li YJ (2019) Interleukin-11 treatment protected against cerebral ischemia/reperfusion injury. Biomed Pharmacother 115:108816. https://doi.org/10.1016/j.biopha.2019.108816
Zhang L, Wang Y, Pan RL, Li Y, Hu YQ, Xv H, Zhu C, Wang X, Yin JW, Ma KT, Zhao D (2021) Neuritin attenuates oxygen-glucose deprivation/reoxygenation (OGD/R)-induced neuronal injury by promoting autophagic flux. Exp Cell Res 407:112832. https://doi.org/10.1016/j.yexcr.2021.112832
Zhao H, Pan W, Chen L, Luo Y, Xu R (2018) Nur77 promotes cerebral ischemia-reperfusion injury via activating INF2-mediated mitochondrial fragmentation. J Mol Histol 49:599–613. https://doi.org/10.1007/s10735-018-9798-8
Zhao S, Li P, Wang P, Yang J, Song P, Zhang D, Zhou G (2019) Nurr1 promotes lung cancer apoptosis via enhancing mitochondrial stress and p53-Drp1 pathway. Open Life Sci 14:262–274. https://doi.org/10.1515/biol-2019-0030
Zhao X, Li S, Mo Y, Li R, Huang S, Zhang A, Ni X, Dai Q, Wang J (2021) DCA Protects against oxidation injury attributed to cerebral ischemia-reperfusion by regulating glycolysis through PDK2-PDH-Nrf2 axis. Oxid Med Cell Longev 2021:5173035. https://doi.org/10.1155/2021/5173035
Zheng L, Tang X, Lu M, Sun S, Xie S, Cai J, Zan J (2020) microRNA-421-3p prevents inflammatory response in cerebral ischemia/reperfusion injury through targeting m6A Reader YTHDF1 to inhibit p65 mRNA translation. Int Immunopharmacol 88:106937. https://doi.org/10.1016/j.intimp.2020.106937
Funding
The present study was supported by the Scientific research Project of Education Department of Hunan Province (grant no. 22A0248) and the Scientific General Program of Hunan Provincial Natural Science Foundation of China (grant no. 2021JJ30529).
Author information
Authors and Affiliations
Contributions
YF contributed to the study design, data collection, statistical analysis, data interpretation, and manuscript preparation. XZ and HZ contributed to data collection and statistical analysis. ZZ contributed to statistical analysis and manuscript preparation. All authors read and approved the final manuscript.
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fan, Y., Zhang, X., Zhou, H. et al. Effects of Liquiritin on Mitochondrial Dynamics in Cerebral Ischemia–Reperfusion Injury by Regulating Nurr1 via the YAP-INF2-Mitochondrial Fission Pathway. Rev. Bras. Farmacogn. 34, 501–510 (2024). https://doi.org/10.1007/s43450-023-00498-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43450-023-00498-0