
Abstract
Angiotensin-converting enzyme (ACE) and its homologue, ACE2, are commonly allied with hypertension, renin–angiotensin–aldosterone system pathway, and other cardiovascular system disorders. The recent pandemic of COVID-19 has attracted the attention of numerous researchers on ACE2 receptors, where the causative viral particle, SARS-CoV-2, is established to exploit these receptors for permitting their entry into the human cells. Therefore, studies on the molecular origin and pathophysiology of the cell response in correlation to the role of ACE2 receptors to these viruses are bringing novel theories. The varying level of manifestation and importance of ACE proteins, underlying irregularities and disorders, intake of specific medications, and persistence of assured genomic variants at the ACE genes are potential questions raising nowadays while observing the marked alteration in response to the SARS-CoV-2-infected patients. Therefore, the present review has focused on several raised opinions associated with the role of the ACE2 receptor and its impact on COVID-19 pathogenesis.
Similar content being viewed by others

Introduction
A storm of enduring coronavirus menace that appeared in late December 2019 in Wuhan, Hubei province of China has now spread all around the world. Despite extreme measures along with the controlled movement of people, the World Health Organization (WHO) announced it as a global health emergency [1, 2]. Eventually, this pertinent infectious pandemic was so-called “Coronavirus Disease-2019” or in short ‘COVID-19’ by WHO [3]. Continuous researches are ongoing to obtain either licensed vaccines to combat this infectious situation or any direct-acting antiviral drugs. Although limited small molecules have revealed efficacy during in vitro studies, not many have proceeded to detailed pre-clinical or randomized human trials, hence, have restricted progress to counteract COVID-19 disease [1, 4,5,6]. Several points, like mode of intracellular penetration of SARS-CoV-2, complicated interaction of renin-angiotensin system (RAS) system including alteration in ACE-2 concentration during COVID disease [4, 5] proposes that ACE2 targeted strategy could be employed as a potential therapeutic intervention for the treatment of COVID-19 disease. Thus, the present article is aimed to discuss the ACE-2 receptors and their role in the pathogenesis of COVID-19 disease. To continue our discussion, the connecting section of the article emphasizes an overview of ACE2 inhibitors.
Overview on ACE-2 receptor
The ACE2 receptors are ubiquitous within the human body, particularly overexpressed on intestinal epithelial cells of the gut, endothelial and smooth cells of the blood vessels, heart (epicardia, adipocytes, fibroblasts, myocytes, coronary arteries), lung (macrophages, bronchial and tracheal epithelial cells, type 2 pneumocytes), brain, testis, and on tubular epithelial cells of kidney [7,8,9,10,11]. It is a homolog of ACE, containing 805 amino acids with carboxy-terminal and amino-terminal domains. Amongst the two domains, the catalytic extracellular domain is the amino acid domain. This receptor is a type I transmembrane protein bearing a catalytic zinc metalloprotein [12, 13] domain in the amino-terminal that shows 41.8% sequence identity with ACE [14,15,16]. The gene encoding ACE2 is situated on the X-chromosome at Xp22.2 [12, 17]. Conjugation of ACE2 to the overexpressed ACE2 receptors brought the conformational change of the receptor and plays a major role in regulating various biological activities. Mainly, ACE2 is involved in counterbalancing the function of ACE [18]. ACE cleaves vasodilator angiotensin I into angiotensin II. ACE2 is also responsible for cleaving several peptide hormones. The most important being the removal of carboxyl-terminal amino acid phenylalanine from vasoconstricting angiotensin II and hydrolyses it back into angiotensin. ACE2 can also participates in cleaving bradykinin, apelin, neurotensin, dynorphin A and ghrelin. Importantly, studies reported that ACE2 receptors serves as an entrance for the access of coronaviruses like HoV-NL63, SARS-CoV, SARS-CoV-2 into human cells [19].
Structural studies of ACE2 receptor
Recently, the affinity of ACE2 towards SARS-CoV-2 S1 S-protein receptor-binding domain (RBD) has been reported to be similar [20] or 10–20 times higher than that of SARS-CoV [21]. One of the structural studies of SARS-CoV-2 showed that RBD has a stronger interaction with ACE2. A distinctive phenylalanine F486 in the flexible loop possibly performs a key function as it is able to penetrate into a long hydrophobic pocket in ACE2. This investigation recommended that ACE2 from animals like fish, amphibians, reptiles, birds to mammals can possibly bind RBD of SARS-CoV-2, permitting these animals as potential natural hosts for transmission of the virus [22,23,24].
The human ACE2 (hACE2) protein has been recognized as a decisive target for the SARS-CoV's S protein that permits the virus to attach to the epithelial cells of a host. Structural analysis involving x-ray crystallographic structures and biophysical attributes of protein–protein interactions disclose a huge interaction surface with high binding-affinity between SARS-CoV-2 and hACE2 (18 interactions) which were at least 15 times intense than between SARS-CoV-1 and hACE2 (8 interactions). These high-affinity interactions between the S protein and ACE2 receptor occur due to numerous interacting residues in SARSCoV2S-ACE2 protein–protein interaction, which form the strong (multi-epitope) adhesion synapse between the viral surface and epithelial layer of the host [25].
Pathogenesis of COVID-19
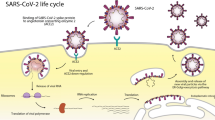
As established, SARS-CoV-2 transmits from person to person in the form of droplets, when an infected person coughs or sneezes [26,27,28]. The SARS-CoV-2 present as droplets in the air enters into the upper respiratory tract, affecting the nasal and pharyngeal epithelia of a healthy individual [29,30,31]. The infection proceeds into the lower respiratory tract, and infects bronchial and alveolar epithelium, generating primary symptoms of cough and fever [26, 32]. According to the previous theory of SARS-CoV, the virus enters the cells via two different pathways, the first one, the pH-independent pathway, by fusion of the viral envelope with the host cell membrane and delivery of the genome into the host cell. The second pathway involves the endocytic pathway upon binding of the virus to the extracellular domain of the ACE2 receptor [33]. However, a recent theory revealed that SARS-CoV-2 enters into the cells via the second pathway, which involves clathrin-mediated endocytic pathway upon binding of the virus to the extracellular domain of ACE2 receptor (Fig. 1) [34, 35]. The second pathway is a pH-dependent pathway, which uses the cells' endocytic mechanism. The activation of the virion occurs in the endosome, where the fusion of the viral particle and endosomal membrane is stimulated by the acidic pH, which resulted in release of the genomic components into the cytoplasm for the multiplication process [33].

Entry of SARS-CoV-2 into the cells via clathrin-mediated endocytosis pathway (Reproduced with permission from Ref. [2])
The virus soon enters into the peripheral bloodstream through the lungs and develops viremia [26, 36]. Here, the ACE2 receptors serve as the gateway for the entry of the virus inside the body. The virus replicates and simultaneously hampers other organs expressing ACE2, such as the heart, kidneys, blood vessels, and gastrointestinal tract [26, 37]. The massive epithelial and endothelial cell death and vascular leakage trigger the production of inflammatory cytokines and chemokines. Hijacking of pulmonary ACE2 receptors, their downregulation, and shedding causes more RAS dysfunction and augment inflammation and vascular permeability, this might be the possible cause behind the acute lung injury (ALI) [38]. The fast approaching cytokine storm further hinders the immune response and causes dysfunction. Hence, the clinical phase evolves from preliminary viremia to acute phase pneumonia, leading to serious ailments like acute respiratory distress syndrome (ARDS), ALI, and ultimately multi-organ collapse [37] (Fig. 2).

Pathogenesis of SARS-CoV-2 virus, in relation to ACE2 receptors
Cell signaling pathways contributing to COVID-19 pathogenesis
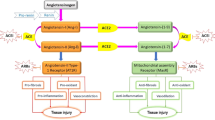
Several host cells signaling pathways start on the binding of coronaviruses to ACE2 of the host. The RAS is a signaling pathway known for maintaining homeostatic regulation. In general, ACE and angiotensin II are accountable for clinical problems, particularly ARDS in patients. ACE2 possesses only one active site and it is responsible for the conversion of angiotensin II to angiotensin (1–7). ACE2 also converts angiotensin I to angiotensin (1–9) and ultimately stimulate Mas-related G protein-coupled receptors [39, 40].
As SARS-CoV-2 enters the cells through ACE2 present on the cell surfaces, the S protein of the virus binds to the catalytic component of ACE2. The Janus kinase/signal transducers and activators of the transcription (JAK-STAT) signaling pathway are associated with inflammatory conditions. The cytokine signaling pathways are controlled by different processes, together with suppressors of cytokine signaling (SOCS) proteins, responsible for the suppression of signaling to JAK or cytokine receptors. JAK signaling pathway is also inhibited by SOCS1 and SOCS3 via stimulation of their kinase inhibitory domain [39,40,41,42,43,44].
An Interaction of ACE2 with virus surface spike protein or its RBD region
A spike of SARS-CoV-2 is a homo-trimer composed of glycoprotein. It contains three similar copies of the same protein chain attached together. A spike consists of two subunits i.e., S1 and S2. The S1 subunit is essential for receptor binding as it possesses the RBD. While S2 subunit contains the fusion machinery required for integration of the viral membrane with the host cell membrane. SARS-CoV-2 begins its entry into the host cell after the spike protein present on the envelope attaches to ACE2. The S protein is cleaved into S1 and S2 by a human cell-derived protease i.e., Furin. The S1 then binds to ACE2, while another fragment, S2, is cleaved by a human cell surface protease i.e., TMPRSS2 (transmembrane protease serine 2 precursors) leading to membrane fusion (Fig. 3) [45,46,47,48].

Spike protein of SARS-CoV-2 and its interaction with ACE2 receptor of host. a Structure of SARS-CoV-2 and structural subunits of spike protein; b Interaction of spike protein of SARS-CoV-2 with ACE2 receptor indicating receptor binding domain
An interactive study between the SARS-CoV-2 S glycoprotein and the ACE2 receptor was accomplished using single-molecule force spectroscopy. It showed that a specific binding mechanism between the S1 subunit and the ACE2 receptor that both domains interact with similar kinetic and thermodynamic properties. Furthermore, this study also exhibited that RBD binds the ACE2 receptor with a remarkably elevated intrinsic affinity (~ 120 nM) [49].
The reported binding mechanisms of human ACE2 and S proteins of both SARS-CoV and SARS-CoV-2 indicating that the S protein of coronaviruses possesses affinity towards ACE2 due to electrostatic forces, applicable even at dissimilar distances. SARS-CoV-2 possesses a higher electric field line density when compared to SARS-CoV, thus there is obviously a stronger interaction between SARS-CoV-2 and ACE2 when compared to that of SARS-CoV. The latter fact may be the major reason behind the fast-spreading of SARS-CoV-2 as compared to SARS-CoV [50,51,52].
SARS-CoV-2 binds with the ACE2 to enter the host cell, where the S protein is primed by serine protease TMPRSS2, which releases the S protein subunit S2 to fuse the viral and cellular membrane [52]. Thus, the viral gene enters into the cell and multiplication of viruses occurs. Therefore, drug design or vaccines against SARS-CoV-2 S protein might be a potential approach to achieve protection and treatment against COVID-19 disease.
Recent research advancement ofACE-2 related to COVID-19
Multiple novel research approaches on utilizing ACE2 core for COVID-19 treatment (Fig. 4) are yet to bear fruits in terms of either novel drugs or vaccine or recombinant biologicals being progressed to market, but initial results and reports tend to develop a strong belief that ACE2 receptor might play an integral role in coping with the pandemic. ACE2 receptors are bringing and moreover, becoming a perfect storm of many different advances for reaching the ultimate apogee in COVID-19 treatment.

Different therapeutic approaches to overcome COVID-19 pandemic
Consequences of SARS-CoV-2 and ACE2 binding
According to the recent research, SARS-CoV and SARS-CoV-2 conjugate with ACE2 receptors on the lung epithelial membrane and enters the cell thereafter replicates and results in downregulation of these receptors. The physiological function of these receptors is to degrade angiotensin II. The downregulation of ACE2 by virus causes an upsurge in angiotensin II, which by acting on AT1 receptor causes systemic injury [49], pulmonary fibrosis, pulmonary inflammation, and in severe cases of the deadly complication of COVID-19 i.e., ARDS [66]. Invitro studies in different pulmonary injury models revealed that multiple inflammatory bruises like thickening of alveolar wall, edema, bleeding, inflammatory cell infiltrates are activated in the respiratory tree due to downregulation of ACE2 receptors [4]. The results of another study revealed that simultaneous injection of recombinant ACE2 and AT1 receptor blockers shows effectiveness in weakening the magnitude of lung injury [55]. The isolated viral spike protein of SARS-CoV-2 was notably reported by few authors in their studies as being responsible to induce downregulation of ACE2 and the over-activity of angiotensin II and AT1 bringing damage to the lung tissues and trigger severe pulmonary inflammatory lesions. This model also explained that AT1 receptor blockers might help to attenuate the complication provoked by the spike viral protein [4, 56].
It is imperative to consider a point that ACE2 is markedly expressed in pneumocytes type II. Pneumocytes are the small cylindrical or cuboidal surface epithelial cells of the alveoli. Pneumocytes type II represent only 5% of all pneumocytes but are more diffuse than type I cells [4, 57]. They seem foamier because of phospholipid multi-lamellar bodies, the precursor to pulmonary surfactant. Hence, they participate in the production of alveolar surfactant and simultaneously, function as stem cells, precursor of pneumocytes type I. Pneumocytes type I constitute 95% of all pneumocytes and play a vital role in gaseous exchange. Downregulation of ACE2 receptors due to binding of coronavirus might hinder the expression of pneumocytes type II cells, slowing the rate of gaseous exchange [4, 58]. Overall, the interaction of coronavirus to ACE2 receptors is destructive due to the four underlying reasons. These are (a) rise of inflammatory lesions due to downregulation of ACE2 receptors; (b) local unrestricted angiotensin II and AT1 receptor over-activity; (c) insufficient surfactant by bruised pneumocytes type II causing pulmonary compliance and amplified surface tension; and (d) reduced generation and repair of pneumocytes type I impair gaseous exchange along with alveolar-capillary diffusion capacity and fibrosis [4, 59].
Furnishing disproportionate soluble form of ACE2 in disrupting viral spread and infection
Clinically in normal cells, angiotensin 1–7 and/or ACE2 effectively reduces pulmonary damage and ARDS, which is being the major complications in COVID-19 disease. Recent studies on developing and testing the recombinant human ACE2 (rhACE-2; APN01, GSK258.6881) [60, 61] from the supernatant of ACE2 transfected cells has grasped researcher attention, as it might lessen plasma angiotensin II and escalate angiotensin 1–7, hence preventing angiotensin II prompted myocardial hypertrophy, diastolic dysfunction and pulmonary fibrosis [62]. The administration of rhACE2 in ARDS diagnosed patients and healthy subjects in human studies were found safe and have a tendency todecline interleukin-6 (IL-6) concentration [63], diminishing the potency of cytokines storm. It is supposed that a marked soluble form of ACE2 may enable delayed entry and further spreading of the virus. Hence, from various pilot studies, rhACE2 may be deliberated as a promising therapeutic alternative in COVID-19 treatment [64].
Inhibiting the binding of SARS-CoV-2 with ACE2 receptor
Certain compounds, such as opioids (e.g., morphine and codeine) have been suggested for directly binding to ACE2 receptors with high affinity, hence limiting further binding of SARS-CoV-2 with the extracellular domain of ACE2. These opiate alkaloids proved beneficial in reports released by the National Health Service, England [64, 65] and by Swedish researchers [64, 66], in managing shortness of breath, coughing, and pain in diseased patients. Experimental findings further revealed that morphine treatment considerably suppressed some inflammatory cytokines, including IL-6, IFNs, and tumor necrosis factor-alpha (TNF-α), as well as improved hyper-inflammatory status that otherwise might bring more impairment to host cells than foreign invaders. Opioids also potentiate the immune response through the central nervous system. Morphine and codeine might, hypothetically, serve a potential role in the treatment of patients with COVID-19 [64].
Different impact of SARS-CoV-2 expression on ACE-2 due to gender-related dissimilarities
The recent SARs-CoV-2 pandemic has infected a wide age group of people, principally elderly individuals, with increased percentage in males [67]. It seems that men are more vulnerable to novel coronavirus due to high blood levels of gateway molecules that enable more infection in healthy cells than women do. Few of the available reports revealed the presence of hormonal protection apart from protective behavior in women (e.g., washing hands) along with better lifestyle (less smoking and drinking alcohol) resulted in the women more powerful in fighting against the virus [67, 68]. With the speed and scale of the pandemic worldwide, viral genetic variants known to influence disease progression have received much interest. The data from WHO and Chinese scientists showed that 1.7% female mortality was reported when compared to 2.8% of men mortality by the influence of SARS-CoV-2 virus. The SARs-CoV-2 plays an essential role by forming a durable contact with human ACE2 and facilitates the entry into cells along with TMPRSS2. However, it is remarkable to consider that the ACE2 gene lays on X-chromosome, making females possibly heterozygous and differently varied as compared to those with homozygous males [69]. Second, estrogen physiologically is supposed to upregulate the expression of ACE2, serving advantage of two alleles for premenopausal women, decreasing the deficiency of ACE2 in response to viral attack [70]. In addition, several other genes are supposed to be located on X-chromosomes, like surplus numbers of immune-related genes, which can work in innate and adaptive responses towards infections. There are also certain genes; basically, not being part of RAS pathway, but may directly or indirectly sway the ACE-1/ACE-2 balance by persuading its major factors like ABO locus, SRY, SOX3, ADAM17, etc. However, X-heterozygous females might also motivate a miscellany advantage and reveal further distinct sex-related differences leading to sex dimorphism, moreover, esteeming them in counteracting the advancement of the SARS-CoV-2 infection [69].
Polymorphisms of ACE and ACE2 influencing RAS equilibrium
The pragmatic variances in COVID-19 death rates might lie in significant alterations in equilibrium of ACE:ACE2 system associated with gender, racial, and age differences in genetic ACE and ACE2 polymorphism and environmental aspects manipulating ACE2 expression [70,71,72]. The physiological task of ACE genotype is to cause breakdown of angiotensin I into angiotensin II. A study performed by Chem and co-workers in 2020 showed that the lower ACE2 expression and concentration in tissues in Asian males compared to females and other cultural groups as well as type II diabetic patients. The study also displayed a negative relationship between ACE2 concentration in tissues at the molecular level and COVID-19 mortality, in both populations. The decrease in ACE2 expression was also reported to be dependent on age-related factors. The loci for higher ACE2 expression, about 100% in East Asians, 30% greater than other cultural groups [70]. Furthermore, an insertion (I) or deletion (D) in a sequence of ACE2 gene due to polymorphism might cause twice an increase in ACE activity levels than ACE2 in DD carriers. Any I or D in the gene sequence of ACE2 might impose twice an increase in ACE activity when compared to ACE2 in DD carriers. The physiological function of ACE is to facilitate the breakdown of angiotensin I into angiotensin II. This breakdown ability is impaired in persons exposed to COVID-19 disease, causing a greater circulating quantity of angiotensin II and more noticeable RAS system imbalance leading to severe diseases. At the same time, the downregulation of ACE2 receptors on the lungs membrane and decrease binding affinity of ACE2 with viral spike proteins might be instigated by genetic and environmental factors. The ACE and ACE-2 balance may be disturbed by ACE-2 polymorphism, especially, in the endothelium of the lungs. Since, it is the site where the virus attacks first and thus, the disabled ACE2-mediated repair system eventually contributes more in raising the disease severity [70].
Infection induced ACE2 downregulation initiating ACE:ACE2 imbalance
Considering the significance of ACE:ACE2 imbalance in age, gender, and racial alterations and the differences amongst circulating and tissue bound ACE2 may prove an important factor in raising disease severity. A suitable correlation of serum angiotensin II concentration has been established, due to ACE:ACE2 imbalance, with both viral load and pulmonary damage in COVID-19 infected patients [61, 70, 72]. Though ACE inhibitors may hamper the formation of angiotensin II from I, non-ACE pathways (such as chymase) contribute to the formation of nearly 40% of angiotensin II [70, 73]. ACE inhibitory drugs could solitarily chunk 60% creation of angiotensin II. This makes ACE inhibitors significantly less operational than angiotensin receptor blockers (ARB) in decreasing the serum concentration of angiotensin II. Angiotensin II primarily acts on angiotensin 1(AT1) receptors, so ARB effectively blocks angiotensin II induced systemic and pulmonary responses and stimulates a vital counter-regulatory anti-inflammatory cascade to reestablish ACE:ACE2 equilibrium. Simultaneously the circulating angiotensin II being incapable to attach with AT1, desire more binding with AT2 and Mas receptors, stimulating counter-regulatory anti-inflammatory cascade more and upregulating ACE2 expression to eliminate surplus circulating serum angiotensin II [70, 75].
A comparatively higher ACE2 level in the younger population, premenopausal women, and perhaps Asian population benefits in having low disease compliance and mortality. This, on the other hand, is hard on the European population with greater percentage of the older population with threats of diabetes, hypertension, other CVS and kidney-related disorders, showing high severity and morbidities, as these diseases permit more ACE2 shedding, eventually higher circulating ACE-2 [70]. While considering disease severity on a gender basis, it seems that males with single X-linked ACE-2 allele make them more susceptible to phenotypes causing lessened ACE-2 regulation. The same is when considered with respect to age, it seems that older patients with a history of diabetes or hypertension exhibit higher incidence of ACE polymorphisms leading to an up-rise in angiotensin II level and intensified angiotensin II response because of the viral attack. Finally, when considered on the genetic ground, the news telecasts on high rate of tragic striking of coronavirus infection in royal families are well sufficient to prove its substantial role in COVID-19 pathophysiology [70, 76].
ACE2 deficiency signing a noteworthy role in attracting COVID-19 disease
Various studies reported major involvement of ACE2 deficiency in hiking the spread and prolongation of viral infection in COVID-19 pathogenesis, due to several other health problems. The downregulation of ACE2 by viral attack could be unfavorable in persons with baseline ACE2 deficiency, hypertension, diabetes, and past heart ailments. Many cases revealed, for example, aging, especially in men, a marked decrease in ACE2 expression [4, 77]. More glycosylation in diabetes mellitus also displayed reduced ACE2 expression. ACE2 deficiency is also thought to be associated with the inhibition of gene sequence in hypertension and cardiac hypertrophy. This ACE2 deficiency might even worsen heart failure. A heterozygote loss of ACE2 might be adequate to rise exposure towards heart disease [4]. In individuals with different health issues, the viral invasion may take full advantage of this deficiency for causing inflammation and pulmonary complications and augment the unevenness between the ACE → angiotensin II → AT1 receptor pole (detrimental) and the ACE-2 → angiotensin1-7 → AT2 and Mas receptor pole (protective) [4, 78]. Hence, ACE-2 inhibitors and AT1 blockers might be established as promising therapeutic strategies in COVID-19 treatment. AT1 blocking drug, losartan, is currently under clinical trials in hospitalized (NCT04312009) [79] or not hospitalized (NCT04311177) [79] patients. However, current evidence neither deny nor approves the continuation of these medications for COVID-19 infected patients [4].
Acute lung injury by means of ACE-2 downregulation and possible consequences of SARS-CoV-2 prompted ALI
The hidden function of renin–angiotensin–aldosterone system (RAAS) behind pathophysiology of ALI
Understanding the pathophysiology of ALI imparts the significant role of the RAAS system. Severe pulmonary damage, caused by lung infection, sepsis, aspiration, and trauma, confirms an increased capillary permeability with the existence of neutrophils, macrophages, and protein-rich fluid in considerable amounts in alveolar cavity, along with the foundation of hyaline membranes [12, 80, 81]. Since 2003, various studies have been conducted on portraying the role of RAAS in SARS-CoV-2 induced ARDS and ALI. ALI persuaded by acid aspiration showed raised angiotensin II levels in lungs and plasma of mouse model and in contrast, ACE deactivation diminished angiotensin II. Liu et al. recently reported in their studies that the patients with SARS-CoV-2 having high angiotensin II levels. In addition, the severity of lung injury is linked with the expression of ACE. ALI was less complicated in complete knockout (Acee/e) mice and AT1 receptor knockout mice compared to partial ACE knockout (Aceþ/e) mice and wild-type mice, respectively. Injection of recombinant SARS spike protein along with AT1 blockers elevated the expression of angiotensin II leading to ARDS in mice [12].
Polymorphism showing, I or D in gene sequence of ACE was allied with vulnerability and prognosis of ARDS. Hence, the proportion of D/D genotype carrying ARDS patients was higher than control individuals with D/D genotype. The morbidity rate of ARDS patients together with the D/D genotype was more when compared to that of ARDS patients together with the I/I genotype [12, 82]. These outcomes presented that ACE/Ang-II/AT1R might facilitate ALI pathogenesis [12].
Role of ACE/angiotensin II/AT1 receptor and ACE-2/angiotensin1-7/Mas receptor disequilibrium in reducing the elevated ACE-2 induced ALI
Severe ALI is significantly noticeable in the SARS-CoV-2 infected individuals with ACE2 receptors downregulation. ACE-2 involvement in pathophysiology of SARS-COV and SARS-CoV-2 infection became a topic of interest among researchers. Imai et al. studied that clinical pathological report of ALI persuaded by acid aspiration and sepsis in ACE-2 knockout mice was poorer than wildtype mice, causing more morbidity. However, angiotensin II in serum and lung of knockout mice were higher than wild-type. The injection of recombinant ACE2 in these critical mice expressively improved lung injury in both groups of mice. This improvement was facilitated through catalytic activity. Thus, showing that ACE2 with catalytic potential has shielding character in ALI. Simultaneously, recombinant ACE2 reduced serum and lung angiotensin II, served more beneficial with AT1 blockers, ultimately improving ALI in ACE2 knockout mice. The result proposed ACE2 negatively regulated ACE/angiotensin II/AT1 receptor to improve ALI [12]. The COVID-19 virus vitiates pulmonary epithelial progenitor cells, affecting the tendency of lung tissue repair subsequent ALI [12, 83].
Is application of ACE-2 inhibitors or AT1 blockers in COVID-19 patients with hypertension effective?
There is always a chance of raising researchable questions while exploring any new topic. In COVID-19 treatment strategy, an interesting topic is gaining significant amount of coverage and discussion, arising multiple objectionable questions. Whether anti-RAS drugs (ACEIs and ARBs) intensify the threat of SARS-CoV-2 infection? Whether is it required to discontinue the ACEIs/ARBs for hypertensive patients who are with or at high threat of SARS-CoV-2 infection? Whether is it required to switch to any other antihypertensive drugs in patients with hypertension or diabetic clinical symptoms? The answers to all these questions are still controversial. However, there are certain points that could prove beneficial in understanding and figuring out conclusion for the above-raised questionnaire. Both ACEIs and ARBs that are, basically, used to treat high blood pressure, though were found useful in upregulating ACE2 expression in rodents, there is not enough evidence of the same in humans. However, lisinopril and losartan work in favor of upregulation at mRNA level of ACE2, contradict to it, ramipril was found to decrease ACE2 protein expression, whereas Olmesartan neither showed any specific change in expression. Hence, it cannot be concluded that ACEIs/ARBs increase the risk of coronavirus infection by upregulating ACE2 expression [84].
Second, ACE2 receptor is not only the sole receptor responsible for increasing the risk of infection. A G-protein-coupled angiotensin type II receptor, transmembrane glycoprotein CD147, intercellular adhesion molecule-3-grabbing integrin (L-SIGN and DC-SIGN), all these have been recently recognized as receptors of SARs-CoV-2. Hence, numerous pathways are available through which virus invades the human body [84].
Third, there is no confirmation that ACEIs/ARBs escalate the possibility of infection in hypertensive and cardiac failure individuals. A systematic review and meta-analysis showed that administration of ACEIs significantly reduced the risk of pneumonia as well as pneumonia-based mortality in patients with higher risk of stroke [84] and heart failure. A comparative study conducted on hospitalized patients with hypertension and were on ACEIs/ARBs versus hospitalized patients with hypertension but not on such medications showed 3.7% and 9.8% mortality rate, respectively, suggesting that ACEIs/ARBs are component of standard therapy in hypertension and may be favorable in fighting against coronavirus infection [26]. However, sudden discontinuation of these drugs may even reverberate hypertension. Hence, currently, none of the literature supports specific discontinuation of these drugs in individuals who are already on these medications [53, 85].
Therapeutic importance of ACE-2 in treating SARS-CoV-2
Researchers and scientists are working extensively with the desire of finding any specific and effective drug or vaccine to deal with this pandemic. As ACE2 acts as a receptor for the entry of viruses into the cell, inducing ALI complications. The drugs targeting ACE2 might serve as a promising scenario for treating patients with COVID-19. In different animal studies, recombinant ACE2 administration established a beneficial role in improving pulmonary blood flow, pulmonary function, elastance, and lessen edema build-up, hypoxemia, and pulmonary hypertension [12, 55, 86]. Studies had established that the recombinant ACE2 is proficient for interacting with the coronavirus directly. In this context, Li et al. showed that S1 domain of virus spike protein might bind with high affinity to the rhACE2 in Vero E6 cells [12, 26]. Similarly, Monteil et al. revealed dose-dependent lessening of virus growth in Vero E6 cells, along with effectively treating viral infection in human blood vessel and kidney organoids, by rhACE2 administration. Hofmann et al. also showed the dose-dependent blocking of the viral spike, causing pseudo-infection in 293 T cells by rhACE2 [12, 87]. But, it is highly important to validate the safety and potency of rhACE2 in healthy individuals and patients with ARDS with minimal side effects on hemodynamic considerations [12, 60, 63, 88]. Catalytic activators like diminazene aceturate potentially increase the protective nature of rhACE2 against COVID-19 related ALI. Presently, the usage of rhACE2 in dealing with COVID-19 patients is under regulatory phases of trials (NCT04287686) [89, 90], thus ACE-2 inhibitors may be a prominent therapeutic target for the treatment of COVID-19 disease [91].
Conclusion and future perspective
Many countries around the world are experiencing extreme social and economic instability because of this COVID-19 pandemic since December 2019. Whilst many treatment options are under trials in laboratories, but currently, none of the approved drugs have been released for use against COVID-19. Thinking of COVID-19 and its relationship with ACE2, an in-depth understanding of pathogenesis might aid in the progress of targeted interventions so as to regulate this pandemic. The highly complex relationship persists between the RAAS and SARS-CoV pathogenicity, and by analogy, SARS-CoV-2 pathogenicity in the circumstances of COVID-19. A significant understanding of viral pathophysiology suggests many therapeutic strategies i.e., inhibition of SARS-CoV2 spike protein activation, inhibition of virus endocytosis, use of a soluble form of ACE2, peptide or non-peptide analogs of ACE2, and sustaining ACE2/Angiotensin-(1–7)/Mas receptor pathway activation. Still, basic research recommends that ACEIs and ARBs specifically have a shielding role. Hence, more researches are necessary before establishing the current treatments of COVID-19 with already marketed AEIs/ARBs or switching to any new medications. Properly-planed prospective and retrospective observational investigations, in addition to randomized controlled clinical trials, are necessary for developing the recommendations for the application of RAAS modulators in SARS-CoV-2 infection. The result from randomized clinical trials will further define the broader role and effectiveness of anti-RAAS agents against COVID-19 pandemic. ACE2-targeted therapeutic strategy against SARS-CoV-2 may lead to novel anti-COVID-19 primary or adjunctive agents. The present information on ACE2 receptors with relation to COVID-19 disease would be beneficial in finding its successful treatment.
References
Dhama K, Sharun K, Tiwari R, Dadar M, Malik YS, Singh KP, et al. COVID-19, an emerging coronavirus infection: advances and prospects in designing and developing vaccines, immunotherapeutics, and therapeutics. Hum Vaccines Immunother. 2020;16:1232–8. https://doi.org/10.1080/21645515.2020.1735227.
Gorain B, Choudhury H, Molugulu N, Athawale RB, Kesharwani P. Fighting strategies against the novel coronavirus pandemic: impact on global economy. Front Publ Health. 2020;8:800. https://doi.org/10.3389/FPUBH.2020.606129.
Gorbalenya AE. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming its SARS-CoV-2. Nat Microbiol. 2020;5:536–44. https://doi.org/10.1038/s41564-020-0695-z.
Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020;76:14–20. https://doi.org/10.1016/j.ejim.2020.04.037.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:280. https://doi.org/10.1016/j.cell.2020.02.052.
Agrawal M, Saraf S, Saraf S, Murty US, Kurundkar SB, Roy D, et al. In-line treatments and clinical initiatives to fight against COVID-19 outbreak. Respir Med. 2020. https://doi.org/10.1016/j.rmed.2020.106192.
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46:586–90. https://doi.org/10.1007/s00134-020-05985-9.
Turner AJ, Hiscox JA, Hooper NM. ACE2: From vasopeptidase to SARS virus receptor. Trends Pharmacol Sci. 2004;25:291–4. https://doi.org/10.1016/j.tips.2004.04.001.
Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/angiotensin 1–7 axis of the renin-angiotensin system in heart failure. Circ Res. 2016;118:1313–26. https://doi.org/10.1161/CIRCRESAHA.116.307708.
Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCov. Am J Respir Crit Care Med. 2020;202:756–9. https://doi.org/10.1101/2020.01.26.919985.
Ortiz-Melo DI, Gurley SB. Angiotensin converting enzyme 2 and the kidney. Curr Opin Nephrol Hypertens. 2016;25:59–66. https://doi.org/10.1097/MNH.0000000000000182.
Liu MY, Zheng B, Zhang Y, Li JP. Role and mechanism of angiotensin-converting enzyme 2 in acute lung injury in coronavirus disease 2019. Chronic Dis Transl Med. 2020;6:98–105. https://doi.org/10.1016/j.cdtm.2020.05.003.
Liu Y, Gayle AA, Wilder-Smith A, Rocklöv J. The reproductive number of COVID-19 is higher compared to SARS coronavirus. J Travel Med. 2020;27:1–4. https://doi.org/10.1093/jtm/taaa021.
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87:E1–9. https://doi.org/10.1161/01.res.87.5.e1.
Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme: cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–43. https://doi.org/10.1074/jbc.M002615200.
Turner AJ, Hooper NM. The angiotensin-converting enzyme gene family: genomics and pharmacology. Trends Pharmacol Sci. 2002;23:177–83. https://doi.org/10.1016/S0165-6147(00)01994-5.
Crackower MA, Sarao R, Oliveira-dos-Santos AJ, Costa JD, Zhang L. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–8. https://doi.org/10.1038/nature00786.
Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, et al. The ACE2/Angiotensin-(1–7)/Mas axis of the renin-angiotensin system: focus on angiotensin-(1–7). Physiol Rev. 2018;98:505–53. https://doi.org/10.1152/physrev.00023.2016.
Sriram K, Insel PA. A hypothesis for pathobiology and treatment of COVID-19: the centrality of ACE1/ACE2 imbalance. Br J Pharmacol. 2020;177:4825–44. https://doi.org/10.1111/bph.15082.
Giron CC, Laaksonen A, Barroso da Silva PA. On the interactions of the receptor-binding domain of SARS-CoV-1 and SARS-CoV-2 spike proteins with monoclonal antibodies and the receptor ACE2. Virus Res. 2020;285: 198021. https://doi.org/10.1016/j.virusres.2020.198021.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–3. https://doi.org/10.1126/science.abb2507.
Chen Y, Guo Y, Pan Y, Zhao ZJ. Structure analysis of the receptor binding of 2019-nCoV. Biochem Biophys Res Commun. 2020;525:135–40. https://doi.org/10.1016/j.bbrc.2020.02.071.
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–8. https://doi.org/10.1126/science.abb2762.
Barrantes FJ. While we wait for a vaccine against SARS-CoV-2, why not think about available drugs? Front Physiol. 2020;11:820. https://doi.org/10.3389/fphys.2020.00820.
Khatri I, Stall FJT, Dongen JJMV. Blocking of the high-affinity interaction-synapse between SARS-CoV-2 spike and human ACE2 proteins likely requires multiple high-affinity antibodies: an immune perspective. Front Immunol. 2020;11: 570018. https://doi.org/10.3389/fimmu.2020.570018.
Bourgonje AR, Abdulle AE, Timens W, Hillebrands JL, Navis GJ, Gordijn SJ, et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J Pathol. 2020;251:228–48. https://doi.org/10.1002/path.5471.
Jayaweera M, Perera H, Gunawardana B, Manatunge J. Transmission of COVID-19 virus by droplets and aerosols: a critical review on the unresolved dichrotomy. Environ Res. 2020;188: 109819. https://doi.org/10.1016/j.envres.2020.109819.
Jarvis MC. Aerosol transmission of SARS-CoV-2: physical principles and implications. Front Publ Health. 2020;8: 590041. https://doi.org/10.3389/fpubh.2020.590041.
Ryu G, Shin HW. SARS-CoV-2 infection of airway epithelial cells. Immune Netw. 2021;21(1): e3. https://doi.org/10.4110/in.2021.21.e3.
Subbarao K, Mahanty S. Respiratory virus infections: understanding COVID-19. Immunity. 2020;52(6):905–9. https://doi.org/10.1016/j.immuni.2020.05.004.
Gallo O, Locatello LG, Mazzoni A, Novelli L, Annunziato F. The central role of the nasal microenvironment in the transmission, modulation, and clinical progression of SARS-CoV-2 infection. Mucosal Immunol. 2021;14:305–16. https://doi.org/10.1038/s41385-020-00359-2.
Sungnak W, Huang N, Becavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. 2020;26:681–7. https://doi.org/10.1038/s41591-020-0868-6.
Wang H, Yang P, Liu K, Guo F, Zhang Y, Zhang G, et al. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008;18:290–301. https://doi.org/10.1038/cr.2008.15.
Bayati A, Kumar R, Francis V, McPherson P. SARS-CoV-2 infects cells following viral entry via clathrin-mediated endocytosis. J Biol Chem. 2021;296: 100306. https://doi.org/10.1016/j.jbc.2021.100306.
Hu TY, Frieman M, Wolfram J. Insights from nanomedicine into chloroquine efficacy against COVID-19. Nat Nanotechnol. 2020;15:247–9. https://doi.org/10.1038/s41565-020-0674-9.
Lin L, Lu L, Cao W, Li T. Hypothesis for potential pathogenesis of SARS-CoV-2 infection—a review of immune changes in patients with viral pneumonia. Emerg Microbes Infect. 2020;9:727–32. https://doi.org/10.1080/22221751.2020.1746199.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–62. https://doi.org/10.1016/S0140-6736(20)30566-3.
Jin Y, Yang H, Ji W, Wu W, Chen S, Zhang W, et al. Virology, epidemiology, pathogenesis, and control of covid-19. Viruses. 2020;12:372. https://doi.org/10.3390/v12040372.
Mohammadpour H, Ziai A, Sadr M, Rezaei M, Marjani M, Tabarsi P. A novel coronavirus disease (COVID-19): a review of host cell signaling pathways. Tanaffos. 2020;19(2):108–11 (PMID: 33262797).
Suryawanshi RK, Koganti R, Agelidis A, Patil CD, Shukla D. Dysregulation of cell signaling by SARS-CoV-2. Trends Microbiol. 2021;29:224–37. https://doi.org/10.1016/j.tim.2020.12.007.
Scialo F, Daniele A, Amato F, Pastore L, Matera MG, Cazzola M, et al. The major cell entry receptor for SARS-CoV-2. Lungs. 2020;198:867–77. https://doi.org/10.1007/s00408-020-00408-4.
Hemmat N, Asadzadeh Z, Ahangar NK, Alemohammad H, Najafzadeh B, Derakhshani A, et al. The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV. Arch Virol. 2021;166(3):675–96. https://doi.org/10.1007/s00705-021-04958-7.
Li-Chin C, Tzu-Jen K, Nam Nhut P, Chung-Chieh C, Meng-Chi Y, Chien-Fu C, et al. Novel signaling pathways regulate SARS-CoV and SARS-CoV-2 infectious disease. Medicine. 2021;100: e24321. https://doi.org/10.1097/MD.000000000002432.
Battagello DS, Dragunas G, Klein MO, Ayub ALP, Velloso FJ, Correa RG. Unpuzzling COVID-19: tissue-related signaling pathways associated with SARS-CoV-2 infection and transmission. Clin Sci (Lond). 2020;134:2137–60. https://doi.org/10.1042/CS20200904.
Huang Y, Yang C, Xu XF, Xu W, Liu SW. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin. 2020;41:1141–9. https://doi.org/10.1038/s41401-020-0485-4.
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271-80.e8. https://doi.org/10.1016/j.cell.2020.02.052.
Lippi G, Lavie CJ, Henry BM, Sanchis-Gomar F. Do genetic polymorphisms in angiotensin converting enzyme 2 (ACE2) gene play a role in coronavirus disease 2019 (COVID-19)? Clin Chem Lab Med. 2020;58:1415–22. https://doi.org/10.1515/cclm-2020-0727.
Davidson AM, Wysocki J, Batlle D. Interaction of SARS-CoV-2 and other coronavirus with ACE (Angiotensin-Converting Enzyme)-2 as their main receptor: therapeutic implications. Hypertension. 2020;76:1339–49. https://doi.org/10.1161/HYPERTENSIONAHA.120.15256.
Yang J, Petitjean SJL, Koehler M, Zhang Q, Dumitru AC, Chen W, et al. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat Commun. 2020;11:4541. https://doi.org/10.1038/s41467-020-18319-6.
Xie Y, Karki CB, Du D, Li H, Wang J, Sobitan A, Teng S, et al. Spike proteins of SARS-CoV and SARS-CoV-2 utilize different mechanisms to bind with human ACE2. Front Mol Biosci. 2020;7: 591873. https://doi.org/10.3389/fmolb.2020.591873.
Tai W, He L, Zhang X, Pu J, Voronin D, Jiang S, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol. 2020;17:613–20. https://doi.org/10.1038/s41423-020-0400-4.
Brielle ES, Schneidman-Duhovny D, Linial M. The SARS-CoV-2 exerts a distinctive strategy for interacting with the ACE2 human receptor. Viruses. 2020;12:497. https://doi.org/10.3390/v12050497.
Bansal S, Agstam S, Gupta A, Gupta P, Isser HS. Continue ACE inhibitors/ARB’S till further evidence in coronavirus disease 2019 (COVID-19). Indian Heart J. 2020;72:212–3. https://doi.org/10.1016/j.ihj.2020.04.007.
Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care. 2020;24:422. https://doi.org/10.1186/s13054-020-03120-0.
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–6. https://doi.org/10.1038/nature03712.
Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–9. https://doi.org/10.1038/nm1267.
Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ, Goor HV. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–7. https://doi.org/10.1002/path.1570.
Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–36. https://doi.org/10.1172/JCI68782.
Rivellese F, Prediletto E. ACE2 at the centre of COVID-19 from paucisymptomatic infections to severe pneumonia. Autoimmun Rev. 2020;19: 102536. https://doi.org/10.1016/j.autrev.2020.102536.
Zhang H, Baker A. Recombinant human ACE2: acing out angiotensin II in ARDS therapy. Crit Care. 2017;21:305. https://doi.org/10.1186/s13054-017-1882-z.
Guo J, Huang Z, Lin L, Lv J. Coronavirus Disease 2019 (COVID-19) and cardiovascular disease: a viewpoint on the potential influence of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers on onset and severity of severe acute respiratory syndrome coronavirus 2 Infection. J Am Heart Assoc. 2020;9: e016219. https://doi.org/10.1161/JAHA.120.016219.
Colafella KMM, Uijl E, Danser AHJ. Interference with the renin-angiotensin system (RAS): classical inhibitors and novel approaches. In: Encycl Endocr Dis. Elsevier; 2018. p. 523–30. https://doi.org/10.1016/B978-0-12-801238-3.65341-2.
Khan A, Benthin C, Zeno B, Albertson TE, Boyd J, Christie JD, et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit Care. 2017;21:234. https://doi.org/10.1186/s13054-017-1823-x.
Elfiky AA, Mahdy SM, Elshemey WM. Quantitative structure-activity relationship and molecular docking revealed a potency of anti-hepatitis C virus drugs against human corona viruses. J Med Virol. 2017;89:1040–7. https://doi.org/10.1002/jmv.24736.
Adedeji AO, Severson W, Jonsson C, Singh K, Weiss SR, Sarafianos SG. Novel inhibitors of severe acute respiratory syndrome coronavirus entry that act by three distinct mechanisms. J Virol. 2013;87:8017–28. https://doi.org/10.1128/jvi.00998-13.
Ting R, Edmonds P, Higginson IJ, Sleeman KE. Palliative care for patients with severe COVID-19. BMJ. 2020;370: m2710. https://doi.org/10.1136/bmj.m2710.
Bwire GM. Coronavirus: why men are more vulnerable to COVID-19 than women? SN Compr Clin Med. 2020;2:874–6. https://doi.org/10.1007/s42399-020-00341-w.
Chang WH. Understanding the COVID-19 pandemic from a gender perspective. Taiwan J Obstet Gynecol. 2020;59:801–7. https://doi.org/10.1016/j.tjog.2020.09.004.
Gemmati D, Bramanti B, Serino ML, Secchiero P, Zauli P, Tisato V. COVID-19 and individual genetic susceptibility/receptivity: Role of ACE1/ACE2 genes, immunity, inflammation and coagulation might the double x-chromosome in females be protective against SARS-COV-2 compared to the single x-chromosome in males? Int J Mol Sci. 2020;21:3474. https://doi.org/10.3390/ijms21103474.
Arnold RH. COVID-19—does this disease kill due to imbalance of the renin angiotensin system (RAS) caused by genetic and gender differences in the response to viral ACE 2 attack? Heart Lung Circ. 2020;29:964–72. https://doi.org/10.1016/j.hlc.2020.05.004.
Patel SK, Wai B, Ord M, MacIsaac RJ, Grant S, Velkoska E, et al. Association of ACE2 genetic variants with blood pressure, left ventricular mass, and cardiac function in caucasians with type 2 diabetes. Am J Hypertens. 2012;25:216–22. https://doi.org/10.1038/ajh.2011.188.
Luo Y, Liu C, Guan T, Li Y, Lai Y, Li F, et al. Association of ACE2 genetic polymorphisms with hypertension-related target organ damages in south Xinjiang. Hypertens Res. 2019;42:681–9. https://doi.org/10.1038/s41440-018-0166-6.
Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63:364–74. https://doi.org/10.1007/s11427-020-1643-8.
Wevers BA, Hoek LVD. Renin-angiotensin system in human coronavirus pathogenesis. Future Virol. 2010;5:145–61. https://doi.org/10.2217/fvl.10.4.
Gurwitz D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev Res. 2020;81:537–40. https://doi.org/10.1002/ddr.21656.
Lowe A, Chang DD, Creek G. Multipte fatalities in a family cluster of COVID-19 with acute respiratory distress syndrome. Ochsner J. 2020;20:134–8. https://doi.org/10.31486/toj.20.0056.
Saponaro F, Rutigliano G, Sestito S, Bandini L, Storti B, Bizzarri R, et al. ACE2 in the era of SARS-CoV-2: controversies and novel perspectives. Front Mol Biosci. 2020;7:271. https://doi.org/10.3389/fmolb.2020.588618.
Okamoto H, Ichikawa N. The pivotal role of the angiotensin-II–NF-κB axis in the development of COVID-19 pathophysiology. Hypertens Res. 2020;44:126–8. https://doi.org/10.1038/s41440-020-00560-7.
Rothlin RP, Duarte M, Pelorosso FG, Nicolosi L, Salgado MV, Vetulli HM, et al. Angiotensin receptor blockers for COVID-19: pathophysiological and pharmacological considerations about ongoing and guture prospective clinical trials. Front Pharmacol. 2021;12:603736.
Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–49. https://doi.org/10.1056/NEJM200005043421806.
Nagase T, Uozumi N, Ishii S, Kume K, Izumi T, Ouchi Y, et al. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat Immunol. 2000;1:42–5. https://doi.org/10.1038/76897.
Marshall RP, Webb S, Bellingan GJ, Montgomery HE, Chaudhari B, McAnulty RJ, et al. Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2002;166:646–50. https://doi.org/10.1164/rccm.2108086.
Ling TY, Kuo MD, Li CL, Yu AL, Huang YH, Wu TJ, et al. Identification of pulmonary Oct-4+ stem/progenitor cells and demonstration of their susceptibility to SARS coronavirus (SARS-CoV) infection in vitro. Proc Natl Acad Sci USA. 2006;103:9530–5. https://doi.org/10.1073/pnas.0510232103.
Bian J, Zhao R, Zhai S, Li Z. Anti-RAS drugs and SARS-CoV-2 infection. Acta Pharm Sin B. 2020;10:1251–2. https://doi.org/10.1016/j.apsb.2020.04.013.
Hariyanto TI, Japar KV, Damay V, Kwenandar F, Sieto NL, Kurniawan A. The use of ACE inhibitor/ARB in SARS-CoV-2 patients: a comprehensive narrative review. Asian J Med Sci. 2020;11:113–20. https://doi.org/10.3126/ajms.v11i6.29911.
Treml B, Neu N, Kleinsasser A, Gritsch C, Finsterwalder T, Geiger R, et al. Recombinant angiotensin-converting enzyme 2 improves pulmonary blood flow and oxygenation in lipopolysaccharide-induced lung injury in piglets. Crit Care Med. 2010;38:596–601. https://doi.org/10.1097/CCM.0b013e3181c03009.
Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181:905-913.e7. https://doi.org/10.1016/j.cell.2020.04.004.
Haschke M, Schuster M, Poglitsch M, Loibner H, Salzberg M, Bruggisser M, et al. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin Pharmacokinet. 2013;52:783–92. https://doi.org/10.1007/s40262-013-0072-7.
Shenoy V, Gjymishka A, Jarajapu YP, Qi Y, Afzal A, Rigatto K, et al. Diminazene attenuates pulmonary hypertension and improves angiogenic progenitor cell functions in experimental models. Am J Respir Crit Care Med. 2013;187:648–57. https://doi.org/10.1164/rccm.201205-0880OC.
Augoustides JGT. The renin-angiotensin-aldosterone system in coronavirus infection-current considerations during the pandemic. J Cardiothorac Vasc Anesth. 2020;34:1717–9. https://doi.org/10.1053/j.jvca.2020.04.010.
Chatterjee B, Thakur SS. ACE2 as a potential therapeutic target for pandemic COVID-19. RSC Adv. 2020;10:39808–13. https://doi.org/10.1039/d0ra08228g.
Acknowledgements
Vijay K Patel is grateful to Indian Council for Medical Research (ICMR), New Delhi for providing financial support in the form of Research Associateship (Grant number: 45/05/2019-PHA-BMS). One of the authors, Harish Rajak is thankful to ICMR, New Delhi, India for providing financial assistance in the form of an Extra-Mural Research project (ICMR/2020/2817).
Funding
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial conflict with the subject matter or materials discussed in the manuscript. No writing assistance was utilized in the production of this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There is no conflict of interest and disclosures associated with the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shirbhate, E., Pandey, J., Patel, V.K. et al. Understanding the role of ACE-2 receptor in pathogenesis of COVID-19 disease: a potential approach for therapeutic intervention. Pharmacol. Rep 73, 1539–1550 (2021). https://doi.org/10.1007/s43440-021-00303-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43440-021-00303-6