
Abstract
In-situ zeolites NaY and Na[B]Y were synthesized on microspherical matrices of kaolin to obtain FCC catalysts. An alkaline pretreatment of the matrix was investigated in order to evaluate its effect on matrix properties and crystallization of the in-situ synthetized zeolites. Catalysts were characterized by SEM, TEM, Ar adsorption, XRD and NH3-TPD. It was observed an increase in the surface area and mesoporosity of the alkaline treated catalysts either synthetized with the presence of boron or with no boron in the hydrothermal reaction mixture. Ammonia TPD analyses have shown an increase in the amount and strength of the acidity of the catalysis with the zeolites crystallized on the pretreated matrices and exchanged with lanthanum ions. Thus, a combination between higher concentration of stronger acid sites and higher proportion of mesoporous generated in the matrices treated with alkaline solution had resulted in more active catalyst as shown by the triisopropylbenzene cracking experiments conducted here.
Graphical abstract

Similar content being viewed by others

Introduction
The zeolite Y is a microporous crystalline synthetic aluminosilicate with a pore opening of 0.74 nm, acidity adjustable due to the Si/Al ratio and high thermal and hydrothermal stability, making these materials suitable for their application in heterogeneous catalysis, especially in fluid catalytic cracking (FCC) (Baerlocher et al. 2007; Karami and Rohani 2009; Vermeiren and Gilson 2009). Nowadays, great difficulties have been generated in the FCC due to the changing quality of the feedstocks, affecting mainly the catalyst performance considering their higher metal content and coke precursors. The average boiling point and molecular diameter of the heavy hydrocarbon molecules involved have led to higher coke formation with the concomitantly pore blocking and decrease in conversion (Chen et al. 2013; Pan et al. 2019; Primo and Garcia 2014). Therefore, strategies to achieve an efficient catalyst have been designed to improve mass transport, the adsorption/desorption of bulky molecules and promoting reaction at active sites located on the internal surface of the catalyst. These strategies include the partial destruction of zeolite (demetallation) or adding templates that are removed after synthesis to introduce mesoporous (Wei et al. 2015; Schwieger et al. 2016). Thus, hybrid properties of microporous zeolites (shape selectivity) and mesoporous materials (diffusion of bulky reactants) are encompassed in the catalyst.
Post-synthesis treatments as dealumination by steaming or acid leaching and desilication by alkaline solutions to improve diffusion properties in zeolites have been extensively studied. These methods produce mesopores with heterogeneous distribution, and occasionally it could lead to damage of structure (Silaghi et al. 2014, 2016). Alternative methods used to generate mesoporous based on the use of boron in the synthesis gel aimed at isomorphic substitution of Al3+ ions by B3+ at T sites of the structure have been reported (Koller et al. 2015). However, isomorphic substitution of boron in large pore zeolites such as faujasite Y has been unsuccessful and post-synthesis methods have been also evaluated (Han et al. 1994; Gaffney et al. 1989). Mi et al. synthesized zeolite Y with a B/Al molar ratio of 0.01 and suggested that boron insertion in this structure promotes hydrolysis of the Si–O–Al and Si–O–B bonds during steaming which results an interconnected mesoporous system better than that obtained by conventional dealumination and generating higher catalytic activity compared with zeolite without boron (Mi et al. 2017).
The performance of the FCC catalyst also depends on the properties of the matrix. In that way, the nature and composition of the matrix is decisive, since it provides primary acid sites to crack bulky molecules into smaller products that can reach then the active sites of the zeolite (Gamero et al. 1997). Besides, the matrix imparts attrition-resistance, tolerance to metals and, based on in-situ manufacturing of the FCC catalyst, it supports and disperses the zeolite (Clough et al. 2017). The in-situ synthesis route is a method where the zeolite grows directly on a pre-formed kaolin matrix, which provides silicon and aluminum sources for direct crystallization avoiding the use of binders. Due to the important contribution of the matrix on the catalyst, in recent years manufacturers have been directed efforts towards improving its properties. In particular, Engelhard corporation (now BASF) has designed a Distributed Matrix Structures (DMS) technology with a macropore system that decreases coke selectivity and favors a rapid diffusion of feed and products during the catalytic reaction (Clough et al. 2017; Pan et al. 2015).
As mentioned before, the conversion of heavy feedstocks into desired products has been the current challenges of the FCC process, because of the conventional catalysts mainly presents restrictions on diffusion properties. Consequently, in this research a set of catalysts have been designed that combine functionalities inside both matrix and zeolite phases, in order to increase interconnectivity and facilitate the diffusion of molecules and reaction products. Thus, four samples were prepared by the in-situ synthesis methodology. In this respect, two FCC-matrices were prepared using mixtures of thermal- and chemically treated kaolins with the presence of mesoporosity in the microspheres prior to crystallization of the zeolite. The zeolite NaY and Na[B]Y were hydrothermally synthesized directly on the matrices using boric acid as source of boron. Subsequently, the Na-samples were treated by steaming to modify the textural properties of zeolites by dealumination. Finally, the catalysts after ammonium and lanthanum exchange were evaluated in the catalytic cracking of 1,3,5-triisopropylbenzene at 300 °C in a MAT unit.
Experimental section
Matrix preparation
The starting material was a kaolin K obtained from Caolines de Vimianzo. The kaolin was calcined in a furnace at 1000 °C and 1100 °C during 1 h using a heating rate of 5 °C/min. The samples were named K-1000 and K-1100, respectively. After calcination, K-1000 was alkaline treated at 95 °C for 24 h using an aqueous solution 3.5 M of NaOH (5 mL/g of calcined kaolin) and identified as K-1000A (Padilla et al. 2020).
Two different aqueous slurries A and B with 40 wt% of solid content using a sodium silicate solution (28.5 wt% SiO2, 8.5 wt% Na2O; Merck) as dispersant agent and deionized water were prepared. The slurry A was obtained mixing mass percentages 50 K:35 K-1000:15 K-1100 and slurry B 50 K:35 K-1000A:15 K-1100.
The matrix microspheres were obtained by spray drying of the slurries in a pilot scale unit using a flow of 3 mL/min and adjusting the inlet temperature between 135 and 140 °C, air pressure 0.59 MPa, atomization air flow 8–10 L/min and dried air flow 70 L/min. The microspheres obtained from slurries A and B were sieved between 40 and 90 µm, calcined in furnace at 750 °C for 3 h and labeled as M and M-Na, respectively.
Synthesis in situ of NaY and Na[B]Y
The crystallization of Na[B]Y zeolite on matrices were carried out by hydrothermal synthesis. As boron source in synthesis gel was used boric acid (H3BO3, Merck) dissolved in water and alkalinized with sodium hydroxide (NaOH, Merck) up to pH 13. A seed solution was prepared according to the stereochemistry reported by Qiang, Li. and was aged at 22 °C for 6 h (Qiang et al. 2010). The synthesis of Na[B]Y/M-Na is illustrated as an example. First, the boron solution and M-Na were added to sodium silicate solution to obtain a molar composition of 1.3Na2O:4.6SiO2:Al2O3:0.059B2O3:63.2H2O. Then, seeds were added to the reaction mixture to adjust a molar ratio of 1.9Na2O:5.1SiO2:Al2O3:0.056B2O3:74.1H2O, and was left to age at 54 °C during 1 h. Subsequently, hydrothermal crystallization was carried out at 100 °C for 21 h in Teflon reactors with a stainless steel cover. Solid product was separated by filtration, washed abundantly with deionized water and dried for 12 h at 90 °C. Additionally, the samples were sieved to remove fines less than 40 µm. The samples of NaY on matrices were prepared following the same procedure without adding the boron solution.
Catalysts preparation
The ammonium form of the samples was obtained by ion exchange at 85 °C for 1 h using a NH4NO3 aqueous solution 15 wt% stirring at 50 rpm. After the exchange step, the samples were washed and dried. NH4[B]Y and NH4Y samples on the M-Na and M matrices were calcined in a quartz reactor at 600 °C for 1 h under dry air flow (100 mL/min). Successively, air flow was stopped and maintaining temperature was performed an hydrothermal treatment for 5 h with steam flow and N2 flow (0.3 mL/min) to keep the catalyst fluidized. The catalysts obtained were labeled as HY/M-Na, H[B]Y/M-Na, HY/M and H[B]Y/M.
Additionally, catalysts were exchanged once with an aqueous solution 0.2 M of La(NO3)3·6H2O at 85 °C during 1 h under stirring. This samples were denoted as La,HY/M-Na, LaH[B]Y/M-Na, La,HY/M and La,H[B]Y/M.
Characterization methodologies
The XRD patterns were collected in a RIGAKU Smartlab SE Advance powder diffractometer, using CuKα radiation. The samples were scanned at 1.2 º/min between 2° and 70° 2θ. The zeolite NaY crystallinity in the samples was determined according to ratio between peak height at 22°–24° 2θ of sample/reference and the percentage of zeolite was calculated with a Rietveld refinement using TOPAS software.
For determination of bulk elemental composition, the samples were prepared by acid digestion in a closed system (microwave) for quantification of Aluminum, Silicon and Sodium and open system (heating plate) for analysis of Boron and Lanthanum. The solutions obtained were analyzed by Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES) using an equipment Optima 8300 and external standardization.
The morphological characteristics of samples were studied with a broad range of microscopic techniques that include transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The SEM images were collected using the QUANTA 450 and LEO 1450 VP electron microscopes equipped with the OXFORD scattered energy X-ray system, operated in the high vacuum and low vacuum mode. A Tecnai F20 Super Twin TMP microscope was used to obtain TEM micrographs.
The textural properties were measured by argon adsorption at − 186 °C in an automated analyzer Micromeritis 3FLEX™, after sample degasification at 300 °C under vacuum (6 Pa). The data analysis was carried out in the software 3FLEX V.4.03 by Micromeritics. The porosity distribution by Non-Local Density Functional Theory (NLDFT) was studied with Ar@87-Zeolites, Me-Form and Ar@87-Zeolites, H-Form models for Na-samples and H-samples, respectively.
The acid properties of catalysts were determined by temperature programmed desorption of ammonia (NH3-TPD) in an apparatus Micromeritics AutoChem II equipped with a thermal conductivity detector. A sample of ~ 0.5 g was charged in the quartz tube. The catalyst was heated in situ at a rate of 10 °C/min until 600 °C for 1 h under He flow before saturation with NH3 at 150 °C for 30 min. The NH3-TPD profile was recorded from 150 to 600 °C.
Catalytic testing
The catalysts were evaluated in 1,3,5-triisopropylbenzene (TIPB) cracking at 300 °C, C/O = 5, under atmospheric pressure using a fixed-bed reactor in a microactivity test (MAT) unit. Before starting test, 2.5 g of catalyst was loaded in the reactor and heated at 300 °C under N2 flow (30 mL/min) at least for 4 h. Afterward, 0.5 g of TIPB (95 wt%, Aldrich) were injected during 90 s and N2 flow was timed for an additional 15 min to recover products. The liquid products were condensed and analyzed on an Agilent 6890 chromatograph according to ASTM D6729 “Determination of Individual Components in Spark Ignition Engine Fuels by 100 Meter Capillary High Resolution Gas Chromatography” for quantification of paraffins, iso-paraffins, aromatics, naphthenes and olefins, and simulated distillation analysis on an Agilent 7890B chromatograph following ASTM D7213 “Boiling Range Distribution of Petroleum Distillates in the Boiling Range from 100 to 615 °C by Gas Chromatography”. The gas products were analyzed by gas chromatography in an Agilent 6890 equipment according to the UOP 359 standard “Gas Refinery Analysis by GC”, and coke deposited on catalyst after reaction was measured by combustion in a LECO carbon analyzer C230 equipped with an IR detector to quantify CO2 generated. A commercial FCC catalyst (labelled as CAT-COM) was used for the comparison of results of the catalytic evaluation.
Results
In situ crystallization of NaY and Na[B]Y on matrices M-Na and M
The zeolite NaY crystals were grown under hydrothermal conditions on two kaolin matrices with different textural properties and composition. The matrix M-Na was prepared with a mixture of calcined kaolin, where a 35 wt% of matrix was a K-1000A fraction corresponding to a kaolin calcined at 1000 °C and treated with sodium hydroxide to extract soluble silicon species. As shown in our previous report, the alkaline treatment leads to an increasing of the external surface area and the amount of mesopores between 3 and 10 nm in the matrix (Fig. 1) (Padilla et al. 2020). Thus, the matrix M-Na with only 7.4% of silicon moles removed, have shown a BET area of 24 m2/g and 5.5 times more pore volume compared with the matrix M (Table 1). An indirect measure of the voids inside matrix microspheres is related to skeletal densities. In this regard, the Matrix M showed a higher density value indicating a more compact material than M-Na.

Argon adsorption–desorption isotherms at – 186 °C and BJH desorption pore distribution plot of matrices M-Na and M
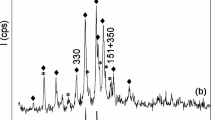
The X-ray diffraction profiles of the matrices showed patterns with high content of amorphous material and characteristic reflections of quartz and mica phases (Fig. 2). In the case of the M-Na matrix, an additional phase was identified and assigned as muscovite 2M2, Na, that could be related to the alkaline treatment of the K-1000A phase and the remaining percentage of sodium in the matrix after preparation of microspheres (Table 1). On the other hand, the loss of the crystalline ordering on kaolin occurs during the calcination of the microspheres at 750 °C, due to the dehydroxylation process of kaolinite. The gradual removal of water molecules induces a coordination change in the octahedral (AlVI) towards tetrahedral (AlIV) aluminum environments (Sperinck et al. 2011; Yan et al. 2017; White et al. 2010). This AlIV is used as an active source of aluminum during the in-situ synthesis of the zeolite NaY (Padilla et al. 2020). Additionally, the content of amorphous material in matrices is related to the mixture of kaolin phases calcined at different temperatures that are linked with sodium silicate acting as a binder.

XRD profiles of matrices, NaY and Na[B]Y on matrices. Qz quartz, Ms muscovite, MsNa sodium muscovite, A γ-Alumina or Al-Si spinel, Y zeolite Y and P: zeolite P
The growth of faujasites NaY and Na[B]Y on matrices after the in-situ synthesis was confirmed by XRD and Argon physisorption. According to the surface area results of synthesized samples, it was found that ~ 80% of surface area corresponded to microporous and attributed to the presence of the crystallized zeolite (Table 2). The quantitative analysis of the diffraction profiles revealed that during the synthesis of the zeolite NaY, the zeolite NaP was also crystallized, and the amount of each zeolite phases in the microspheres could depend on the matrix used (Fig. 2). In the case of the samples obtained with the matrix M, they reached a higher content of NaY zeolite compared to the M-Na matrix samples (Table 2). However, NaY/M and Na[B]Y/M showed lower zeolite crystallinity, and a broad band between 20° 2θ and 35° 2θ in the XRD traces associated with amorphous material.
In the Fig. 3 are shown the SEM micrographs of the in-situ synthesis of NaY/M-Na and NaY/M. The heterogeneous appearance inside the particles is due to the mode of aggregation of the kaolin particles during the spray drying process (Padilla et al. 2021). Images of the matrix microspheres showed a moderately smooth surface (Fig. 3a, e), whereas after the hydrothermal synthesis, the microspheres appear to be covered by a layer of pseudo-cubic crystallites. The NaY/M sample exhibits a mostly compact-looking surface with scarce pores, which forms a crust with a thickness of less than 1 µm (Fig. 3f–h). Furthermore, the sample NaY/M-Na presents a surface with voids between 120 and 3 µm. For both samples, the zeolite crystals grown on matrix particles are also observed inside microsphere (Fig. 3d).

SEM images of a matrix M-Na; b microsphere, c surface of microsphere, d inside the microsphere of NaY/M-Na; e matrix M and f microsphere, g surface of microsphere, h inner view of microsphere of NaY/M
Taking into account that zeolites supported on the silica-alumina matrices have not enough contrast between particles for acquisition of TEM images, samples were sonicated to release the zeolite crystals (Fig. S1). Thus, allowing an indirect determination of the crystal size distribution of the zeolites crystallized on matrices. It was found that for all samples the sizes are > 300 nm. The NaY/M and NaY/M-Na samples showed most of the crystals between 800–1000 and 600–800 nm, respectively. Furthermore, the Na[B]Y/M sample displayed a Gaussian crystal size distribution with a maximum at 600–800 nm. Especially, the Na[B]Y/M-Na sample exhibited a wide range of particle sizes from 600 nm, where 17% of crystals were > 1000 nm (Fig. 4).

TEM images of Na[B]Y/M-Na sample
Properties of the samples in the acid form
Samples of NaY and Na[B]Y synthesized on the matrices were exchanged with NH4NO3(ac), followed by steaming at 600 °C to obtain H-form of zeolite in the catalysts particles. Some of the samples were further exchanged with lanthanum in order to evaluated its effect on acidity and activity of the catalysts.
Surface area and pore distribution in FCC catalysts play a very important role in its catalytic activity, since the reactant molecules must diffuse through cavities to reach the active sites where the cracking reactions take place. As shown in the Fig. 5, the catalysts prepared by the in-situ method and steaming-treated exhibited a type IV argon adsorption–desorption isotherms, where the presence of mesopores were attributed to the hysteresis loop with p/p0 between 0.4 and 1.0. These type of isotherms have been previously reported in studies of zeolites embedded in matrix particles (Patrylak et al. 2001; Zheng et al. 2017, 2015). In contrast, the commercial FCC catalyst used here that is manufactured by an incorporation methodology showed a different isotherm with no defined steps (Fig. S2).

Argon adsorption/desorption isotherms (top) and Pore size distributions derived from NLDFT method applied to argon at – 186 °C isotherms for the prepared samples after steaming (bottom)
On the other hand, the pore size distribution plot of the in-situ catalysts showed two main groups of pore size (Fig. 5). The first one corresponds to a sharp micropore distribution in the range between 0.6 nm and 0.8 nm and another less intense with larger pore diameter between 3 nm and 15 nm. The maximum at 0.74 nm is associated with the micropores of zeolite Y, whereas the mesopores could be formed inter-crystal or as a result of the treatment with steam. The catalysts synthesized with the M-Na exhibited more volume of mesopores with 7.0 nm and 9.0 nm of diameter, compared to those prepared with M (Fig. 5). After calcination and steaming, it was also found that the percentage of microporous area decreased by 6–10% in acid samples compared with Na-samples (Table 3).
Unlike molecular acids, the zeolites have a distribution of acid sites with different local environment, composition, arrangement, dimensions and geometry that will determinate acid strength for each site. Likewise, the acid amount and acid strength of the catalyst strongly depend on the kinetic diameter and basicity of the probe used in the characterization (Primo and Garcia 2014; Sandoval-Díaz et al. 2015). Ammonia is a basic molecule with a kinetic diameter of 0.16 nm that can diffuse into the cavities of zeolite Y (pore opening) and permitting the determination of the majority of the zeolite Y acid sites. NH3 adsorption temperature was set at 150 °C with the purpose to be adsorbed on the acid sites with enough strength to react this probe molecule at the condition. Thus, ammonia desorption thermograms were recorded from 150 to 600 °C for the determination of acidity of the catalysts (Fig. 6). According to NH3-TPD results, a correlation between the micropore surface area and the total acidity value of H-form of the catalysts determined at 150 °C was found with R2 = 0.9273. Besides, the samples exchanged with La3+ ions showed an increase of about 85 μmolNH3/g in comparison to the non-lanthanum exchanged samples (Table 4). The distribution of acid sites was classified by integrating of NH3-TPD signal between 150–300 °C and 300–600 °C, corresponding to with weak acid (Wa) and strong acid (Sa) sites, respectively. It was found that exchange with La3+ ions decrease the Wa/Sa ratio in samples with no boron used in the synthesis mixture. In the samples with boron this ratio remained almost unchangeable.

NH3-TPD profiles and percentage of sites between 150–300 °C and 300–600 °C of catalysts obtained by in-situ synthesis and commercial catalyst
Cracking of 1,3,5-Triisopropylbenzene (TIPB)
The TIPB is a symmetrical branched hydrocarbon with a kinetic diameter of 0.95 nm and widely used as a probe molecule for the evaluation of the activity of cracking catalysts (Qi et al. 2015). Due to its molecular size, the TIPB is convenient to estimate the contribution of the external surface area of the zeolite in the cracking of bulky molecules. According to the MAT experiments, it was found that the synthesized catalysts have a TIPB weight conversion over 85% (Fig. 7). The distribution of products after the catalytic reaction is presented in Table 5. Usually, in catalytic cracking reactions, the content of coke and gas are increased at a higher density of acidic sites in the catalyst (Fig. S3), since the coke formation reaction is started at these acidic sites (Al-Khattaf 2002). Consequently, the samples exchanged with La3+ exhibited higher gas and coke yields.

Results of the catalytic cracking TIPB of deactivated catalyst at 600 °C, 5 h and 100% steaming. The TIPB conversion and reaction yields (left) and selectivity of liquid products (right)
Discussion
In situ synthesis of zeolites
In previous reports, the use of kaolin as a precursor for the synthesis of zeolites has been demonstrated usually from a single phase of calcined kaolin (Patrylak et al. 2001; Zhang and Xiong 2012). However, the current study choices each of the matrix components based on a preliminary analysis of the structural changes of raw kaolin (Padilla et al. 2020). Thus, the mass ratio of different kaolin phases (K, K-1000, K-1000A and K-1100) used to obtain the matrices M and M-Na, aimed the fact that K-1100 provides mechanical resistance to the particle, while K-1000 is a soluble silica-rich phase which can be added as a source of silicon to reaction mixture or partially extracted with alkali (K-1000A) prior to zeolite synthesis to increase the external area of the matrix. With respect to the calcination of the matrices at 750 °C after the formation of the microsphere, it was determined from 27Al-NMR spectra of raw kaolin calcined at this temperature which indicated a ~ 47% of tetrahedral aluminum species required for zeolite crystallization.
In general, the zeolite crystallization model proposes the formation of a supersaturated solution with silicate and aluminate nuclei of critical size, which are agglomerated to be part of the crystalline structure (Wright 2008). On the contrary, the in-situ synthesis have a concentration gradient between the surface of the kaolin microsphere and the alkaline solution (Zheng et al. 2005), i.e., as the alkaline solution diffuses and reaches the active SiO2 and the active Al2O3 in the matrix, a surface gel is formed which reacts to form dispersed zeolite crystals. In addition, for the zeolitization process of kaolin to occur, a thermal transformation of the raw material is required, for instance, the complete calcination of kaolin ≧ 1000 °C produces the Si-Al spinel rich in active silica (Padilla et al. 2020; Rocha and Klinowski 1990).
In case of the matrix M-Na, the quantity of active silica was reduced due to the alkaline treatment carried out on fraction of kaolin calcined at 1000 °C, and simultaneously, its sodium content was increased (Table 1). The silica leaching provided an increase in M-Na surface area (Fig. 1), that have led to the formation of cavities with a larger diameter and high external surface, and more accessibility of the alkaline solution allowing a proper dispersion of zeolite crystals in both surface and inside of the microspherical particle (Fig. 3). However, in the samples obtained with M-Na, the percentage of NaY zeolite was ~ 19% lower than the samples from M (Table 2). Considering that the concentration gradient of silicon and aluminum for the growth of the particles is not instantaneous, the difference between the matrices during synthesis is the gel formation rate. Thus, the matrix M with a greater quantity of active SiO2 has the ability to form the solid–liquid phase on the surface faster, increasing the number of crystal nuclei, and hence, favoring the synthesis of NaY.
During hydrothermal synthesis, other phases can also crystallize, such as: zeolites A, X, P and SOD, which can be crystallized in reaction media with SiO2/Al2O3 ratio < 5 and, the crystallization control will depend on different parameters such as: alkalinity, synthesis time and temperature, aging and nature of the reagents (Garcia et al. 2018; Lutz 2014). According to Oswald's Law, the zeolite NaY as a metastable structure can transform into a more thermodynamically stable phase, like zeolite NaP (Johnson and Bin Arshad 2016; Oleksiak and Rimer 2014). In the XRD patterns of the synthesized samples (Fig. 2), the zeolite P was identified as a secondary phase, because of pure reagents and organic templates were not used in the procedure (Garcia et al. 2018). Therefore, the comparison with catalysts focuses on the content of zeolite Y, which is the phase required for hydrocarbons cracking.
In the zeolites with large pores such as faujasites, under hydrothermal conditions, the isomorphic substitution of boron during crystallization is unsuccessful. This is because zeolite Y pore system is unable to accommodate a significant amount of smaller B3+ ions in the framework (Han et al. 1994; Occelli and Robson 1989). In addition, the level of boron substitution in the faujasite framework contracts the unit cell, as the Al–O and B–O bonds have a length of 0.175 nm and 0.147 nm, respectively (Gaffney et al. 1989; Mi et al. 2017). The samples Na[B]Y/M-Na and Na[B]Y/M showed a slight decrease in the length of a0 value, despite of the low boron content (Table 2). The structural tetrahedral boron in zeolites on matrices could not be detected by 11B NMR. No evidence was found to indicate that hydrolyzed or oxidized boron species could be as exchange cations in the structure.
Furthermore, it has been shown that adding of B2O3 in the synthesis gel inhibits the nucleation or crystallization of the zeolite, as was observed in sample Na[B]Y-M (Han et al. 1994; Occelli and Robson 1989). In this sample a loss of crystallinity and surface area was evidenced (Table 2). In the synthesis of zeolites, the alkalinity can be defined in terms of the ratio H2O/Na2O. An increasing of alkalinity reduces the induction and nucleation periods, while the crystallization is accelerated (Johnson and Arshad 2014). Thus, the zeolites synthetized on M-Na, with and without boron, shown higher crystallinity values which could be associated with the sodium content in the matrix (Table 1).
As mentioned above, the surface chemistry plays a key role to form a concentrated gel on the surface of the matrix during synthesis. In general, the monomeric species of silica and alumina produce amorphous aggregates which form larger crystals, as has been shown in the TEM images of the matrices M and M–Na (Fig. S1) (Grand et al. 2016). However, with a single phase precursor matrix composed only of metakaolin calcined at 750 (M-750), a highly reactive and uniform source, were obtained the zeolites with a narrow size distribution < 300 nm (Fig. S4), indicating the importance of the nature of the precursor on the zeolite crystallization.
Catalysts characterization
In the catalyst samples differences in mesopores volume associated with the alkaline pretreatment applied to the kaolin calcined at 1000 °C of the matrix M-Na were found. The silicon leaching provides mesoporosity and voids to matrix during the preparation of the microspheres. These findings are in line with the pore volumes (Table 3), where the HY/M-Na and H[B]Y/M-Na catalysts have shown a greater fraction of mesopore surface area and total pore volume.
On the other hand, the rare earth metals (RE) added to FCC catalyst mainly improve the structural stability of the zeolite, as a consequence of formation of bridges-type bonds –RE–O–RE– inside the cavities (Zhang et al. 2019; Sousa-Aguiar et al. 2013). However, it has also been revealed that a moderate content of RE3+ ions increases the acid properties and the activity of the catalyst. Although each La3+ cation is able to replace 3H+, it has been proposed that the increase in acidity is due to a partial hydrolysis of hydrated La3+ ions forming stronger Brønsted acid sites than those removed during exchange (Sousa-Aguiar et al. 2013; Cerqueira et al. 2008). Additionally, the increase in acidity is related to replacement of Na+ ions remaining after exchange with NH4NO3 by La3+ (Table 4), or polarization induced by the RE cations to protonic acid sites, or even by hydrolysis of rare earth cations (Sousa-Aguiar et al. 2013; Cerqueira et al. 2008).
In aluminosilicate zeolites, the strength of the Brønsted acid sites ≡Si–O(H)–Al≡ is closely associated to the electronegativity of the T atom in the second coordination sphere around the bringing OH group (Corma et al. 1987). Thus, the strongest acid sites will be those with 6 silicon atoms in the second coordination sphere and as these silicon atoms are replaced with aluminum, acid strength will decrease. Therefore, despite of the low boron content in the zeolites H[B]Y and La,H[B]Y synthesized on matrices (Table 4), those boron atoms in T positions of the second coordination sphere around the acidic OH group could induce an increase in the strength of these Brønsted acid sites.
Catalytic performance of samples
Activity, selectivity and accessibility are important properties for the FCC catalyst, since its performance in feedstocks conversion to desired molecules will mainly depend on them. The catalytic cracking of hydrocarbons occurs at the acidic sites of the catalyst through a chain reaction, involving the formation of carbenium or carbonium ions by hydrogen donation from Brønsted acid site or hydride abstraction from Lewis acid site (Primo and Garcia 2014; Vogt and Weckhuysen 2015; Kotrel et al. 2000).
The main component of the gas obtained from the cracking of TIPB was propylene (C3=), that is formed by the three sequential events starting with the TIPB adsorption on the Brønsted acid site that leads to the formation of the planar carbenium ion ([TIPB]+) on the catalyst surface, then, followed a bimolecular reaction that leads to the abstraction of an hydride and breaks the C–C bond between the benzene ring and the isopropyl substituent through β scission (Sanchez et al. 2019; Qi et al. 2011). However, other secondary gas products such as propane (C3), iso-butane (iC4) and iso-pentane (iC5) were obtained (Table 5). The hydrogen transfer reactions are usually measured in terms of the paraffin/olefin ratio, since the transferred hydrogen saturates an olefin to produce a paraffin (Al-Khattaf 2002; Sedran 1994). In this study, the C3/C3= ratio was calculated and used as an indication of the ability of the catalysts to promote the hydrogen transfer reactions. As it can be seen in Table 5 the presence of La3+ ions in the catalysts has favored this type of reactions. The samples La,H[B]Y/M-Na and La,HY/M have shown ratios C3/C3= practically equal to 1 indicating that one of two propylene molecules formed from the TIPB cracking is saturated to obtain propane. Likewise, larger yields of isoparaffins such as iC4 and iC5 that are originated from secondary reactions of polymerization and alkylation were produced in the catalysts exchanged with lanthanum, showing the effect of having an increase in the total acidity of the catalysts treated with lanthanum (Cuquerella 2010).
In case of the liquid product, it was found that more than 50% of the reaction products were recovered in the liquid. Three main reaction products corresponding to the loss of the isopropyl substituent, such as 1,3-diisopropylbenzene (1,3-DIPB), isopropylbenzene (IPB) and benzene were identified, and other aromatic compounds with lower yields such as ethylbenzene (C8H10) and 1,2-dimethyl-3-ethylbenzene (C10H14) were also analyzed. Following the scheme of successive dealkylation reactions of TIPB for the production of propylene molecules, 1,3-DIPB is formed after the rupture of one σ bond (Sanchez et al. 2019; Al-Khattaf and Lasa 2002; Falco et al. 2006); however, the 1,4-DIPB isomer was also identified in the PIANO analysis as a secondary product of isomerization. According to data presented in Table 5 1,3-DIPB yields are approximately three times higher than 1,4-DIPB, indicating that approximately a 25% of the 1,3-DIPB was isomerized. It is reported that this linear isomer has a better diffusion rate through the cavities of the catalyst, increasing the probability of being cracked to obtain IPB (Bazyari et al. 2009).
The H[B]Y/M sample showed the lowest conversion, which can be related to its lower external surface area (Table 2), however, this catalyst revealed a better selectivity towards 1,3-DIPB (Fig. 7). Considering that the kinetic diameter of 1,3-DIPB is 0.84 nm (Al-Khattaf and Lasa 2002) and that in this catalyst sample micropores > 0.8 nm were generated (Fig. 5), very likely due to the structural defects in the zeolite associated with its low crystallinity and the boron presence (Table 1), shorter contact times of products in the catalyst are expected, leading to a decrease in the over-cracking of the 1,3-DIPB molecule (Fig. 7). In contrast, it was found that benzene selectivity increases > 3% after lanthanum exchange of the catalyst, probably related to the higher number of acid sites per unit surface area in these catalysts. Besides, the samples La,HY/M and CAT-COM showed the highest benzene selectivity > 5%, which is associated with the presence of stronger acid sites (Fig. 6).
Despite of the fact that dealkylation reactions are dominant in the catalytic cracking process, there are some secondary reactions related to isomerization, disproportionation, condensation and hydrogen transfer that affect the distribution of the products (Tsai et al. 1999; Corma and Wojciechowski 1982; Thakur et al. 2016). In that sense, coke formation is the result of several properties of the catalyst, mainly the density of the acidic sites in the catalyst (Fig. S3). The RE-exchanged catalysts lead to a concomitant increase in the number of acid sites, so this also favors the increase in hydrogen transfer reactions and coke formation. Taking into account that catalytic cracking involves carbenium ions as intermediates, these cations play an important role in the formation of catalytic coke by bimolecular reactions, considering a high Si/Al ratio produces strong acid sites, and at these sites coking is accelerated (Cumming and Wojciechowski 1996). It should be noted that zeolite catalysts with medium pores (e.g. ZSM-5) have low tendency to coke formation, since in their small cavities with geometric restriction limits the formation of polynuclear aromatics. While larger pore zeolites, such as faujasite, are more prone to coking (Stern et al. 2008). Bimolecular processes between ions require at least two neighboring adsorption sites. Thus, the rate of coke formation would be linked to the density of acid sites in the catalyst.
The presence of paired aluminum atoms, typically of the class 3-NNN and 4-NNN, are related to weak acid sites which strongly adsorb carbenium ions, extending the residence time on the surface of catalysts and giving rise to coke formation via oligomerization, elimination and disproportionation reactions (Al-Khattaf 2002; Pine et al. 1984). Thus, a higher density of acid sites in the catalysts should increase coke yields during reaction. As it can be seen in Tables 4 and 5, catalysts exchanged with lanthanum have shown the higher total acidity, therefore leading to more active catalyst with the concomitant increase of coke yields. In FCC processing of gasoils, RE-exchanged catalysts not only show an increase in Brønsted acid sites, but also have an effect on catalyst activity and selectivity. High density of acid sites increases activity but leads to low coke selectivity, and hydrogen transfer reactions reduce olefin concentration (Sousa-Aguiar 2016; Akah 2017).
Based on the above discussion, it can be considered that the set of modifications made to the final catalyst, such as the composition of the matrix, addition of boron and exchange with lanthanum ions, confer properties that modify the distribution of reaction products. In spite of the kinetic diameter of the reactant TIPB molecule is larger than the majority of the zeolite pores, the mesopore system of the catalysts and the good dispersion of the zeolite on the spherical matrix particle which contribute to a larger external surface available for the reaction have facilitated the diffusion of reactants and products during the catalytic reaction.
Conclusions
The highest mesoporosity generated by the sodium hydroxide treatment of the matrix conducted in this study introduces a greater crystallinity and good dispersion of the in situ synthetized zeolite on the surface of the matrix microspheres. Likewise, despite of that is reported that boron addition to the synthesis gel inhibits the nucleation of the zeolite, it was found that the matrix M-Na previously treated with sodium hydroxide avoids this effect and, hence, allowing the growth of both zeolites NaY and Na[B]Y with relative high crystallinity. Furthermore, the higher proportion of mesoporous between 4 and 10 nm generated in the matrices favors the diffusion of the reaction products and therefore diminishing the over-cracking of main products.
On the other hand, as shown by the ammonia TPD analysis the amount and strength of acid sites was considerably improved with the ion exchange of the samples with La3+ after hydrothermal deactivation while textural properties are not modified. This increase in quantity and strength of the acid sites of the catalysts has improved their catalytic performance and therefore showing higher TIPB conversions during the catalytic test with the La treated samples, as well as, an increased selectivity in the cracking reaction towards isopropylbenzene and benzene products that are a consequence of the presence and higher concentration of stronger acid sites in the sodium hydroxide treated catalysts. In addition, the presence of lanthanum promotes the hydrogen transfer reactions as measured by the increase in propane yields.
References
Akah A (2017) Application of rare earths in fluid catalytic cracking: A review. J Rare Earths 35–10:941–956. https://doi.org/10.1016/S1002-0721(17)60998-0
Al-Khattaf S (2002) Appl Catal A Gen 231:293–306
Al-Khattaf S, De Lasa H (2002) Appl Catal A Gen 226:139–153
Baerlocher C, McCusker LB, Olson D (2007) Atlas of zeolite framework types, 6th edn. Elsevier, Oxford
Bazyari A, Khodadadi AA, Hosseinpour N, Mortazavi Y (2009) Fuel Process Technol 90:1226–1233
Cerqueira HS, Caeiro G, Costa L, Ribeiro FR (2008) J Mol Catal A Chem 292:1–13
Chen W, Han D, Sun X, Li C (2013) Fuel 106:498–504
Clough M, Pope JC, Tan L, Lin X, Komvokis V, Pan SS, Yilmaz B (2017) Microporous Mesoporous Mater 254:45–58
Corma A, Wojciechowski BW (1982) Catal Rev 24:1–65
Corma A, Fornés V, Melo FV, Herrero J (1987) Zeolites 7:559–563
Cumming KA, Wojciechowski BW (1996) Hydrogen transfer, coke formation, and catalyst decay and their role in the chain mechanism of catalytic cracking. Catal Rev 38(1):101–157. https://doi.org/10.1080/01614949608006455
Cuquerella JM (2010) Craqueo térmico y catalítico, con y sin vapor de aga, de alcanos sobre zeolitas. Cinética, desactivación y estabilización del catalizador. Universitat De València
Falco M, Morgado E, Amadeo N, Sedran U (2006) Appl Catal A Gen 315:29–34
Gaffney TR, Pierantozzi R, Seger MR (1989) Zeolite synthesis (ACS Symp. Ser.). American Chemical Society, Washington, DC, pp 374–392
Gamero PM, Maldonado CM, Moreno JC, Guzman OM, Mojica EM, Gonzalez RS (1997) Stability of an FCC catalyst matrix for processing gas oil with resid. In: Bartholomew CH, Fuentes GA Studies in surface science and catalysis. https://doi.org/10.1016/S0167-2991(97)80177-2
Garcia G, Cabrera S, Hedlund J, Mouzon J (2018) J Cryst Growth 489:36–41
Grand J, Awala H, Mintova S (2016) Cryst Eng Comm 18:650–664
Han S, Schmitt KD, Schramm SE, Reischman PT, Shihabi DS, Chang CD (1994) J Phys Chem 98:4118–4124
Johnson EBG, Arshad SE (2014) Appl Clay Sci 97–98:215–221
Johnson EBG, Arshad SEB (2016) Int J Eng Technol 13:33–39
Karami D, Rohani S (2009) Ind Eng Chem Res 48:4837–4843
Koller H, Chen CY, Zones SI (2015) Top Catal 58:451–479
Kotrel S, Knözinger H, Gates BC (2000) Microporous Mesoporous Mater 35–36:11–20
Lutz W (2014) Adv Mater Sci Eng 2014:1–20
Mi S, Wei T, Sun J, Liu P, Li X, Zheng Q, Gong K, Liu X, Gao X, Wang B, Zhao H, Liu H, Shen B (2017) J Catal 347:116–126
Occelli ML, Robson HE (1989) Zeolite synthesis (ACS Symposium Series). American Chemical Society, Washington, DC
Oleksiak MD, Rimer JD (2014) Rev Chem Eng 30:1–49
Padilla J, Guzman A, Molina D, Poveda-jaramillo JC (2020) Clay Miner 55:293–302
Padilla J, Ramirez I, Guzman A, Poveda-Jaramillo J (2021) Braz J Chem Eng 38:945–956
Pan SS, Lin LTX, Komvokis V, Spann A, Clough M, Yilmaz B (2015). In: Louise J, Bashir L (eds) ACS symp. ser. American Chemical Society, Washington, DC, pp 3–18
Pan M, Zheng J, Liu Y, Ning W, Tian H, Li R (2019) J Catal 369:72–85
Patrylak L, Likhnyovskyi R, Vypyraylenko V, Leboda R, Skubiszewska-zi J (2001) Adsorpt Sci Technol 19:525–540
Pine LA, Maher PJ, Wachter WA (1984) J Catal 85:466–476
Primo A, Garcia H (2014) Chem Soc Rev 43:7548–7561
Qi J, Zhao T, Xu X, Li F, Sun G (2011) J Porous Mater 18:69–81
Qi J, Jin Q, Zhao K, Zhao T (2015) J Porous Mater 22:1021–1032
Qiang L, Ying Z, Zhijun C, Wei G, Lishan C (2010) Influence of synthesis parameters on the crystallinity and Si/Al ratio of NaY zeolite synthesized from kaolin. Pet Sci 7:403–409. https://doi.org/10.1007/s12182-010-0085-x
Rocha J, Klinowski J (1990) Angew Chemie Int Ed English 29:553–554
Sanchez A, Ramirez S, Silva W, Espinal JF (2019) Mol Catal 466:13–18
Sandoval-Díaz LE, González-Amaya JA, Trujillo CA (2015) Microporous Mesoporous Mater 215:229–243
Schwieger W, Machoke AG, Weissenberger T, Inayat A, Selvam T, Klumpp M, Inayat A (2016) Chem Soc Rev 45:3353–3376
Sedran UA (1994) Catal Rev 36:405–431
Silaghi MC, Chizallet C, Raybaud P (2014) Microporous Mesoporous Mater 191:82–96
Silaghi MC, Chizallet C, Sauer J, Raybaud P (2016) J Catal 339:242–255
Sousa-Aguiar EF, Trigueiro FE, Zotin FMZ (2013) Catal Today 218–219:115–122
Sousa-Aguiar EF (2016). Y zeolites as a major component of FCC catalysts: main challenges in the modification thereof. In: Sels BF, Kustov LM (eds) kustovzeolites and zeolite-Like materials. https://doi.org/10.1016/B978-0-444-63506-8.00007-0.
Sperinck S, Raiteri P, Marks N, Wright K (2011) J Mater Chem 21:2118–2125
Stern DL, Brown SH, Beck JS (2008) Isomerization and transalkylation of alkylaromatics. In: Ertl G, Knözinger H, Schüth F, Weitkamp J (eds) Handbook of Heterogeneous Catalysis. https://doi.org/10.1002/9783527610044.hetcat0160
Thakur R, Barman S, Gupta RK (2016) Braz J Chem Eng 33:957–967
Tsai TC, Liu SB, Wang I (1999) Appl Catal A Gen 181:355–398
Vermeiren W, Gilson J-P (2009) Top Catal 52:1131–1161
Vogt ETC, Weckhuysen BM (2015) Chem Soc Rev 44:7342–7370
Wei Y, Parmentier TE, De Jong KP, Zec J (2015) Chem Soc Rev 44:7234–7261
White CE, Provis JL, Proffen T, Riley DP, van Deventer JSJ (2010) J Phys Chem A 114:4988–4996
Wright PA (2008) Microporous framework solids. The Royal Society of Chemistry, Cambridge
Yan K, Guo Y, Fang L, Cui L, Cheng F, Li T (2017) Appl Clay Sci 147:90–96
Zhang Y, Xiong C (2012) Catal Sci Technol 2:606–612
Zhang L, Qin Y, Zhang X, Gao X, Song L (2019) Ind Eng Chem Res 58:14016–14025
Zheng S, Sun S, Zhang Z, Gao X, Xu X (2005) Bull Catal Soc India 4:12–17
Zheng S-Q, He L-J, Ren S, Yu H-X, Zhu W (2015) Kem u Ind 64:603–610
Zheng S, He L, Yao H, Ren S, Zhang J (2017) China Pet Process Petrochem Technol 19:19–25
Acknowledgements
The authors would like to acknowledge Professor Jose Antonio Henao from the X-ray diffraction laboratory UIS-PTG for his support with the analysis of XRD profiles. Special thanks to Catalysis laboratory of the Centro de innovación y tecnología—ICP for the willingness and help during the development of this research.
Funding
Open Access funding provided by Colombia Consortium. This work was supported by Colciencias-Ecopetrol-UIS Agreement 437 of 2012/Contract number 403-2013 and Fund 8821 of Vicerrectoria de Investigación of Universidad Industrial de Santander (Colombia), Mining and Energy Program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Padilla, J., Guzman, A. & Poveda-Jaramillo, J.C. Synthesis and catalytic behavior of FCC catalysts obtained from kaolin by the in-situ method. Braz. J. Chem. Eng. 40, 803–816 (2023). https://doi.org/10.1007/s43153-022-00280-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43153-022-00280-0