
Abstract
Verrucosidins, a methylated α-pyrone class of polyketides rarely reported upon, have been implicated in one or more neurological diseases. Despite the significance of verrucosidins as neurotoxins, the absolute configurations of most of the derivatives have not been accurately characterized yet. In this study, three pairs of C-9 epimeric verrucosidin derivatives, including the known compounds penicyrones A and B (1a/1b) and 9-O-methylpenicyrones A and B (2a/2b), the new compounds 9-O-ethylpenicyrones A and B (3a/3b), together with the related known derivative verrucosidin (4), were isolated and identified from the culture extract of Penicillium cyclopium SD-413, which was obtained from the marine sediment collected from the East China sea. Their structures were established based on an in-depth analysis of nuclear magnetic resonances (NMR) and mass spectroscopic data. Determination of the absolute configurations of these compounds was accomplished by Mosher’s method and time-dependent density functional theory (TDDFT) calculations of electronic circular dichroism (ECD) and optical rotation (OR). The configurational assignment of penicyrone A demonstrated that the previously reported C-6 absolute configuration of verrucosidin derivatives needs to be revised from (6S) to (6R). The 9R/9S epimers of compounds 1–3 were found to exhibit growth inhibition against some pathogenic bacteria, indicating that they have potential as lead compounds for the creation of antimicrobial agents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
According to numerous studies (Cao et al. 2020; Hai et al. 2021; Meng et al. 2021; Song et al. 2018; Wang et al. 2018; Zhou et al. 2022), marine-derived fungi have demonstrated to be a significant source of natural products with diverse chemical structures such as alkaloids, phenols, polyketides, terpenoids, and benzenes, which have remarkable potential as drug candidates in clinical applications (Carroll et al. 2022; EI-Kashef et al. 2019). Verrucosodin (4), the first example of a rarely reported class of methylated α-pyrone polyketide derivative, was initially isolated from Penicillium verrucosum var cyclopium in 1983 (Burka et al. 1983) and was found to possess the inhibitory activity for the expression of glucose-regulated protein 78 (GRP78) under strict hypoglycemic conditions and led to selective cell death of glucose-deprived HT-29 human colon cancer cells (Park et al. 2007). The importance of verrucosidin as a neurotoxin sparked a great deal of interest in characterizing its biosynthetic genes by clustered regularly interspaced short palindrome repeats (CRISPR) technology (Valente et al. 2021).
As part of our attempts to discover naturally occurring compounds with distinctive structures and physiologically activities from marine-derived fungi (Chi et al. 2021; Du et al. 2022; Hu et al. 2021; Li et al. 2016, 2020; Yan et al. 2022), we conducted chemical investigations on the culture extract of Penicillium cyclopium SD-413, a fungus isolated from marine sediment that was collected from the East China Sea. As a result, three pairs of verrucosidin epimers, including the known compounds penicyrones A and B (1a/1b) and 9-O-methylpenicyrones A and B (2a/2b), the new compounds 9-O-ethylpenicyrones A and B (3a/3b), together with the related known derivatives verrucosidin (4) (Fig. 1A), were isolated and identified from the fungus P. cyclopium SD-413. The analysis of NMR data and ECD spectra were primarily used to clarify the planar structures and configurations of isolated compounds. Based on the configurational determination of penicyrone A (1a) by a combination of Mosher’s method and TDDFT calculations of ECD and OR, the absolute configurations at C-6 in the previously reported verrucosidin derivatives need to be revised (Bu et al. 2016). Antimicrobial properties against human- and aquatic-pathogenic bacteria, and plant-pathogenic fungi, were assessed for each isolated compound. In this work, the obtained compounds are described together with their isolation, structural identification, chiral HPLC resolution, stereochemical assignment, and biological activities.

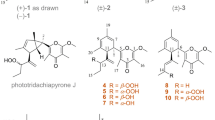
A Chemical structures of compounds 1–4. B Key NOE correlations of compounds 1–3 (solid lines: β-orientation; dashed lines: α-orientation). C ΔδS–R values for Mosher esters (S)-1a/(R)-1a and (S)-1b/(R)-1b
Results and discussion
The fermentation culture of P. cyclopium SD-413 was extensively extracted with EtOAc to produce an extract, which was then purified using column chromatography over Si gel, Lobar LiChroprep RP-18, Sephadex LH-20, and semi-preparative HPLC to yield compounds 1–4. However, two sets of signals in the 13C NMR spectra of compounds 1–3 for chemical shifts of C-7 (Δδ 0.5, 0.6, 0.6, respectively) through C-10 (Δδ 0.2, 0.5, 1.3, respectively), C-11 (Δδ 0.7, 1.0, 1.0, respectively), C-13 (Δδ 0.1, 0.2, 0.1, respectively), C-19 (Δδ 0.5, 0.4, 0.5, respectively), and C-20 (Δδ 0.4, 0.3, 0.4, respectively) were observed (Table 1), indicating the presence of three pairs of epimers for compounds 1–3. Each of the epimeric mixtures was further separated using chiral HPLC to successfully obtain individual epimeric isomers 1a and 1b, 2a and 2b, and 3a and 3b.
Compound 1 was initially obtained as yellowish oil. Based on the positive HRESIMS data, the identified molecular formula was C24H34O7, indicating eight degrees of unsaturation. The NMR spectral data of compound 1 (Table 1) were found to be almost identical to those of penicyrones A and B, which were a pair of C-9 epimers and separated by repeated HPLC with the eluent CH3OH-H2O (3:2) (Bu et al. 2016). The relative configurations of C-6 and C-9 were determined by NOESY experiments in the literature, and the absolute configurations of penicyrones A (1a) and B (1b) were assigned by comparison of their OR values to those of known related compounds, as well as by biosynthetic considerations. Although the experimental ECD spectra were recorded, no ECD calculations were performed to verify these results. As the hydroxy groups or methyl groups at C-6, C-9 and C-12 are not sufficiently bulky to suppress the free rotations of the related single bonds in compounds 1a and 1b, the correlation of the C-6 and C-9 chirality centers had large uncertainty. Thus the absolute configurations of C-6 and C-9 in penicyrones A (1a) and B (1b) were determined independently in the present work by the Mosher’s method and TDDFT-ECD and -OR calculations.
The relative configuration of the tetrahydrofuran ring of compound 1a was established by the key NOESY enhancements from H3-22 to H-13 and H3-23 and from H-15 to H3-21. Analysis of the NOEs from H-9 to H-7 and H-11 revealed that the geometries of double bonds at C-7 and C-10 were E (Fig. 1B).
The absolute configuration of C-6 of compound 1a was determined by the solution TDDFT-ECD method (Mándi et al. 2019). In a 21 kJ/mol energy window, the Merck Molecular Force Field (MMFF) conformational search was performed on the arbitrarily selected (6S, 9R,12S,13S,14R,15R)-1a, which resulted in 3038 conformer clusters. The α-pyrone chromophore and its contiguous C-6 chirality center govern the ECD spectrum (Suárez-Ortiz et al. 2017), and to analyze fewer conformers, an AM1 level re-optimization was carried out before the DFT reoptimization (Csupor et al. 2020), and reclustering was applied to neglect the rotation of the flexible side-chain and the condensed tetrahydrofuran ring beyond C-9 (Zhou et al. 2014). The resulting 87 conformers were re-optimized at the ωB97X/TZVP (Chai et al. 2008) PCM/MeCN and ωB97X/TZVP PCM/MeOH levels for the subsequent ECD and OR calculations, respectively. The TDDFT-ECD calculations performed at four different levels for conformers over 1% Boltzmann-population gave good mirror-image agreement with the experimental spectrum indicating (6R) absolute configuration, opposite to that reported in the literature (Supplementary Fig. S46).
To further verify the absolute configuration of C-6, OR calculations (Polavarapu et al. 2008; Sun et al. 2016) were also carried out at four different levels of theory for the ωB97X/TZVP PCM/MeOH conformers. The B3LYP/TZVP PCM/MeOH, BH&HLYP/TZVP PCM/MeOH, CAM-B3LYP/TZVP PCM/MeOH and PBE0/TZVP PCM/MeOH calculations gave large negative OR values ranging from 78 to 83 for (6S,9R,12S,13S,14R,15R)-1a [experimental value: [α] 25D + 110 ( c 0.1, MeOH)] (Supplementary Table S1). In contrast to the ECD calculations, the OR calculation was not decisive for the determination of the C-6 chirality center even with the good agreement in hand, since the contribution of the other five chirality centers to the OR was not known. The (6S) absolute configuration of 1a described in the literature was confirmed to be wrong (Bu et al. 2016). It is important to note that Bu et al. applied a low-level conformational analysis to correlate the experimental NOE peaks with the distances of protons and determine the relative configuration of C-6 and C-9. The relative configuration of the C-6 and C-9 centers was determined on the basis of the H-7/H3-18 NOE correlation, which was feasible for both the (6R) and (6S) epimers, and hence it is not decisive.
The absolute configuration at C-9 was established by the modified Mosher’s method (Seco et al. 2012). The Δδ values obtained for the (S)- and (R)-MTPA esters [(R)-1a and (S)-1a, respectively] of 1a allowed determining the absolute configuration of C-9 as (R) (Fig. 1C).
With the consideration of the biogenetic origin and co-isolation of verrucosidin (4), whose absolute configuration was determined by single crystal X-ray diffraction (Burka et al. 1983), the chirality of C-12 in 1a was suggested to be (S), the same as that in compound 4 (Burka et al. 1983). Finally, the absolute configuration of compound 1a was established as (6R, 9R,12S,13S,14R,15R).
The NMR and positive HRESIMS data of compound 1b were almost identical to those of 1a (Table 1), indicating that they share the same planar structure. Key NOESY enhancements in compound 1b from H3-22 to H-13 and H3-23 and from H-15 to H3-21 assigned 1b possessing the same relative configuration of the tetrahydrofuran ring as that in compound 1a. The geometries of the double bonds at C-7 and C-10 were also confirmed to be E by analysis of the NOEs from H-9 to H-7 and H-11, which were also well matched with those of compound 1a (Fig. 1B).
The experimental ECD curve of compound 1b well matched that of compound 1a (Supplementary Fig. S10), suggesting that the absolute configuration of C-6 in compound 1b was also (R). By applying the modified Mosher's method, the absolute configuration of C-9 was determined to be (S) (Seco et al. 2012) (Fig. 1C). Considering the biosynthetic pathway, the absolute configuration of compound 1b was assigned as (6R,9S,12S,13S,14R,15R).
Compound 2 was also obtained as a yellowish oil. According to the positive HRESIMS data, its molecular formula was determined to be C25H36O7 (eight unsaturations). The NMR spectral data of compound 2 (Table 1) were found to be nearly identical to those of methyl penicyrone, which was isolated as a mixture of two epimers being epimeric at C-9 (Pan et al. 2018).
In the present work, methyl penicyrone (2) was purified by chiral HPLC to yield the optically active derivatives compounds 2a and 2b (Supplementary Fig. S30). The NOESY and NMR data of compounds 2a and 2b were well matched to that of compounds 1a and 1b (Fig. 1B and Table 1). The double bonds at C-7 and C-10 were also deduced as E based on NOEs from H-9 to H-7 and H-11 (Fig. 1B). Since compounds 1b, 2a and 2b had similar ECD curves, the absolute configuration of C-6 is expected to be the same.
The absolute configurations of C-9 in compounds 2a and 2b were confirmed by NMR data and chiral HPLC analysis, in which the (9S)-epimer possessed a larger retention time than that of the (9R)-epimer, as also observed for compounds 1a and 1b (Table 1 and Supplementary Fig. S30). Considering the biosynthetic pathway, the absolute configurations of compounds 2a and 2b were elucidated as (6R,9R,12S,13S,14R,15R) and (6R,9S,12S,13S,14R,15R), respectively.
Compound 3 was also obtained as a yellowish oil and was successfully separated by chiral HPLC to yield two epimers 3a and 3b (Supplementary Fig. S47).
Based on the positive HRESIMS data, compound 3a had the molecular formula of C26H38O7 (eight degrees of unsaturation). The 1H, 13C, and HSQC NMR data of compound 3a (Table 2) displayed resonances of 10 methyls (including one methoxy), one oxygenated methylene, five methines (including two olefinic and three oxygenated), and 10 nonprotonated (including one carbonyl, six olefinic and three sp3 oxygenated) carbons, and one exchangeable proton (6-OH). Compound 3a was found to be a derivative of verrucosidin that was comparable to compound 1 by thorough investigation of the 1H and 13C NMR spectroscopic data (Bu et al. 2016). However, the -OH group at C-9 in compound 1 was changed to an -OCH2CH3 group in compound 3a, and COSY and HMBC experiments which were shown in Supplementary Fig. S48 corroborated this deduction. Thus, it was established that compound 3a had the planar structure as depicted in Fig. 1A.
Based on the positive HRESIMS data, the molecular formula of compound 3b was confirmed as C26H38O7 (8 degrees of unsaturation), same as that of 3a. The NMR spectra for 3a and 3b were almost identical, which showed they share the same planar structure.
The NOESY and NMR data of compounds 3a and 3b matched well with that of compound 1a (Fig. 1B). The ECD behaviors of compounds 3a and 3b were similar to those of compounds 1a and 1b (Supplementary Fig. S10 and S46), indicating that their absolute configurations at C-6 were both (R), the same as that of compound 1a. Considering the NMR data, retention time in chiral HPLC chromatograms, and the biosynthetic pathway, the absolute configurations of compounds 3a and 3b were tentatively determined to be (6R,9R,12S,13S,14R,15R) and (6R,9S,12S,13S,14R,15R), respectively, and the geometry of C-7 and C-10 in these two compounds were also confirmed to be E based on NOEs as shown in Fig. 1B.
All of the isolated pure epimers were evaluated for antibiotic efficacy against one human pathogenic bacterium (Escherichia coli EMBLC-1), 10 aquatic pathogens (Aeromonas hydrophila QDIO-1, Edwardsiella ictaluri QDIO-9, E. tarda QDIO-2, Micrococcus luteus QDIO-3, Pseudomonas aeruginosa QDIO-4, Vibrio alginolyticus QDIO-5, V. anguillarum QDIO-6, V. harveyi QDIO-7, V. parahemolyticus QDIO-8, and V. vulnificus QDIO-10), and seven plant-pathogenic fungi (Bipolaris sorokiniana QDAU-3, Ceratobasidium cornigerum QDAU-6, Colletotrichum glecosporioides QDAU-2, Fusarium graminerum QDAU-4, F. oxysporum QDAU-5, Penicillium digitatum QDAU-14 and Physalospora piricola Nosa QDAU-15). As shown in Table 3, the epimer 2a showed activity against human pathogenic bacterium E. coli and aquatic pathogens P. aeruginosa and Ed. ictaluri, with MIC values of 4, 8, and 8 μg/mL, respectively, while 2b showed moderate inhibitory activities against E. coli with an MIC value of 8 μg/mL. Epimers 3a and 3b showed activity against A. hydrophilia, each with an MIC value of 8 μg/mL.
Conclusion
In summary, three pairs of verrucosidin derivatives (1–3) representing an uncommonly described class of methylated α-pyrone polyketides, together with one related known derivative verrucosidin (4), were isolated from the culture extract of Penicillium cyclopium SD-413 obtained from the marine sediment. The optically pure epimers for each mixed epimers were successfully separated by chiral HPLC resolution, allowing the absolute configuration to be determined by Mosher’s method and TDDFT-calculations and comparisons. The epimer 2a showed to be more effective than epimer 2b in inhibiting one human- and two aquatic-pathogens, with MIC values ranging from 4 to 8 g/mL. This information may be helpful in the development of new antibacterial agents.
Material and methods
General experimental procedures
The instruments and chemical materials employed in this investigation are the same as those used in our earlier work (Du et al. 2022; Hu et al. 2021).
Fungal material
The fungus Penicillium cyclopium SD-413 was obtained from a marine sediment sample, which was collected from the East China Sea in May 2017. It was identified by DNA amplification and the sequencing of the ITS region utilizing a molecular biological procedure described in our prior research (Wang et al. 2006). GenBank has received the sequenced data of the fungal strain (accession no. MN818582.1). The sequence was most similar (99%) to the sequence of Penicillium cyclopium (accession no. MT990551.1), according to the results of a BLAST search.
Fermentation
The fresh mycelia of P. cyclopium were grown on PDA medium at 28 °C for five days and then were inoculated into 1 L conical flasks (100 flasks) with solid rice medium at room temperature for 30 days. Each flask contained 70 g of rice, 0.1 g of corn flour, 0.3 g of peptone, 0.1 g of sodium glutamate, and 100 mL naturally sourced and filtered seawater, which was obtained from the Huiquan Gulf of the Yellow Sea near the campus of IOCAS.
Extraction and isolation
The fermented cultures (98 flasks) were extracted four times with EtOAc, and the solvents were then evaporated under reduced pressure to afford an extract (71.2 g), which was fractionated using vacuum liquid chromatography (VLC) on silica gel while eluting with various solvents of increasing polarity from petroleum ether (PE) to MeOH to obtain nine fractions (Frs. 1–9). Fr. 5 (6.65 g), eluted with PE-EtOAc (2:1), was further purified by column chromatography (CC) over Lobar LiChroprep RP-18 with a MeOH-H2O gradient (from 10:90 to 100:0) to afford 10 subfractions (Fr. 5.1–5.10). Fr. 5.4 (610 mg) was further purified by CC on silica gel eluting with a CH2Cl2-MeOH gradient (from 100:1 to 20:1) and then by CC on Sephadex LH-20 (MeOH) to yield compounds 1 (14.6 mg) and 2 (32.1 mg). The optical resolution of compound 1 by chiral HPLC (CHIRALPAK IG column, n-hexane–isopropanol 75:25, 1.0 mL/min) afforded optical pure epimers 1a (3.1 mg) and 1b (5.9 mg); whereas, the optical resolution of compound 2 by chiral HPLC (CHIRALPAK IG column, n-hexane–isopropanol 85:15, 1.0 mL/min) afforded optical pure epimers 2a (5.6 mg) and 2b (3.1 mg). Purification of Fr. 5.6 (596 mg) by CC on silica gel eluting with a CH2Cl2-MeOH gradient (from 150:1 to 100:1) and by preparative TLC (pTLC) (plate: 20 cm × 20 cm, developing solvents: PE-acetone 5:1) and then by CC on Sephadex LH-20 (MeOH) yielded the mixture of compound 3 (50.4 mg), which was further separated by chiral HPLC (CHIRALPAK IG column, n-hexane–isopropanol 80:20, 1.0 mL/min) to afford optical pure isomers 3a (7.2 mg) and 3b (6.9 mg).
Penicyrone A (1a)
Yellowish oil; [α] 25D + 110 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 300 (3.96), 203 (4.42) nm; ECD (1.87 × 10–4 M, MeCN) λmax (Δε) 297 (+ 9.07), 212 (–27.07); ECD (3.94 × 10–4 M, MeOH) λmax (Δε) 298 (+ 12.78), 212 (–42.22); 1H and 13C NMR data, Table 1; HRESIMS m/z 435.2379 [M + H]+ (calcd for C24H35O7, 435.2377).
Penicyrone B (1b)
Yellowish oil; [α] 25D + 100 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 300 (3.80), 203 (4.24) nm; ECD (2.73 × 10–4 M, MeCN) λmax (Δε) 299 (+ 7.65), 211 (–24.34); ECD (3.82 × 10–4 M, MeOH) λmax (Δε) 300 (+ 11.77), 213 (–40.24); 1H and 13C NMR data, Table 1; HRESIMS m/z 435.2368 [M + H]+ (calcd for C24H35O7, 435.2377).
(9R)-O-Methylpenicyrone (2a)
Yellowish oil; [α] 25D + 104 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 300 (3.99), 203 (4.43) nm; ECD (2.51 × 10–4 M, MeCN) λmax (Δε) 297 (+ 10.89), 213 (–32.57); ECD (3.79 × 10–4 M, MeOH) λmax (Δε) 298 (+ 11.90), 214 (–37.50); 1H and 13C NMR data, Table 1; HRESIMS m/z 449.2539 [M + H]+ (calcd for C25H37O7, 449.2534).
(9S)-O-Methylpenicyrone (2b)
Yellowish oil; [α] 25D + 92 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 300 (4.01), 203 (4.46) nm; ECD (2.51 × 10–4 M, MeCN) λmax (Δε) 298 (+ 10.07), 213 (–29.62); ECD (3.63 × 10–4 M, MeOH) λmax (Δε) 299 (+ 10.44), 214 (–32.11); 1H and 13C NMR data, Table 1; HRESIMS m/z 449.2539 [M + H]+ (calcd for C25H37O7, 435.2534).
(9R)-O-Ethylpenicyrone (3a)
Yellowish oil; [α] 25D + 90 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 300 (3.79), 203 (4.24) nm; ECD (3.22 × 10–4 M, MeOH) λmax (Δε) 299 (+ 10.50), 215 (–32.26); 1H and 13C NMR data, Table 1; HRESIMS m/z 463.2688 [M + H]+ (calcd for C26H39O7, 463.2690).
(9S)-O-Ethylpenicyrone (3b)
Yellowish oil; [α] 25D + 75 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 300 (4.00), 203 (4.44), nm; ECD (3.22 × 10–4 M, MeOH) λmax (Δε) 295 (+ 10.92), 214 (–33.04); 1H and 13C NMR data, Table 1; HRESIMS m/z 463.2677 [M + H]+ (calcd for C26H39O7, 463.2690).
Preparation of MTPA esters of 1a and 1b
Preparation of the (R)- and (S)-MTPA esters of 1a and 1b. To the stirred solutions of 1a (1.1 mg) and 1b (1.2 mg) in pyridine (400 μL), respectively, each were given an addition of 2.0 mg of 4-(dimethylamino) pyridine and 10 μL of (S)-( +)-α-methoxy-α-(trifluoromethyl) phenylacetyl (MTPA). The mixtures were stirred at 25 ℃ for 12 h before adding 0.2 mL of H2O to cease the reactions. The reaction mixtures were subjected to pTLC with CH2Cl2-MeOH (20:1) as developing solvents to obtain the (R)-Mosher ester R-1a (0.8 mg) and R-1b (0.9 mg). Treatment of 1a (1.0 mg) and 1b (1.3 mg) with (R)-MTPA (10 μL) with the same procedure was used to obtain the corresponding (S)-Mosher ester S-1a (0.8 mg) and S-1b (1.0 mg) (see Supplementary data for NMR data) (Suárez-Ortiz et al. 2017).
Antimicrobial assay
The microplate assay with three repetitions was used to evaluate the effectiveness of the antibiotics against human pathogenic bacteria, aquatic pathogens, and plant-pathogenic fungi (Pierce et al. 2008). The Institute of Oceanology, Chinese Academy of Sciences (IOCAS) provided the pathogenic bacterial and aquatic pathogenic strains, and Qingdao Agricultural University provided the plant pathogenic fungal strains. Positive controls against bacteria and fungi were chloramphenicol and amphotericin B, respectively.
Computational section
Mixed torsional/low-mode conformational searches were carried out with the Macromodel 10.8.011 software using the MMFF with an implicit solvent model for CHCl3 applying a 21 kJ/mol energy window (MacroModel. 2015). Geometry reoptimizations of the resultant conformers (AM1, and ωB97X/TZVP with PCM solvent model for MeCN and MeOH), TDDFT-ECD and OR calculations were performed with Gaussian 09 software package (Frisch et al. 2013). Distance based reclustering was performed with a 0.5 Å cut off for the non-hydrogen atoms plus the 6-OH hydrogen taking into account the left part of the molecule until C-9. For the ECD and OR calculations the B3LYP/TZVP, BH&HLYP/TZVP, CAM-B3LYP/TZVP, and PBE0/TZVP levels were applied with the same solvent model as in the preceding DFT optimization level. ECD spectra were generated as the sum of Gaussians with 3000 cm−1 half-height widths, using dipole-velocity-computed rotational strength values (Stephens et al. 2010). Boltzmann distributions were estimated from the ωB97X energies. The MOLEKEL program was used for visualization of the results (Varetto et al. 2009).
Data availability
The data that supports the findings of this study are included in this published article (and its supplementary information file).
References
Bu YY, Yamazaki H, Takahashi O, Kirikoshi R, Ukai K, Namikoshi M (2016) Penicyrones A and B, an epimeric pair of α-pyrone-type polyketides produced by the marine-derived Penicillium sp. J Antibiot 69:57–61
Burka LT, Ganguli M, Wilson BJ (1983) Verrucosidin, a tremorgen from Penicillium verrucosum var cyclopium. J Chem Soc Chem Commun. https://doi.org/10.1039/c39830000544
Cao J, Wang BG (2020) Chemical diversity and biological function of indolediketopiperazines from marine-derived fungi. Mar Life Sci Technol 2:31–40
Carroll AR, Copp BR, Davis RA, Keyzers RA, Prinsep MR (2022) Marine natural products. Nat Prod Rep 39:1122–1171
Chai JD, Head-Gordon MJ (2008) Systematic optimization of long-range corrected hybrid density functionals. J Chem Phys 128:84106
Chi LP, Li XM, Wan YP, Li YH, Li X, Wang BG (2021) Two new phenol derivatives from the cold seep-derived fungus Aspergillus insuetus SD-512. Chem Biodivers 18:e2100512
Csupor D, Kurtán T, Vollár M, Kúsz N, Kövér KE, Mándi A, Szűcs P, Marschall M, Tahaei SAS, Zupkó I, Hohmann J (2020) Pigments of the moss paraleucobryum longifolium: isolation and structure elucidation of prenyl-substituted 8,8’-linked 9,10-phenanthrenequinone dimers. J Nat Prod 83:268–276
Du FY, Mándi A, Li XM, Meng LH, Kurtán T, Wang BG (2022) Experimental and computational analysis of the solution and solid-state conformations of hexadepsipeptides from Beauveria felina. Chin J Chem 40:378–384
El-Kashef DH, Daletos G, Plenker M, Hartmann R, Mándi A, Kurtán T, Weber H, Lin WH, Ancheeva E, Proksch P (2019) Polyketides and a dihydroquinolone alkaloid from a marine-derived strain of the fungus Metarhizium marquandii. J Nat Prod 82:2460–3246
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich A, Bloino J, Janesko BG, Gomperts R, Hratchian HP, Ortiz JV, Izmaylov AF, et al (2013) Gaussian 09, Revision E. 01; Gaussian: Wallingford, CT, USA
Hai Y, Wei MY, Wang CY, Gu YC, Shao CL (2021) The intriguing chemistry and biology of sulfur-containing natural products from marine microorganisms (1987–2020). Mar Life Sci Technol 3:488–518
Hu XY, Wang CY, Li XM, Yang SQ, Li X, Wang BG, Si SY, Meng LH (2021) Cytochalasin derivatives from the endozoic Curvularia verruculosa CS-129, a fungus isolated from the deep-sea squat lobster Shinkaia crosnieri living in the cold seep environment. J Nat Prod 84:3122–3130
Li XD, Li XM, Li X, Xu GM, Liu Y, Wang BG (2016) Aspewentins D-H, 20-nor-isopimarane derivatives from the deep sea sediment-derived fungus Aspergillus wentii SD-310. J Nat Prod 79:1347–1353
Li YH, Li XM, Yang SQ, Shi XS, Li HL, Wang BG (2020) Antibacterial alkaloids and polyketide derivatives from the deep sea-derived fungus Penicillium cyclopium SD-413. Mar Drugs 18:553
MacroModel software. Schrödinger, LLC (2015) Available online: http://www.schrodinger.com/MacroModel.
Mándi A, Kurtán T (2019) Applications of OR/ECD/VCD to the structure elucidation of natural products. Nat Prod Rep 36:889–918
Meng ZH, Sun TT, Zhao GZ, Yue YF, Chang QH, Zhu HJ, Cao F (2021) Marine-derived fungi as a source of bioactive indole alkaloids with diversified structures. Mar Life Sci Technol 3:44–61
Pan CQ, Shi YT, Auckloo BN, Chen CTA, Chen XG, Wu XD, Wu B (2018) Four verrucosidin derivatives isolated from the hydrothermal vent sulfur-derived fungus Penicillium sp. Y-50-10. Chem Nat Compd 54:253–256
Park HR, Ryoo IJ, Choo SJ, Hwang JH, Kim JY, Cha MR, Shin-Ya K, Yoo ID (2007) Glucose-deprived HT-29 human colon carcinoma cells are sensitive to verrucosidin as a GRP78 down-regulator. Toxicology 229:253–261
Pierce CG, Uppuluri P, Tristan AR, Wormley FL, Mowat E, Ramage G, Lopez-Ribot JL (2008) A simple and reproducible 96-well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat Protoc 3:1494–1500
Polavarapu PL (2008) Why is it important to simultaneously use more than one chiroptical spectroscopic method for determining the structures of chiral molecules? Chirality 20:664–672
Seco JM, Quiñoá E, Riguera R (2012) Assignment of the absolute configuration of polyfunctional compounds by NMR using chiral derivatizing agents. Chem Rev 112:4603–4641
Song YP, Fang ST, Miao FP, Yin XL, Ji NY (2018) Diterpenes and sesquiterpenes from the marine algicolous Fungus Trichoderma harzianum X-5. J Nat Prod 81:2553–2559
Stephens PJ, Harada N (2010) ECD cotton effect approximated by the Gaussian curve and other methods. Chirality 22:229–233
Suárez-Ortiz GA, Cerda-García-Rojas CM, Fragoso-Serrano M, Pereda-Miranda R (2017) Complementarity of DFT calculations, NMR anisotropy, and ECD for the configurational analysis of brevipolides K-O from Hyptis brevipes. J Nat Prod 80:181–189
Sun P, Yu Q, Li J, Riccio R, Lauro G, Bifulco G, Kurtán T, Mándi A, Tang H, Zhuang CL, Gerwick WH, Zhang W (2016) Bissubvilides A and B, cembrane–capnosane heterodimers from the soft coral Sarcophyton subviride. J Nat Prod 79:2552–2558
Valente S, Piombo E, Schroeckh V, Meloni GR, Heinekamp T, Brakhage AA, Spadaro D (2021) CRISPR-Cas9-based discovery of the verrucosidin biosynthesis gene cluster in Penicillium polonicum. Front Microbiol 12:660871
Varetto U, MOLEKEL 5.4 (2009) Swiss National Supercomputing Centre: Manno, Switzerland
Wang S, Li XM, Teuscher F, Li DL, Diesel A, Ebel R, Proksch P, Wang BG (2006) Chaetopyranin, a benzaldehyde derivative, and other related metabolites from Chaetomium globosum, an endophytic fungus derived from the marine red alga Polysiphonia urceolata. J Nat Prod 69:1622–1625
Wang JJ, Chen FM, Liu YH, Liu YX, Li KL, Yang XL, Liu SW, Zhou XF, Yang J (2018) Spirostaphylotrichin X from a marine-derived fungus as an anti-influenza agent targeting RNA polymerase PB2. J Nat Prod 81:2722–2730
Yan LH, Li PH, Li XM, Yang SQ, Liu KC, Wang BG, Li X (2022) Chevalinulins A and B, proangiogenic alkaloids with a spiro[bicyclo[2.2.2]octane-diketopiperazine] skeleton from deep-sea cold-seep-derived fungus Aspergillus chevalieri CS-122. Org Lett 24:2684–2688
Zhou ZF, Kurtán T, Yang XH, Mándi A, Geng MY, Ye BP, Taglialatela-Scafati O, Guo YW (2014) Penibruguieramine A, a novel pyrrolizidine alkaloid from the endophytic fungus Penicillium sp. GD6 associated with Chinese mangrove Bruguiera gymnorrhiza. Org Lett 16:1390–1393
Zhou GL, Zhu TJ, Che Q, Zhang GJ, Li DH (2022) Structural diversity and biological activity of natural p-terphenyls. Mar Life Sci Technol 4:62–73
Acknowledements
Financial support by programs from the National Natural Science Foundation of China (U2006203) and from the Senior User Project of RV KEXUE (KEXUE2020GZ02) is gratefully acknowledged. B.-G.W. appreciates the support of the RV KEXUE of the National Major Science and Technology Infrastructure from the Chinese Academy of Sciences (for sampling). The research work of the Hungarian authors was supported by the National Research, Development, and Innovation Office (K138672, FK134653). The Governmental Information-Technology Development Agency (KIFÜ) is acknowledged for CPU time.
Author information
Authors and Affiliations
Contributions
BW and TK conceived and supervised the work and approved the manuscript; YHL performed the experiments; YHL and AM analyzed the data and prepared the manuscript, XML performed the 1D and 2D NMR measurement; HLL, XL, LHM, SQY, and XSS partially participated in the experiments and were involved the result discussion.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest. Author Bin-Gui Wang is one of the Editorial Board Members, but he was not involved in the journal’s review of, or decision related to, this manuscript.
Animal and human rights statement
This article does not contain any studies with human participants and animal experiments.
Additional information
Edited by Chengchao Chen.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, YH., Mándi, A., Li, HL. et al. Isolation and characterization of three pairs of verrucosidin epimers from the marine sediment-derived fungus Penicillium cyclopium and configuration revision of penicyrone A and related analogues. Mar Life Sci Technol 5, 223–231 (2023). https://doi.org/10.1007/s42995-023-00173-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42995-023-00173-2