
Abstract
Aberrant DNA methylation is a hallmark of acute myeloid leukemia (AML). Various studies showed that t(8;21) AML presented a distinct DNA methylation profile and could be categorized into a separate cluster according to DNA methylation sequencing. Yet, there is still a lack of understanding regarding the causes and mechanisms of this phenomenon. Knowing how the DNA methylation is regulated in t(8;21) AML would enhance our understanding of leukemogenesis and may assist clinical decision-making regarding DNA methylation-targeted therapy. Herein, we summarized our current knowledge concerning DNA methylation regulation in t(8;21) AML and discussed their potential clinical significance in this article.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myeloid leukemia (AML) is a highly heterogeneous disease at cytogenetic and molecular levels (Dou et al., 2019). A primary cause of the heterogeneity is genetic variation (Jan & Majeti, 2013), which is implicated in its initiation and progression (Bayani et al., 2007). Nearly all de novo AML cases carry at least one driver mutation, and more than half of patients are accompanied by chromosome aberrations (Cancer Genome Atlas Research et al., 2013). These recurrent chromosomal variations and mutations are well established as diagnostic and prognostic markers in AML.
The t(8;21)(q22;q22) translocation is the most prevalent chromosome aberration, accounting for 15–20% of non-M3 AML patients (Liu et al., 2019). Although patients with t(8;21) translocation have been shown to have good clinical outcomes (Liu et al., 2019), as many as 30–40% of patients eventually relapsed, resulting in decreased long-term survival (Guo et al., 2020; Martino et al., 2013; Scalea et al., 2019). Especially for patients who are not eligible for standard chemotherapy using cytarabine and anthracyclines. For these patients, hypomethylating agents (HMAs) in combination with low-dose cytarabine were considered the frontline alternative.
Accompanied by the rapid development of sequencing technology, genetic studies in AML have revealed the recurrent somatic mutations implicated in DNA methylation. To be noticed, the recurrent mutations are much more prevalent in t(8;21) AML, even when compared with normal and complex karyotypes (Cancer Genome Atlas Research et al., 2013). According to a large cohort study, epigenetic modification-related genes, including DNMT3A, TET2, IDH1/2, EZH2, ASXL2, and ASXL1 constitute the second most frequent mutation subgroup in t(8;21) AML (Christen et al., 2019). The finding that t(8;21) AML presents with more DNA methylating mutations not only provides insight into mutation target treatment, but also emphasizes the importance of reversing perturbed DNA methylation profile derived from mutant DNA methylation regulators.
t(8;21) AML harboring distinct DNA methylation signature
DNA methylation is typically linked to tissue differentiation and homeostasis under physiological conditions. As it has long been established, AML is characterized by altered DNA methylation distribution (Qu et al., 2015; Zhang et al., 2016). AML cells are prone to global hypomethylation along with localized hypermethylation (Ketkar et al., 2020). Quite different from those of normal hemopoietic cells, altered DNA methylation leads to inappropriate chromatin accessibility and abnormal expression of modified genes, which are involved in the initiation, progression, and maintenance of malignant phenotypes (Ketkar et al., 2020).
Importantly, AML could be recategorized according to different DNA methylation patterns of promoter regions. Using Roche NimbleGen microarray analysis, researchers managed to distinguish t(8;21) AML from other subtypes of leukemia (Figueroa et al., 2010b). Meanwhile, another study has also confirmed the distinct DNA methylation profile of t(8;21) AML by employing DNA methylation microarray 450 K (Li et al., 2017).
Despite the fact that numerous studies have demonstrated the specificity of the DNA methylation signature of t(8;21) AML in both cell lines and bone marrow samples, no consensus has yet been reached regarding its DNA methylation profile. The lack of a platform that integrates diverse data generated by multiple sequencing technologies was a major reason. Furthermore, the small number of t(8;21) AML bone marrow samples collected in each medical center also made it challenging to identify the specific aberrant methylation signature associated with this disease. Thus, the underlying mechanism and clinical significance of aberrant DNA methylation profile attracted much more attention.
As presented here, to better understand the underlying molecular biology of the specific DNA methylation signature of t(8;21) AML, we focused on the biological effect of AML1-eight-twenty-one (AML1-ETO), DNA methylation regulated genes, and their interaction in AML. We also discussed the significance of methylation-guided therapy in AML.
AML1-ETO phenocopy DNA methylation
The DNA methylation is dynamically regulated by DNA methyltransferases (DNMTs), and demethylase TET2 and IDH1/2 (Delhommeau et al., 2009). However, patients with t(8;21), inv(16), t(15;17), and aberrant CEBPA but not mutant DNMTs, TET2, or IDH1/2 are identified as distinct subgroups carrying unique DNA methylation profiles. Furthermore, the DNA methylation profile of SKNO-1, an AML1-ETO positive cell line, changed significantly following inhibition of the expression of AML1-ETO (Cancer Genome Atlas Research et al., 2013; Yang et al., 2020). Taking all of these phenomena into account, it appears that the AML1-ETO plays an important role in the development of an aberrant methylation profile in patients with t(8;21) AML.
AML1-ETO is a fusion protein generated by t(8;21) translocation and plays a critical role in the development and progression of t(8;21) AML. AML1-ETO is well known for being a clinical biomarker that can be used for stratification and relapse prediction (Zhu et al., 2013). Though AML1-ETO plays an indispensable role in maintaining the malignant phenotype of t(8;21) AML, AML1-ETO alone is not sufficient to trigger leukemogenesis (Mandoli et al., 2016). In both the AML1-ETO knock-in mouse model and transgenic mice expressing AML1-ETO, an additional mutagenic event is required to induce AML (Wiemels et al., 2002).
Molecularly, consisting of the N-terminal portion of AML1 and the majority of the ETO protein, the AML1-ETO acquired DNA-binding ability from AML1 and transcriptional repressive ability from ETO, which was mainly mediated by the recruitment of the corepressors NCoR/SMRT, mSin3a, and HDAC (Cancer Genome Atlas Research et al., 2013; Stengel et al., 2020). In addition, various studies indicated that AML1-ETO possesses a transcriptional activating effect through its interaction with other transcriptional activators. Coactivators such as p300 and PRMT1 can be recruited by AML1-ETO to activate target genes, including ID1, CDKN1A, and EGR1 (Cancer Genome Atlas Research et al., 2013; Li et al., 2016b). According to genome-wide analysis, it appears that the relative binding signal of wild-type AML1 and AML1-ETO on target genes played a decisive role in determining whether the gene is repressed or activated by AML1-ETO (Li et al., 2016b). In addition to its transcriptional regulation activity, AML1-ETO also contributes to the formation of the transcription complex, namely, the AML1-ETO-containing transcription factor complex (Mandoli et al., 2016). One of the multiprotein complexes, composed of AML1-ETO, CBFβ, E proteins like HEB, E2A, LYL1, LMO2, and its interacting partner LDB1, has been proved to be essential in the maintenance of leukemia phenotype (Liu et al., 2019).
The multifaceted effect of AML1-ETO on transcription regulation resembles those of DNA methylation on modified genes. Likewise, modification of AML1-ETO on the promoter regions mainly repressed the expression of target genes. A number of classically repressed genes identified in t(8;21) AML include CEBPA, IL-3, IL-6, CDKN2A, LAT2, NF1, and SP1 (Breig et al., 2014; Liu et al., 2012; Lo et al., 2013; Mandoli et al., 2016; Micol et al., 2017). These genes are involved in regulating hematopoiesis, immunity, cell cycle, and Ras signal transduction in leukemia cells. Additionally, various global analyses have confirmed that AML1-ETO is capable of remodeling genome-wide transcriptional profiles. Enrichment of AML1-ETO to ETS motifs decreased the acetylation of histones H3, and H4, and subsequently reduced the expression of numbers of genes (Mandoli et al., 2016).
In summary, accumulation of AML1-ETO exerts a regulation effect similar to that of DNA methylation on the transcriptome in t(8;21) AML, and is indirectly involved in DNA methylating regulation.
Mutations in DNA methylation contribute to the abnormal methylation signature of t(8;21) AML
It is well established that AML cases have a much lower mutation burden than solid tumors (Cancer Genome Atlas Research et al., 2013; Kandoth et al., 2013). Nevertheless, recurrent mutations are considerably common in t(8;21) AML and always confined to a few clusters (Kandoth et al., 2013). The most common mutation cluster consists of a mutations in Ras/RTK-related genes, followed by mutations in epigenetic modification-related genes which account for 45% of t(8;21) AML cases (Christen et al., 2019). Though the cooccurrence of AML1-ETO and mutant DNA methylation-related genes has been confirmed by numerous studies, the underlying mechanism remains unclear.
Mutations in DNA methylation-related genes are identified as an early event in leukemogenesis (Christen et al., 2019). Recurrent mutations of DNA methylation-related genes are frequently observed in myelodysplastic syndromes (MDS) (Delhommeau et al., 2009), which is regarded as a preleukemia disease. Furthermore, these mutations can also be found in healthy and aging individuals without hematopoietic malignancies (Delhommeau et al., 2009; Zebisch et al., 2013). Mutations in DNA methylation-related genes are therefore considered as tumor-initiating events in leukemia (Welch et al., 2012) contribute to leukemia pathogenesis (Li et al., 2016a). Moreover, these mutations are stable from AML diagnosis through relapse (Greif et al., 2018), further emphasizing the importance of DNA methylation regulators in AML.
The DNA methyltransferases (DNMT), consisting of DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L enzymes, are widely regarded as the major enzymes in DNA methylation (McCabe & Caudill, 2005). Currently, only DNMT1, DNMT3A, and DNMT3B have been proved to be essential for the establishment and maintenance of DNA methylation (Ley et al., 2010). DNMT1 is responsible for maintaining CpG methylation, whereas DNMT3A and DNMT3B are capable of de novo methylation. DNMT3A is the most predominant methyltransferase in AML, carrying out nearly all of the de novo methylation reactions (Spencer et al., 2017). Furthermore, altered hypermethylation of CpG islands has been linked to abnormal expression and dysregulation of DNMT1 and DNMT3A (Lund et al., 2014).
Mutations of DNMT3A and DNMT1 are the primary cause of methylation dysfunction in most AML patients. DNMT3A is the third most mutated gene in AML, following FLT3 and NPM1. It occurs in 20–25% of de novo cases (Metzeler et al., 2016). While in patients with t(8;21), DNMT3A is the most common mutant methyltransferase with the highest variant allele frequency (VAF) (Christen et al., 2019). More than 50% of mutant DNMT3A exhibits a substitution of arginine 882 (R882), which is located in the methyltransferase activity domain of DNMT3A and results in defective methylation. In spite of the fact that DNMT3A mutations are usually heterozygous, they affect the homodimeric interfaces that bind to DNA, which further reduces the DNA methylation activity (Spencer et al., 2017; Zhang et al., 2018). In vitro and in vivo studies revealed that mutant DNMT3A abrogates almost 80% of methylation activity (Ketkar et al., 2020; Spencer et al., 2017), which is in line with the observation of global genomic hypomethylation in AML patients (Spencer et al., 2017).
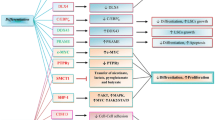
DNA methylation is a highly dynamic biological process that regulated and maintained by DNA methyltransferases and demethylases. TET enzymes, including TET1, TET2, and TET3, play a critical role in the initial step of DNA demethylation (Khwaja et al., 2016). TET2 mutations are more common in AML than TET1 or TET3, and mutant TET2 is associated with a poor overall survival rate (Shih et al., 2015). TET2 mutations are found in 10–20% of all AML patients (Cimmino et al., 2015; Prada-Arismendy et al., 2017) and in 8% of patients with t(8;21)(Christen et al., 2019). Generally, TET2 protein converts 5-methylcytosines (5mC) into 5-hydroxymethylcytosine (5hmC) in an α-Ketoglutarate (αKG)-dependent manner, resulting in DNA demethylation (Fig. 1). Mutations in TET2 are often heterozygotes, including deletions, nonsense mutations, and missense (Shih et al., 2015). Intriguingly, mutations of the TET2 gene are restricted through its coding region without any hotspots. TET2 mutations abrogate the initial demethylation step and cause a hypermethylated DNA phenotype (Inoue et al., 2016). Patients with mutant TET2 display diminished DNA demethylation activity and increased DNA methylation.
IDH1/2 mutations are detected in 20% of all AML patients (Cancer Genome Atlas Research et al., 2013; Contieri et al., 2020; Figueroa et al., 2010a), as well as 3% of t(8;21) cases. Mutant IDH1/IDH2 are commonly found in R132 and R172, respectively (Figueroa et al., 2010a). These proteins do not directly interact with DNA in the same way that DNMT3A or TET2 do. They are involved in DNA methylation modification by catalyzing the conversion of isocitrate to α-KG, which is required for TET2 demethylation. However, mutant IDH1/2 gains extra ability to convert α-KG to 2-hydroxyglutarate (2-HG), a structural analog of α-KG and a well-known oncometabolite. Consequently, the accumulation of 2-HG competes with α-KG for α-KG-dependent enzymes, thereby impairing the DNA demethylation process. Since IDH1/2 mutations are heterozygous, leukemia cells retain one wild-type allele and partially maintain the activity to produce α-KG. Briefly, heterozygous mutations in IDH1/IDH2 catalyze isocitrate to produce α-KG and catalyze the α-KG into 2-HG simultaneously (Fig. 1). Therefore, TET2 function may not be completely abolished by the mutations of IDH1/2, which is in line with the different clinical features of hematopoietic disease caused by mutant IDH1/2 and TET2 (Inoue et al., 2016).
AML patients with mutant IDH1/2 exhibit aberrant DNA hypermethylation, affecting the overall promoter regions (Figueroa et al., 2010a). However, the subsequent effect on the transcriptome is limited compared with mutant DNMT3A. The perturbed methylation profiles in mutant IDH1/2 and TET2 are largely overlapping (Figueroa et al., 2010a). Further establish a similar mechanism between mutant IDH1/2 and TET2 in the regulation of demethylation.
Despite the fact that each of the aforementioned DNA methylation regulators contributes only to a small proportion of patients with t(8;21) AML, approximately 45% of patients carry at least one of these mutant genes. According to a large cohort study, mutant DNMT3A often co-occurs with mutant TET2 or IDH1/2 in t(8;21). While mutations of TET2 and IDH1/2 are mutually exclusive (Patel et al., 2012), indicating that one mutation in this pathway is adequate for AML pathogenesis.
The interaction between AML1-ETO and DNA methylation genes influences the distinctive DNA methylation signature of t(8;21) AML. In particular, both DNMT3A (Zhou et al., 2018) and DNMT1 (Liu et al., 2005) can be recruited by AML1-ETO to diverse promoter regions. For instance, abnormal recruitment of DNMT1 and DMT3A results in hypermethylation silencing of IL-3 (Liu et al., 2005) and BASP1 (Zhou et al., 2018) in t(8;21) AML cells, respectively (Fig. 1). AML1-ETO, on the other hand, was able to transactivate the DNMT3A gene and disrupt DNA methylation profiles by collaborating with HIF1α (Gao et al., 2015). To summarize, AML1-ETO regulates DNA methylation indirectly through its interaction with DNMT1 and DNMT3A. However, the correlation between AML1-ETO and mutant DNA methylation-related genes required further study.
Clinical significance of restoring DNA methylation in t(8;21) AML
The reversibility of DNA methylation and the significance of aberrant methylation in AML make remodeling DNA methylation a promising therapeutic approach (El-Osta, 2004; Gozzini & Santini, 2005; Ketkar et al., 2020).
DNA methyltransferase inhibitors (DNMTi), also known as HMAs, have been widely explored for treating AML. Since first reported in dealing with MDS patients in 1993 (Suarez & Gore, 2013), research on HMAs in AML has been intensified. First-generation HMAs, including 5-azacitidine (AzaC) and 5-aza-2'-deoxycytidine (decitabine, DAC), act as cytosine analogs that irreversibly incorporate into DNA in the S phase, resulting in trapped DNMTs and hypomethylation of DNA. Meanwhile, the trapped DNMTs are degraded by proteasomes leading to passive hypomethylation in subsequent replication cycles (Issa et al., 2004). Resumption of hypomethylation in the promoter is accompanied by the reactivation of gene expression (Suarez & Gore, 2013). For decades, HMAs has been demonstrated to be an effective and tolerable alternative to intensive chemotherapy for patients who are not eligible for such treatment (Ganetsky, 2012; Issa et al., 2004; Martino et al., 2013).
According to in vitro and in vivo studies, HMAs activates tumor suppression genes and exerts various antineoplastic effects on t(8;21) AML cells. The most commonly used HMAs in AML, decitabine, inhibits proliferation, induces differentiation, increases apoptosis, and arrests cell cycle in t(8;21) cells by restoring the expression of EYA4, AKAP12, THAP10, and BASP1 (Flotho et al., 2007; Huang et al., 2016; Li et al., 2017; Zhou et al., 2018). According to a large number of studies, low doses of decitabine reverse aberrant DNA methylation, whereas high doses are always cytotoxic.
Although monotherapy with HMAs has been reported to be effective in relapsed/refractory (R/R) AML patients with a 30% remission rate (Khan et al., 2017), combined therapy offers better clinical outcomes. When combined with chidamide and the CAG regimen (cytarabine, aclarubicin, and granulocyte colony-stimulating factor), the overall response rate in R/R AML was 46.2% (Xu et al., 2020b). While the overall response rate in patients with R/R t(8;21) AML was 59%, significantly higher than that in patients with other karyotypes. It seemed that t(8;21) AML appeared to be more susceptible to the HMAs combined regimen, which may owe to their distinct DNA methylation profiles.
More recently, an increasing number of studies have shown significant improvement in remission rates for de novo patients treated with venetoclax and decitabine or azacytidine. The complete response (CR)/complete response with incomplete recovery of peripheral blood counts (CRi) rates for venetoclax in combination with azacytidine and decitabine were 71% and 74%, respectively. And the median overall survival (OS) time was 16.4 months and 16.2 months, respectively. Consequently, venetoclax, an oral inhibitor of B-cell lymphoma 2 (BCL2), combined with decitabine or azacytidine is considered the first-line therapy for patients with unfavorable risk.
Although improved remission rates and overall survival have been observed in AML and MDS patients treated with an HMAs-based regimen, durable efficacy is lacking due to a short plasma half-life (Contieri et al., 2020). Guadecitabine, formerly known as SGI-110, is a second-generation HMAs that releases decitabine gradually, allowing leukemia cells exposed to its active ingredient for a longer period of time (Ferrara, 2017; Lichtenegger et al., 2017). In phase I/II study, 80% of R/R AML patients benefit from guadecitabine monotherapy (Chung et al., 2019). In phase 3 randomized clinical trials (NCT02348489), guadecitabine monotherapy was found to have similar efficacy to standard chemotherapy in AML. However, it fails to meet the primary endpoint of significant advantages over standard chemotherapy.
It is possible that a certain subgroup of patients will benefit more from HMAs. Since the alterations in the transcriptome failed to predict treatment response (Chung et al., 2019), it is presumed that the clinical response to HMAs may be related to the methylation signature of promoters. However, no specific promoter methylation change was found to be correlated with therapy response to HMAs (Issa et al., 2004). While a retrospective study of intermediate-risk patients confirmed a significant positive relationship between epigenetic modifier gene mutations and clinical response to HMAs (Xu et al., 2020a). Consistent with the phenomenon that genetic mutations of DNA methylation regulators contribute to a remarkable change in DNA methylation profiles (Delhommeau et al., 2009). To further discriminate AML patients who will benefit from HMAs, it is necessary to investigate the global changes in mutation, methylation, and corresponding expression profiles in patients.
Recurrent mutations of IDH1/IDH2 are rational therapeutic targets. Ivosidenib (AG-120) is a small molecule that targets mutant IDH1. It has been approved by the FDA for treating R/R AML patients with mutant IDH1, including t(8;21) AML. Ivosidenib inhibits mutant IDH1 expression in AML patients, and hence reduces plasma 2-HG to a level comparable to that of healthy individuals (Roboz et al., 2020). According to a phase 1 clinical trial conducted in newly diagnosed AML patients that ineligible for standard treatment, a CR/CRi rate of 42.4% has been achieved by ivosidenib (Roboz et al., 2020). When combined with intensive chemotherapy, the CR/CRi rate was 72% (Stein et al., 2021). Meanwhile, a prolonged disease-free remission has been observed in patients whose IDH1 mutations have been clarified (Roboz et al., 2020).
Enasidenib (AG-221), an oral inhibitor targeting mutant IDH2, has been approved for treating R/R AML patients with mutant IDH2. A phase 1/2 clinical trial of enasidenib for R/R AML patients reported a CR/CRi rate of 40.3%. It is important to note that the overall response and survival rates in IDH2-R140 and IDH2-R172 are comparable, indicating that enasidenib has the same inhibitory effect on both mutation types. Enasidenib can reduce the formation of 2-HG in the way similar to ivosidenib. However, there are few patients with reduced 2-HG who failed to reach clinical response. Indicating that 2-HG is ineligible to serve as a predictive marker for enasidenib treatment (Chung et al., 2019). Considering the observation of recurrent mutations of IDH1/2 in t(8;21) AML, as well as their critical biological functions, it seems reasonable to assume that (8;21) AML would benefit from these inhibitors, although further clinical trials are required.
Conclusion
AML1-ETO, DNA methylation regulators, and their interactions contribute to the specific DNA methylation profile of t(8;21) AML. However, there are still 50% of t(8;21) AML patients without any mutant DNA methylation-related genes. In addition, the relationship between wild-type DNMT3A and AML1-ETO is insufficient to explain the distinctive methylation signature in t(8;21) as it could be impaired by mutant DNMT3A. Therefore, clarifying the relationship between AML1-ETO and mutant DNA methylation regulators would be helpful for understanding the underlying mechanism of aberrant DNA methylation.
For decades, methylation sequencing has been mainly conducted using methylation arrays, which generally target promoter regions and do not reveal the full signature of specific methylation profiles. With the application of whole-genome methylation sequencing, it is possible to examine alterations in methylation throughout the genome. It has been reported that DNA methylation in gene bodies is positively correlated with the expression level of corresponding genes, contrary to the regulation effect of DNA methylation in promoters. Meanwhile, DNMT3B has been shown to participate in gene bodies methylation (Yang et al., 2014), though the exact mechanisms remain unclear. Notably, methylation activation in gene bodies may be a valuable therapeutic target for DNA demethylation therapy, which may lead to the downregulation of oncogenes. The opposite pathogenic effect of methylation on promoters and gene bodies necessitates a thorough examination of methylation patterns throughout the genome.
From a clinical perspective, a better understanding of the mechanisms behind abnormal DNA methylation in t(8;21) AML may facilitate the development of new treatment options. It may also provide insight into the progression of t(8;21) AML oncogenesis and identify prognostic indicators associated with epigenetic targeting therapy.

DNA methylation modulation in t(8;21) AML
Data Availability
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
References
Bayani, J., Selvarajah, S., Maire, G., Vukovic, B., Al-Romaih, K., Zielenska, M., & Squire, J. A. (2007). Genomic mechanisms and measurement of structural and numerical instability in cancer cells. Seminars in Cancer Biology, 17, 5–18.
Breig, O., Bras, S., Martinez Soria, N., Osman, D., Heidenreich, O., Haenlin, M., & Waltzer, L. (2014). Pontin is a critical regulator for AML1-ETO-induced leukemia. Leukemia, 28, 1271–1279.
Cancer Genome Atlas Research, N, Ley, T. J., Miller, C., Ding, L., Raphael, B. J., Mungall, A. J., Robertson, A., Hoadley, K., Triche, T. J., Laird, P. W., et al. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England Journal of Medicine, 368, 2059–2074.
Christen, F., Hoyer, K., Yoshida, K., Hou, H. A., Waldhueter, N., Heuser, M., Hills, R. K., Chan, W., Hablesreiter, R., Blau, O., et al. (2019). Genomic landscape and clonal evolution of acute myeloid leukemia with t(8;21): An international study on 331 patients. Blood, 133, 1140–1151.
Chung, W., Kelly, A. D., Kropf, P., Fung, H., Jelinek, J., Su, X. Y., Roboz, G. J., Kantarjian, H. M., Azab, M., & Issa, J. J. (2019). Genomic and epigenomic predictors of response to guadecitabine in relapsed/refractory acute myelogenous leukemia. Clinical Epigenetics, 11, 106.
Cimmino, L., Dawlaty, M. M., Ndiaye-Lobry, D., Yap, Y. S., Bakogianni, S., Yu, Y., Bhattacharyya, S., Shaknovich, R., Geng, H., Lobry, C., et al. (2015). TET1 is a tumor suppressor of hematopoietic malignancy. Nature Immunology, 16, 653–662.
Contieri, B., Duarte, B.K.L., and Lazarini, M. (2020). Updates on DNA methylation modifiers in acute myeloid leukemia. Annals of Hematology
Delhommeau, F., Dupont, S., Valle, V. D., James, C., Trannoy, S., Massé, A., Kosmider, O., Le Couedic, J.-P., Robert, F., Alberdi, A., et al. (2009). Mutation inTET2in Myeloid Cancers. New England Journal of Medicine, 360, 2289–2301.
Dou, L., Yan, F., Pang, J., Zheng, D., Li, D., Gao, L., Wang, L., Xu, Y., Shi, J., Wang, Q., et al. (2019). Protein lysine 43 methylation by EZH1 promotes AML1-ETO transcriptional repression in leukemia. Nature Communications, 10, 5051.
El-Osta, A. (2004). The rise and fall of genomic methylation in cancer. Leukemia, 18, 233–237.
Ferrara, F. (2017). Guadecitabine: A new therapeutic option for acute myeloid leukaemia? The Lancet Oncology, 18, 1287–1288.
Figueroa, M. E., Abdel-Wahab, O., Lu, C., Ward, P. S., Patel, J., Shih, A., Li, Y., Bhagwat, N., Vasanthakumar, A., Fernandez, H. F., et al. (2010a). Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell, 18, 553–567.
Figueroa, M. E., Lugthart, S., Li, Y., Erpelinck-Verschueren, C., Deng, X., Christos, P. J., Schifano, E., Booth, J., van Putten, W., Skrabanek, L., et al. (2010b). DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell, 17, 13–27.
Flotho, C., Paulun, A., Batz, C., & Niemeyer, C. M. (2007). AKAP12, a gene with tumour suppressor properties, is a target of promoter DNA methylation in childhood myeloid malignancies. British Journal of Haematology, 138, 644–650.
Ganetsky, A. (2012). The role of decitabine for the treatment of acute myeloid leukemia. Annals of Pharmacotherapy, 46, 1511–1517.
Gao, X. N., Yan, F., Lin, J., Gao, L., Lu, X. L., Wei, S. C., Shen, N., Pang, J. X., Ning, Q. Y., Komeno, Y., et al. (2015). AML1/ETO cooperates with HIF1α to promote leukemogenesis through DNMT3a transactivation. Leukemia, 29, 1730–1740.
Gozzini, A., & Santini, V. (2005). Butyrates and decitabine cooperate to induce histone acetylation and granulocytic maturation of t(8;21) acute myeloid leukemia blasts. Annals of Hematology, 84(Suppl 1), 54–60.
Greif, P. A., Hartmann, L., Vosberg, S., Stief, S. M., Mattes, R., Hellmann, I., Metzeler, K. H., Herold, T., Bamopoulos, S. A., Kerbs, P., et al. (2018). Evolution of cytogenetically normal acute myeloid leukemia during therapy and relapse: an exome sequencing study of 50 patients. Clinical Cancer Research, 24, 1716–1726.
Guo, C., Li, J., Steinauer, N., Wong, M., Wu, B., Dickson, A., Kalkum, M., & Zhang, J. (2020). Histone deacetylase 3 preferentially binds and collaborates with the transcription factor RUNX1 to repress AML1-ETO-dependent transcription in t(8;21) AML. Journal of Biological Chemistry, 295, 4212–4223.
Huang, S., Jiang, M. M., Chen, G. F., Qian, K., Gao, H. H., Guan, W., Shi, J. L., Liu, A. Q., Liu, J., Wang, B. H., et al. (2016). Epigenetic silencing of eyes absent 4 gene by acute myeloid leukemia 1-eight-twenty-one oncoprotein contributes to leukemogenesis in t(8;21) acute myeloid leukemia. Chinese Medical Journal (engl), 129, 1355–1362.
Inoue, S., Li, W. Y., Tseng, A., Beerman, I., Elia, A. J., Bendall, S. C., Lemonnier, F., Kron, K. J., Cescon, D. W., Hao, Z., et al. (2016). Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell, 30, 337–348.
Issa, J. P., Garcia-Manero, G., Giles, F. J., Mannari, R., Thomas, D., Faderl, S., Bayar, E., Lyons, J., Rosenfeld, C. S., Cortes, J., et al. (2004). Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2’-deoxycytidine (decitabine) in hematopoietic malignancies. Blood, 103, 1635–1640.
Jan, M., & Majeti, R. (2013). Clonal evolution of acute leukemia genomes. Oncogene, 32, 135–140.
Kandoth, C., McLellan, M. D., Vandin, F., Ye, K., Niu, B., Lu, C., Xie, M., Zhang, Q., McMichael, J. F., Wyczalkowski, M. A., et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature, 502, 333–339.
Ketkar, S., Verdoni, A. M., Smith, A. M., Bangert, C. V., Leight, E. R., Chen, D. Y., Brune, M. K., Helton, N. M., Hoock, M., George, D. R., et al. (2020). Remethylation of Dnmt3a (-/-) hematopoietic cells is associated with partial correction of gene dysregulation and reduced myeloid skewing. Proceedings of the National Academy of Sciences United States of America, 117, 3123–3134.
Khan, N., Hantel, A., Knoebel, R. W., Artz, A., Larson, R. A., Godley, L. A., Thirman, M. J., Liu, H., Churpek, J. E., King, D., et al. (2017). Efficacy of single-agent decitabine in relapsed and refractory acute myeloid leukemia. Leukaemia & Lymphoma, 58, 1–7.
Khwaja, A., Bjorkholm, M., Gale, R. E., Levine, R. L., Jordan, C. T., Ehninger, G., Bloomfield, C. D., Estey, E., Burnett, A., Cornelissen, J. J., et al. (2016). Acute myeloid leukaemia. Nature Reviews. Disease Primers, 2, 16010.
Ley, T. J., Ding, L., Walter, M. J., McLellan, M. D., Lamprecht, T., Larson, D. E., Kandoth, C., Payton, J. E., Baty, J., Welch, J., et al. (2010). DNMT3A mutations in acute myeloid leukemia. The New England Journal of Medicine, 363, 2424–2433.
Li, S., Garrett-Bakelman, F. E., Chung, S. S., Sanders, M. A., Hricik, T., Rapaport, F., Patel, J., Dillon, R., Vijay, P., Brown, A. L., et al. (2016a). Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nature Medicine, 22, 792–799.
Li, Y., Ning, Q., Shi, J., Chen, Y., Jiang, M., Gao, L., Huang, W., Jing, Y., Huang, S., Liu, A., et al. (2017). A novel epigenetic AML1-ETO/THAP10/miR-383 mini-circuitry contributes to t(8;21) leukaemogenesis. EMBO Molecular Medicine, 9, 933–949.
Li, Y., Wang, H., Wang, X., Jin, W., Tan, Y., Fang, H., Chen, S., Chen, Z., & Wang, K. (2016b). Genome-wide studies identify a novel interplay between AML1 and AML1/ETO in t(8;21) acute myeloid leukemia. Blood, 127, 233–242.
Lichtenegger, F. S., Krupka, C., Haubner, S., Kohnke, T., & Subklewe, M. (2017). Recent developments in immunotherapy of acute myeloid leukemia. Journal of Hematology & Oncology, 10, 142.
Liu, J., Xing, H., Chen, Y., Wang, L., Wang, D., Rao, Q., Tang, K., Tian, Z., He, K., Wang, M., et al. (2012). PIG7, transactivated by AML1, promotes apoptosis and differentiation of leukemia cells with AML1-ETO fusion gene. Leukemia, 26, 117–126.
Liu, N., Song, J., Xie, Y., Wang, X. L., Rong, B., Man, N., Zhang, M. M., Zhang, Q., Gao, F. F., Du, M. R., et al. (2019). Different roles of E proteins in t(8;21) leukemia: E2–2 compromises the function of AETFC and negatively regulates leukemogenesis. Proc Natl Acad Sci U S A, 116, 890–899.
Liu, S., Shen, T., Huynh, L., Klisovic, M. I., Rush, L. J., Ford, J. L., Yu, J., Becknell, B., Li, Y., Liu, C., et al. (2005). Interplay of RUNX1/MTG8 and DNA methyltransferase 1 in acute myeloid leukemia. Cancer Research, 65, 1277–1284.
Lo, M. C., Peterson, L. F., Yan, M., Cong, X., Hickman, J. H., Dekelver, R. C., Niewerth, D., & Zhang, D. E. (2013). JAK inhibitors suppress t(8;21) fusion protein-induced leukemia. Leukemia, 27, 2272–2279.
Lund, K., Cole, J. J., VanderKraats, N. D., McBryan, T., Pchelintsev, N. A., Clark, W., Copland, M., Edwards, J. R., & Adams, P. D. (2014). DNMT inhibitors reverse a specific signature of aberrant promoter DNA methylation and associated gene silencing in AML. Genome Biology, 15, 406.
Mandoli, A., Singh, A. A., Prange, K. H. M., Tijchon, E., Oerlemans, M., Dirks, R., Ter Huurne, M., Wierenga, A. T. J., Janssen-Megens, E. M., Berentsen, K., et al. (2016). The Hematopoietic Transcription Factors RUNX1 and ERG Prevent AML1-ETO Oncogene Overexpression and Onset of the Apoptosis Program in t(8;21) AMLs. Cell Reports, 17, 2087–2100.
Martino, M., Fedele, R., Moscato, T., & Ronco, F. (2013). Optimizing outcomes following allogeneic hematopoietic progenitor cell transplantation in AML: The role of hypomethylating agents. Current Cancer Drug Targets, 13, 661–669.
McCabe, D. C., & Caudill, M. A. (2005). DNA methylation, genomic silencing, and links to nutrition and cancer. Nutrition Reviews, 63, 183–195.
Metzeler, K. H., Herold, T., Rothenberg-Thurley, M., Amler, S., Sauerland, M. C., Görlich, D., Schneider, S., Konstandin, N. P., Dufour, A., Bräundl, K., et al. (2016). Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood, 128, 686–698.
Micol, J. B., Pastore, A., Inoue, D., Duployez, N., Kim, E., Lee, S. C., Durham, B. H., Chung, Y. R., Cho, H., Zhang, X. J., et al. (2017). ASXL2 is essential for haematopoiesis and acts as a haploinsufficient tumour suppressor in leukemia. Nature Communications, 8, 15429.
Patel, J. P., Gönen, M., Figueroa, M. E., Fernandez, H., Sun, Z., Racevskis, J., Van Vlierberghe, P., Dolgalev, I., Thomas, S., Aminova, O., et al. (2012). Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. The New England Journal of Medicine, 366, 1079–1089.
Prada-Arismendy, J., Arroyave, J. C., & Rothlisberger, S. (2017). Molecular biomarkers in acute myeloid leukemia. Blood Reviews, 31, 63–76.
Qu, X., Davison, J., Du, L., Storer, B., Stirewalt, D. L., Heimfeld, S., Estey, E., Appelbaum, F. R., & Fang, M. (2015). Identification of differentially methylated markers among cytogenetic risk groups of acute myeloid leukemia. Epigenetics, 10, 526–535.
Roboz, G. J., DiNardo, C. D., Stein, E. M., de Botton, S., Mims, A. S., Prince, G. T., Altman, J. K., Arellano, M. L., Donnellan, W., Erba, H. P., et al. (2020). Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood, 135, 463–471.
Scalea, S., Maresca, C., Catalanotto, C., Marino, R., Cogoni, C., Reale, A., Zampieri, M., and Zardo, G. (2019). Modifications of H3K4 methylation levels are associated with DNA hypermethylation in acute myeloid leukemia. The FEBS Journal.
Shih, A. H., Jiang, Y., Meydan, C., Shank, K., Pandey, S., Barreyro, L., Antony-Debre, I., Viale, A., Socci, N., Sun, Y., et al. (2015). Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell, 27, 502–515.
Spencer, D. H., Russler-Germain, D. A., Ketkar, S., Helton, N. M., Lamprecht, T. L., Fulton, R. S., Fronick, C. C., O’Laughlin, M., Heath, S. E., Shinawi, M., et al. (2017). CpG Island hypermethylation mediated by DNMT3A is a consequence of AML progression. Cell, 168, 801-816.e813.
Stein, E. M., DiNardo, C. D., Fathi, A. T., Mims, A. S., Pratz, K. W., Savona, M. R., Stein, A. S., Stone, R. M., Winer, E. S., Seet, C. S., et al. (2021). Ivosidenib or enasidenib combined with intensive chemotherapy in patients with newly diagnosed AML: A phase 1 study. Blood, 137, 1792–1803.
Stengel, K.R., Ellis, J.D., Spielman, C.L., Bomber, M.L., and Hiebert, S.W. (2020). Definition of a small core transcriptional circuit regulated by AML1-ETO. Molecular Cell.
Suarez, L., & Gore, S. D. (2013). Demethylation demystification. Blood, 121, 1488–1489.
Welch, J. S., Ley, T. J., Link, D. C., Miller, C. A., Larson, D. E., Koboldt, D. C., Wartman, L. D., Lamprecht, T. L., Liu, F., Xia, J., et al. (2012). The origin and evolution of mutations in acute myeloid leukemia. Cell, 150, 264–278.
Wiemels, J. L., Xiao, Z., Buffler, P. A., Maia, A. T., Ma, X., Dicks, B. M., Smith, M. T., Zhang, L., Feusner, J., Wiencke, J., et al. (2002). In utero origin of t(8;21) AML1-ETO translocations in childhood acute myeloid leukemia. Blood, 99, 3801–3805.
Xu, Q., Li, Y., Jing, Y., Lv, N., Wang, L., Li, Y., & Yu, L. (2020a). Epigenetic modifier gene mutations-positive AML patients with intermediate-risk karyotypes benefit from decitabine with CAG regimen. International Journal of Cancer, 146, 1457–1467.
Xu, W., Li, J., Rong, B., Zhao, B., Wang, M., Dai, R., Chen, Q., Liu, H., Gu, Z., Liu, S., et al. (2020b). Correction to: DNMT3A reads and connects histone H3K36me2 to DNA methylation. Protein & Cell, 11, 230.
Yang, E., Guan, W., Gong, D., Gao, X., Han, C., Zhang, J., Wang, H., Wang, M., Li, Y., & Yu, L. (2020). Epigenetic silencing of miR564 contributes to the leukemogenesis of t(8;21) acute myeloid leukemia. Clinical Science (london, England: 1979), 134, 3079–3091.
Yang, X., Han, H., De Carvalho, D. D., Lay, F. D., Jones, P. A., & Liang, G. (2014). Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell, 26, 577–590.
Zebisch, A., Hoefler, G., Quehenberger, F., Wölfler, A., & Sill, H. (2013). Mutant DNMT3A in acute myeloid leukemia: Guilty of inducing genetic instability? Leukemia, 27, 1777–1778.
Zhang, L. Y., Yuan, Y. Q., Zhou, D. M., Wang, Z. Y., Ju, S. G., Sun, Y., Li, J., & Fu, J. X. (2016). Impact of Global and Gene-Specific DNA Methylation in de Novo or Relapsed Acute Myeloid Leukemia Patients Treated with Decitabine. Asian Pacific Journal of Cancer Prevention, 17, 431–437.
Zhang, Z. M., Lu, R., Wang, P., Yu, Y., Chen, D., Gao, L., Liu, S., Ji, D., Rothbart, S. B., Wang, Y., et al. (2018). Structural basis for DNMT3A-mediated de novo DNA methylation. Nature, 554, 387–391.
Zhou, L., Fu, L., Lv, N., Liu, J., Li, Y., Chen, X., Xu, Q., Chen, G., Pang, B., Wang, L., et al. (2018). Methylation-associated silencing of BASP1 contributes to leukemogenesis in t(8;21) acute myeloid leukemia. Experimental & Molecular Medicine, 50, 44.
Zhu, H. H., Zhang, X. H., Qin, Y. Z., Liu, D. H., Jiang, H., Chen, H., Jiang, Q., Xu, L. P., Lu, J., Han, W., et al. (2013). MRD-directed risk stratification treatment may improve outcomes of t(8;21) AML in the first complete remission: Results from the AML05 multicenter trial. Blood, 121, 4056–4062.
Funding
This study was supported by the Chinese National Major Project for New Drug Innovation (2019ZX09201002003), National Natural Science Foundation of China (82030076, 82070161, 81970151, and 81870134), Shenzhen Science and Technology Foundation (JCYJ20190808163601776, JCYJ20200109113810154), and Shenzhen Key Laboratory Foundation (ZDSYS20200811143757022). Sanming Project of Shenzhen (SZSM202111004).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, S., Zhou, J. & Yu, L. Aberrant DNA methylation in t(8;21) acute myeloid leukemia. GENOME INSTAB. DIS. 3, 209–216 (2022). https://doi.org/10.1007/s42764-022-00074-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42764-022-00074-1