
Abstract
Understanding protein–protein interactions (PPIs) is pivotal for deciphering the intricacies of biological processes. Dysregulation of PPIs underlies a spectrum of diseases, including cancer, neurodegenerative disorders, and autoimmune conditions, highlighting the imperative of investigating these interactions for therapeutic advancements. This review delves into the realm of mass spectrometry-based techniques for elucidating PPIs and their profound implications in biological research. Mass spectrometry in the PPI research field not only facilitates the evaluation of protein–protein interaction modulators but also discovers unclear molecular mechanisms and sheds light on both on- and off-target effects, thus aiding in drug development. Our discussion navigates through six pivotal techniques: affinity purification mass spectrometry (AP-MS), proximity labeling mass spectrometry (PL-MS), cross-linking mass spectrometry (XL-MS), size exclusion chromatography coupled with mass spectrometry (SEC-MS), limited proteolysis-coupled mass spectrometry (LiP-MS), and thermal proteome profiling (TPP).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Biological molecules such as DNA, RNA, proteins, and metabolites play an important role in regulating biological systems. The outcomes of biological processes are determined by the interactions between these molecules. Protein–protein interactions (PPIs) are essential for modulating protein dynamics and characteristics (Arancibia et al. 2007; Jensen and Thomsen 2012; Koyama et al. 2008; Rebsamen et al. 2013; Rual et al. 2005; Thompson et al. 2011). An imbalance in PPIs is linked to a wide range of diseases such as cancer (Jiang et al. 2013; Kanhaiya et al. 2017; Kim et al. 2021; Li et al. 2012), neurodegenerative disorders (George et al. 2019; Karbalaei et al. 2018; Sinsky et al. 2021; Tomkins and Manzoni 2021; Tracy et al. 2022), and innate immune system aberrations (Corleis and Dorhoi 2020; Hornung et al. 2009; Li et al. 2011; Moncrieffe et al. 2020; Muruve et al. 2008; Pleska et al. 2018). This emphasizes the necessity to explore these interactions for potential therapies. In the context of the innate immune system, PPIs are crucial for mediating host–pathogen interactions, regulating inflammatory responses, and controlling immune cell activation. Comprehending the complexities of these interactions is essential for understanding the mechanisms behind innate immune signaling and discovering new therapeutic targets for infectious and inflammatory diseases (Ciuffa et al. 2022; Meng et al. 2021; Pereira and Gazzinelli 2023; Rajpoot et al. 2022; Van Quickelberghe et al. 2018). Studying PPIs is crucial for obtaining an in-depth understanding of biology. Despite the critical significance of PPIs, advancements in this field have been hindered by the requirement for an extensive knowledge of proteins and protein complexes.
Lately, mass spectrometry (MS)-based methods have emerged as effective tools for PPIs with outstanding sensitivity and specificity. These methods allow for the systematic evaluation of protein complexes and the discovery of interacting counterparts in intricate signaling networks. By combining MS-based proteomics with techniques including immunoprecipitation, cross-linking, limited proteolysis, and thermal profiling, researchers can improve their understanding of the dynamics and control of signaling pathways (Fig. 1).

Overview of mass spectrometry-based techniques for protein–protein interactions (PPIs) study
This review offers an extensive overview of mass spectrometry-based methods used to investigate PPIs with the focus on innate immunity. We introduce the principles, methodologies, recent advancements, applications, advantages, and limitations of five main techniques: affinity purification mass spectrometry (AP-MS), proximity labeling mass spectrometry (PL-MS; BioID and its derivatives), cross-linking mass spectrometry (XL-MS), size exclusion chromatography coupled with mass spectrometry (SEC-MS), limited proteolysis-coupled mass spectrometry (LiP-MS), and thermal proteome profiling (TPP) to characterize PPIs resulting in biological processes to understand molecular mechanisms in innate immune system.
Affinity purification mass spectrometry (AP-MS)
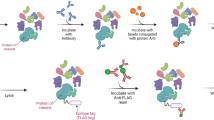
AP-MS is a potent method used to discover and analyze PPIs in biological systems. This method merges the specificity of affinity purification with the sensitivity and accuracy of mass spectrometry to reveal the structural components of protein complexes (Bauer and Kuster 2003; Gingras et al. 2005). Numerous ligands, such as chemicals (Kool et al. 2011; Raida 2011; Rix and Superti-Furga 2009) and biomolecules (Busby et al. 2020; Butter et al. 2009; Faoro and Ataide 2014; Slobodin and Gerst 2011; Tsai et al. 2011), can be used for affinity purification. Following the purification of biological samples, proteins bound to ligands can be separated to simplify the sample or prepared for examination right away with mass spectrometry (Fig. 2). AP-MS has become a fundamental technique for analyzing PPI networks, offering important knowledge about cellular signaling pathways, protein complex formation, and disease processes.

Workflows for affinity purification mass spectrometry (AP-MS), proximity labeling mass spectrometry (PL-MS), and cross-linking mass spectrometry (XL-MS)
AP-MS is based on isolating a specific protein or protein complex from a complex biological material and then identifying and quantifying the proteins that interact with binding partners using mass spectrometry. Affinity purification is commonly done by utilizing antibodies that target the bait protein or using affinity tags fused to the bait protein. This method allows for the separation of proteins that interact with the bait from cell lysates or tissue extracts (Dunham et al. 2012). AP-MS technique has been used to discover new protein–protein interactions that occur under the most significant physiological environments, whereas co-immunoprecipitation (Co-IP) is utilized to investigate the interaction between a known protein and its partners that are expressed in their native physiological conditions. This method uses conjugation with protein A/G beads and target protein-specific antibodies for immunoprecipitation to indirectly capture the proteins associated with the target protein (Lin and Lai 2017; Munoz and Castellano 2018). The interaction between the target protein and the binding protein is confirmed by western blotting. Co-IP is frequently used to identify the molecular pathways involved in inflammation, particularly in the study of the regulation of immunometabolism (Guo et al. 2021; Hu et al. 2021; McGauran et al. 2020), the activation of inflammasomes (Duan et al. 2020; Guan et al. 2021; Niu et al. 2021; Yang et al. 2015), and the networks of Toll-like receptors (Brikos et al. 2007; Kim et al. 2019; Lee et al. 2016; Shahinuzzaman et al. 2022). Both the AP-MS and Co-IP approaches rely on the capacity of the interaction partners to be captured along with the protein of interest.
AP-MS is advantageous because it can capture stable and transient protein interactions, offering a complete view of the protein interactome in specific experimental conditions. AP-MS allows for the purification of protein complexes in their natural shape using antibodies or tags that target the bait protein, thus maintaining the integrity of PPIs. Various adaptations of AP-MS have been created to improve its sensitivity, specificity, and flexibility. Tandem affinity purification (TAP) involves using two different affinity tags in a sequence to purify a protein complex through multiple purification processes, enhancing the purity of isolated proteins (Bian et al. 2022; Burckstummer et al. 2006; Gregan et al. 2007; Kaja et al. 2023; Volkel et al. 2010). Epitope tagging has the huge benefit of being able to tag several proteins with the same epitope and purify them using the same method. Hence, the background contaminants need to be consistent across all purification procedures to enable effective control experiments. AP-MS has been extensively used in diverse biological research fields, such as the investigation of innate immune signaling pathways and host–pathogen interactions (DeBlasio et al. 2021; Gillen and Nita-Lazar 2017; Morwitzer et al. 2019; Terracciano et al. 2021; van Zuylen et al. 2012). This method allowed discoveries of new interactors and regulatory mechanisms involved in innate immune signaling. New modulators of the tumor necrosis factor (TNF)-α/NF-κB pathway have been identified (Bouwmeester et al. 2004) using the tandem affinity purification (TAP) strategy. The authors used an integrated approach, combining large-scale pathway mapping with loss-of-function analysis and created a physical and functional map of the pathway interactions. Stutz et al. identified the components of the NLRP3 inflammasome and the essential novel regulatory phosphorylation sites using AP-MS with FLAG-tagged NLRP3 as a bait (Stutz et al. 2017). A systematic AP-MS approach allowed for the detailed functional analysis of the TBK1/IKKi complex and the associated molecular network (Goncalves et al. 2011).
AP-MS is a potent method for detecting PPIs in complicated biological systems, although it has several limitations. The specificity of the procedure depends on the affinity tag or antibody used for purification, which may result in false positives from non-specific binding or cross-reactivity. Furthermore, AP-MS might not identify weak or transient interactions, and complex samples can mask actual interactions due to background noise. Purifying insoluble or membrane-bound proteins can be difficult, and analyzing the data necessitates advanced computational techniques. Validating interactions is essential but laborious, and AP-MS investigations can be expensive and resource-intensive, which restricts accessibility. Despite its challenges, AP-MS is still an invaluable and widely used tool for studying protein interactions in biological systems when combined with other techniques.
Proximity labeling mass spectrometry (PL-MS)
Proximity labeling mass spectrometry (PL-MS) has advanced the PPIs and spatial proximity in the biological environment (Fig. 2). The major enzyme classes applied for proximity labeling techniques include bacterial biotin ligase (BirA*) and peroxidase-based enzymes such as horseradish peroxidase (HRP) and ascorbate peroxidase (APEX). HRP and APEX proximity labeling depends on fusing the protein of interest with peroxidase-based enzymes. When hydrogen peroxide and biotin-phenol activate these enzymes, they produce short-lived biotin-phenoxyl radicals (Mortensen and Skibsted 1997). The radicals form covalent bonds with biotin atoms that are near proteins, with a specific focus on tyrosine, lysine, and histidine residues located within a range of 10–20 nm. Streptavidin affinity purification is used to concentrate biotinylated proteins, making it easier to identify and measure them by mass spectrometry analysis (Hung et al. 2014; Rhee et al. 2013).
HRP is a well-researched peroxidase enzyme that has been used for proximity labeling. Nevertheless, it showed low labeling efficiency in reducing conditions (Trinkle-Mulcahy 2019). Engineered APEX overcomes this restriction and can be genetically involved as a tag on desired bait proteins. Due to the rapid labeling capabilities of APEX, which are comparable in speed to numerous biological processes, this method is ideally adapted for studying protein interactions that are transient or subject to protein dynamic change. APEX labeling is effective in multiple subcellular locations due to its ability to function in reducing conditions, such as the cytosol (Martell et al. 2012).
BioID is an innovative approach used to characterize PPIs and spatial proximity within cells. BioID uses a proximity-based labeling strategy to capture transient or weak interactions around the bait protein, unlike typical affinity purification methods. This method involves using a fusion protein that includes the protein of interest and an adaptable biotin ligase enzyme like BirA* to facilitate the biotinylation of surrounding proteins. Streptavidin affinity purification isolates the biotinylated proteins for further mass spectrometry analysis. BirA* enzyme converts biotin into a more reactive form, which then combines with primary amines of adjacent proteins, leading to their covalent biotinylation. (Roux et al. 2018).
BioID’s application for the study of temporal protein complexes is restricted as it requires 12–24 h to acquire sufficient labeling signals via mass spectrometry, owing to its slow reaction kinetics. It may result in off-target labeling, high background, and limit the range of experiments suitable for BioID. Furthermore, BioID investigations are restricted to creating static interaction maps because of their temporality. To surpass this restriction, BioID2 was created by modifying the biotin ligase of Aquifex aeolicus through mutations (Kim et al. 2016). This small enzyme greatly reduces the interference with the fusion protein, resulting in enhanced targeting and localization to subcellular compartments (Kim et al. 2017). Nevertheless, it still takes more than 16 h to label. To reduce these experimental limitations, TurboID and miniTurbo were invented. TurboID has 15 mutations and miniTurbo has 13 mutations, and a deletion of the N-terminal domain. These enzymes have a strong affinity to biotin, enabling efficient labeling similar to BioID in less than 10 min. Split proximity labeling methods can be utilized for situations where conventional proximity labeling methodologies are unable to access the region of interest, such as organelle contact sites, to achieve higher targeted specificity. Split proximity labeling methods may also be utilized in situations where large protein fusions are intolerable due to the significantly reduced size of individual fragments. Split forms of APEX (Han et al. 2019), HRP (Heo et al. 2023; Martell et al. 2016), BioID (De Munter et al. 2017; Kwak et al. 2020; Ramirez et al. 2021; Schopp et al. 2017), and TurboID (Cho et al. 2020a, b; Cho et al. 2020a, b; Fujimoto et al. 2023; Schmitt et al. 2021; Shkel et al. 2022; Xu et al. 2021) have been created, where two nonfunctional parts can combine to generate a whole enzyme.
The utilization of PL-MS advanced the process of deciphering the structure and functioning of innate immune signaling networks. BioID was utilized to discover 111 proteins linked to caspase-1 during inflammasome activation in a cell-free system, pointing into the direction of p62/sequestosome1 protein as a regulator of caspase-1-induced inflammation (Jamilloux et al. 2018). A biotin environment scan identified specific interaction partners of the mitochondrial anchored protein ligase (MAPL) a component of MAVS (mitochondrial antiviral signaling) complex in the context of the Sendai virus infection, highlighting a critical role for MAPL and mitochondrial SUMOylation in the early phases of antiviral signaling (Doiron et al. 2017). May et al. used BioID to characterize the SARS-CoV2 virus–host interactome in human lung cancer-derived cells (May et al. 2022). The TurboID technique was utilized to gain a deep understanding of the molecular pathways involved in NLR immune receptor-mediated immunity (Zhang et al. 2019) and SARS-CoV2 suppression (Zhang et al. 2022). Through the process of elucidating the protein interactions that occur during infections, researchers could get a more comprehensive understanding of the host–pathogen interactions. This understanding may lead to the discovery of novel therapeutic targets. These state-of-the-art tools have emerged as a potent tool for understanding the structure and functioning of these signaling networks.
Nevertheless, APEX may lack the precision required for distinguishing direct interactions from proteins merely in proximity due to diffusion. Activating APEX with hydrogen peroxide may induce cellular stress responses and result in non-specific labeling, thereby compromising the accuracy of the outcomes. In addition, the technique might encounter challenges in certain cellular compartments, such as mitochondria, because of the presence of natural biotinylated proteins that could interfere with labeling or complicate analysis. Optimizing APEX labeling may be necessary for different cell types or experimental conditions, which might make its application more complex.
BioID poses challenges due to non-specific biotinylation, leading to false-positive identifications. To address this problem, negative controls and rigorous wash processes should be employed while purifying streptavidin. These approaches guarantee the reliability and precision of BioID-based studies, hence improving its usefulness in biological research.
Cross-linking mass spectrometry (XL-MS)
XL-MS is a method that chemically links amino acid residues in protein complexes and then uses mass spectrometry to analyze and identify the cross-linked peptides, allowing for the interpretation of spatial proximities between residues. The structural information is obtained by detecting adjacent pairs of amino acids, which may include weak or transient interactions, that are connected by a chemical cross-linker of a particular length (Fig. 2). The distance restraint information acquired can help define the structure and positioning of subunits inside the complex and PPI-binding sites (Yu and Huang 2018). The cross-linking process is carried out under the native condition. Enzymatic digestion creates cross-linked peptides which are then enriched, analyzed by MS, and identified through database searching. Data analysis reveals the sequence assignment of cross-linked peptides and identifies the specific cross-linked amino acid residues. Integrative modeling strategies can utilize physical interaction data obtained by XL-MS to enhance structural biology and computational modeling investigations. Restraint information is gathered for proteins that are connected both internally and externally, and used to analyze the arrangement of multiple protein complexes (Shi et al. 2014) and interactions across the entire proteome (Weisbrod et al. 2013). Various cross-linkers are accessible and designed to interact with different amino acid side chains and distances between binding interfaces (Chakrabarty et al. 2020; Gutierrez et al. 2018, 2016; Kao et al. 2011). In addition, several methods have been created to enhance the detection and precision of XL‐MS by enriching cross‐linked peptides. (Chavez and Bruce 2019; Kaake et al. 2014; Leitner et al. 2012; Liu et al. 2020; Rinner et al. 2008; Tang et al. 2005).
XL-MS was used for innate immune signaling studies, especially to map host and pathogen PPIs. XL-MS enabled the identification of functional interactions between bacterial Toll-interleukin receptor (TIR)-domain containing proteins and mammalian receptor TIR domains, revealing differences in binding dynamics between different TIR domain containing proteins and allowing for the further analysis by NMR resulting in the first atomic model of the human TIR domain from IL-1R8 protein (Lee et al. 2022). Intact extracellular human cytomegalovirus virion interactions with host proteins were mapped using XL-MS providing insights into virion organization and revealing crucial PPIs (Bogdanow et al. 2023). XL-MS was used to characterize the interactions between adenovirus C5 hexon and human lactoferrin (hLF), pinpointing major contact sites in hLF and suggesting a mechanism in which the virus uses hLF for cellular entry (Dhillon et al. 2024).
XL-MS has various benefits for analyzing protein complexes, such as its capacity to reveal structure-related information on PPIs within their native cellular environment. XL-MS is capable of capturing transient or weak interactions that could be overlooked in typical biochemical tests, enabling an in-depth examination of dynamic protein complexes participating in signaling networks. Nevertheless, XL-MS has drawbacks such as complex data analysis, probable false-positive identifications, and low spatial resolution of cross-linkers. XL-MS experiments necessitate specialist knowledge in mass spectrometry, chemistry, and bioinformatics, thereby restricting their broad implementation in biological research labs.
Size exclusion chromatography coupled with mass spectrometry (SEC-MS)
Size exclusion chromatography (SEC), also referred to as gel filtration chromatography, is a traditional biochemical technique used to separate protein complexes without bias. This method proves valuable for examining and refining substantial biomolecules, such as proteins, nucleic acids, and polymers. SEC is a technique that separates molecules by their size or hydrodynamic volume. In this process, larger molecules are excluded from the pores of the stationary phase and elute first. Smaller molecules pass through the pores and so have a delayed elution (Fekete et al. 2014; Hartmann et al. 2004). The procedure of SEC-MS involves loading the sample into an SEC column, where the components are segregated based on their size. The eluent from the column is subsequently injected into the MS system, where each of the molecules is ionized and their mass-to-charge ratios are obtained (Havugimana et al. 2022; Skinnider and Foster 2021). The basic concept of SEC-MS is based on the premise that the identification of protein interactions can be determined by analyzing and comparing the elution patterns of individual proteins during SEC fractionation. When two or more proteins coelute, it indicates that there is a protein interaction or complex. However, this evidence needs to be further confirmed by applying statistical filtering and incorporating additional orthogonal information to confirm that the proteins may interact. SEC-MS is frequently used for the analysis of both monoclonal antibodies and biopharmaceuticals (Brusotti et al. 2018; Goyon et al. 2017; Haberger et al. 2016; Murisier et al. 2022; Wehr and Rodriguez-Diaz 2005; Xu et al. 2024). Moreover, this technique can ascertain the molecular weight and purity of biomolecules, detect the presence of aggregates, and identify post-translational modifications (Kirkwood et al. 2013; Kumar et al. 2023).
For the identification of immune complexes, SEC-MS was used to profile protein ADP-ribosylation and protein complexes in LPS-stimulated macrophages (Daniels et al. 2020), to probe the effect of interactomes disassembly to caspase cleavage (Scott et al. 2017), and the structural organization of the TNF-receptor signaling complex (Ciuffa et al. 2022).
To maximize the identification of PPI with SEC-MS and to precisely quantify the proteins in each SEC fraction, researchers applied SEC-MS with SWATH/DIA mass spectrometry approach, and they optimized robust methods for sample preparation and data analysis (Bludau et al. 2020, 2023; Heusel et al. 2020; Rosenberger et al. 2020).
SEC-MS has several key benefits, including the ability to separate without causing damage, to achieve high levels of detail and to conduct extensive analyses. This technology combines the capabilities of both SEC and MS methods to provide in-depth molecular insights. Nevertheless, the method necessitates intricate sample preparation and the use of specialized, expensive equipment. In addition, SEC is restricted to the analysis of soluble compounds and is not appropriate for highly hydrophobic or insoluble substances.
Limited proteolysis-coupled mass spectrometry (LiP-MS)
LiP-MS is a cutting-edge method that has become an effective tool for understanding the intricacies of protein structural changes and interactions in biological systems. This technique depends on the specific digestion of proteins in their native conditions, then using mass spectrometry to analyze the fragments and identify any structural changes. Proteins in the LiP-MS experimental setup undergo digestion with a non-specific protease, usually proteinase K, for a short time. This enzyme cleaves peptide bonds at non-specific protease-binding sites, producing a variety of peptide fragments. Proteolysis is controlled by limiting the digestion of proteins to the protein surface. This regulated digestion offers information on the protein’s structure and conformational changes. The samples are digested with trypsin, producing semi-tryptic and tryptic peptide fragments that exhibit trypsin cleavage sites (Fig. 3). The fragments are injected into an LC–MS/MS system to identify protein conformational changes and understand complex interactions in the biological environment (Malinovska et al. 2023; Schopper et al. 2017).

Workflows for size exclusion chromatography coupled with mass spectrometry (SEC-MS), limited proteolysis-coupled mass spectrometry (LiP-MS) and thermal proteome profiling (TPP)
For successful LiP-MS experiments, high-resolution mass spectrometry instrumentation is required to accurately analyze proteolytic fragments and their cleavage sites. Complex mass spectra interpretation and peptide sequence assignment require advanced bioinformatics algorithms that can efficiently and accurately handle large datasets.
LiP-MS provides unparalleled insights into the dynamic characteristics of proteins in biological systems. This approach provides important insights into protein structural changes and enhances comprehension of complex biological systems. LiP-MS is being used more often to study changes in protein structure caused by interactions with small molecules (Kim et al. 2023; Pepelnjak et al. 2020; Piazza et al. 2018; Sztacho et al. 2021) and disease progression (Liu and Fitzgerald 2016; Mackmull et al. 2022; Shuken et al. 2022).
LiP-MS provides notable benefits for investigating protein structural alterations and interactions, although it also has certain limitations. Using a non-specific protease such as proteinase K for initial cleavage may result in some level of protease specificity, which could cause inadequate digestion or biased cleavage patterns that mask specific protein sequences or interactions. LiP-MS is most efficient when used with pure protein samples. However, in complex biological mixtures like cell lysates or tissue extracts, the presence of many proteins and contaminants makes data interpretation difficult and requires thorough sample preparation and purification. LiP-MS may not be appropriate for insoluble proteins, particularly membrane proteins. LiP-MS offers useful insights into protein conformational changes and interactions, but its structural resolution is inherently limited when compared to techniques such as X-ray crystallography or nuclear magnetic resonance spectroscopy. LiP-MS acts at the peptide level, which makes it difficult to correctly determine protein structures or identify small conformational changes.
Thermal proteome profiling (TPP)
TPP is based on the cellular thermal shift assay (CETSA), which measures the thermal stability of proteins in intact cells or lysates by assessing their susceptibility to thermal denaturation and aggregation (Savitski et al. 2014). Although TPP is technically more demanding, it allows for the identification of multiple targets without prior knowledge. In TPP experiments, cells or lysates are subjected to a temperature gradient, typically ranging from ambient temperature to near the denaturation temperature of most proteins. Following heat treatment, samples are rapidly cooled to halt further denaturation and then lysed to release soluble proteins (Fig. 3). The soluble proteome is then analyzed by mass spectrometry to quantify changes in protein abundance across different temperature conditions (Becher et al. 2016a, b; Franken et al. 2015; Reinhard et al. 2015).
To enable quantitative analysis, TPP experiments often employ isobaric labeling techniques such as tandem mass tags or stable isotope labeling by amino acids in cell culture. Isobaric labeling allows for multiplexed quantification of protein abundance across multiple temperature conditions within a single mass spectrometry experiment, thereby increasing throughput and reducing experimental variability.
A multitude of experimental procedures can be conducted utilizing TPP. Cells are heated to multiple temperatures when a single compound concentration is applied, and this procedure is referred to as the temperature range TPP (TPP-TR) experiment. This method allows for the identification of the majority of targets of a compound present in cell extracts, demonstrating a reproducible change in melting temperature of above 1 °C. There was little association between the Tm shift and the compound’s affinity for each binding protein. The degree of thermal stabilization is influenced by both the ligand’s affinity and the melting thermodynamics of the native protein. A compound concentration range for TPP (TPP-CCR) can be used to calculate affinity estimates through TPP. During TPP-CCR, cells are exposed to various concentrations of a compound and then subjected to a uniform temperature. Finally, a two-dimensional TPP (2D-TPP) method including incubating cells with various chemical concentrations and subjecting them to different temperatures has become accessible (Becher et al. 2016a, b; Mateus et al. 2020). This expansion enables a quick assessment of compound affinity towards the target and is far more effective at pinpointing targets.
TPP has been used in the innate immune system to study structural changes in heat shock proteins of activated macrophages, in combination with LiP revealing that the structures of molecular chaperones such as HSP60 can vary greatly during macrophage activation (Zhang et al. 2021). It enabled identification of antiviral inhibitor targets with broad range of activity against RNA viruses (Tampere et al. 2020), and analysis of protein complex formation and dissociation during human cytomegalovirus infection. (Hashimoto et al. 2020; Justice et al. 2021). These investigations have shown that protein–ligand interactions are altered depending on temperature, offering an understanding of the molecular processes involved in innate immune responses.
The TPP has numerous benefits for investigating protein stability and interactions. This method provides a comprehensive way to analyze protein–ligand interactions and complex formation in reaction to thermal stress across the whole proteome. TPP experiments can be conducted in intact cells or lysates to analyze protein stability under physiological conditions.
Nevertheless, TPP has drawbacks such as the possibility of non-specific protein aggregation and the requirement for specialized mass spectrometry instrumentation and skills in proteomics and bioinformatics. TPP experiments may be prone to artifacts and false-positive identifications, requiring thorough validation and controls to guarantee result reliability.
Data analysis and visualization
Using these five methodologies, we can acquire valuable insights into protein complexes. In terms of MS/MS peptides quantification, MaxQuant (Cox and Mann 2008) and Proteome Discoverer are widely used. In addition, in-house developed R or Python packages, such as MSstatsLiP (Malinovska et al. 2023), Rtpca (Kurzawa et al. 2021), NPARC (Childs et al. 2019), or Tapioca (Reed et al. 2024), can be used according to their respective objectives. To characterize protein networks, PPI databases (e.g., BioGRID (Oughtred et al. 2021), Reactome (Milacic et al. 2024), STRING (Szklarczyk et al. 2023), CORUM (Giurgiu et al. 2019) and IntAct (Del Toro et al. 2022)) that suggest a complex interaction are commonly used. The program known as Cytoscape is extensively employed to analyze expression data and genetic interactions, as well as PPIs, and for visualization of biological networks (Shannon et al. 2003).
The purpose of functional annotation is to gather details regarding the established biological function of PPIs. Gene Ontology (GO) is most often used for annotation, offering an extensive set of terms, which, however, might lead to an overwhelming number of terms connected with a certain protein (Ashburner et al. 2000).
The purpose of enrichment analysis is to ascertain if a portion of the network is enriched in a related function. After adding GO functional annotation to the network, enrichment analysis can be performed on each of the bait protein's interaction partners. If the proteins exhibit a greater probability of sharing a certain set of GO terms than what would be anticipated by a random assignment of all terms in the network, it is probable that these proteins are implicated in the corresponding biological process or cellular function, dependent on the branch of GO. Ontology-based enrichment analysis can be conducted using many tools, such as web-based platforms such as DAVID (Sherman et al. 2022) and several Cytoscape applications including BiNGO (Maere et al. 2005), ClueGO (Bindea et al. 2009), NOA (Zhang et al. 2013) and ReactomeFIViz (Wu et al. 2014).
Conclusion and perspective
In this review, we presented various mass spectrometry-based techniques for characterizing PPIs and their applications to understanding biological processes (Table 1).
Through the exploration of these techniques, it is evident that mass spectrometry has become an indispensable tool for unraveling the complexities of molecular interactions within biological systems. From elucidating signaling cascades triggered by endotoxins to identifying drug-target interactions, the ability to characterize PPIs with high sensitivity and specificity has greatly enhanced our understanding of fundamental biological processes.
Looking ahead, continued advancements in mass spectrometry instrumentation, data analysis algorithms, and sample preparation methodologies hold promise for further expanding the capabilities of PPI characterization techniques. Integration with other omics approaches such as genomics, transcriptomics, and metabolomics will enable a more holistic understanding of cellular processes and disease mechanisms.
Moreover, the development of multiplexed and high-throughput screening methods will facilitate the rapid and comprehensive analysis of PPI networks under diverse conditions. These advancements will not only deepen our understanding of basic biology but also pave the way for the discovery of novel therapeutic targets and the development of precision medicine strategies.
Data availability
No datasets were generated or analysed during the current study.
References
Arancibia SA, Beltran CJ, Aguirre IM, Silva P, Peralta AL, Malinarich F, Hermoso MA (2007) Toll-like receptors are key participants in innate immune responses. Biol Res 40(2):97–112. https://doi.org/10.4067/s0716-97602007000200001
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25(1):25–29. https://doi.org/10.1038/75556
Bauer A, Kuster B (2003) Affinity purification-mass spectrometry. Powerful tools for the characterization of protein complexes. Eur J Biochem 270(4):570–578. https://doi.org/10.1046/j.1432-1033.2003.03428.x
Becher I, Werner T, Doce C, Zaal EA, Tögel I, Khan CA, Rueger A, Muelbaier M, Salzer E, Berkers CR (2016a) Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat Chem Biol 12(11):908–910. https://doi.org/10.1038/nchembio.2185
Becher I, Werner T, Doce C, Zaal EA, Tögel I, Khan CA, Rueger A, Muelbaier M, Salzer E, Berkers CR, Fitzpatrick PF, Bantscheff M, Savitski MM (2016b) Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nature Chem Biol 12(11):908. https://doi.org/10.1038/Nchembio.2185
Bian W, Jiang H, Feng S, Chen J, Wang W, Li X (2022) Protocol for establishing a protein-protein interaction network using tandem affinity purification followed by mass spectrometry in mammalian cells. STAR Protoc 3(3):101569. https://doi.org/10.1016/j.xpro.2022.101569
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J (2009) ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25(8):1091–1093. https://doi.org/10.1093/bioinformatics/btp101
Bludau I, Heusel M, Frank M, Rosenberger G, Hafen R, Banaei-Esfahani A, van Drogen A, Collins BC, Gstaiger M, Aebersold R (2020) Complex-centric proteome profiling by SEC-SWATH-MS for the parallel detection of hundreds of protein complexes. Nat Protoc 15(8):2341–2386. https://doi.org/10.1038/s41596-020-0332-6
Bludau I, Nicod C, Martelli C, Xue P, Heusel M, Fossati A, Uliana F, Frommelt F, Aebersold R, Collins BC (2023) Rapid profiling of protein complex reorganization in perturbed systems. J Proteome Res 22(5):1520–1536. https://doi.org/10.1021/acs.jproteome.3c00125
Bogdanow B, Gruska I, Muhlberg L, Protze J, Hohensee S, Vetter B, Bosse JB, Lehmann M, Sadeghi M, Wiebusch L, Liu F (2023) Spatially resolved protein map of intact human cytomegalovirus virions. Nat Microbiol 8(9):1732–1747. https://doi.org/10.1038/s41564-023-01433-8
Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, Croughton K, Cruciat C, Eberhard D, Gagneur J, Ghidelli S, Hopf C, Huhse B, Mangano R, Michon AM, Schirle M, Schlegl J, Schwab M, Stein MA, Bauer A, Superti-Furga G (2004) A physical and functional map of the human TNF-α NF-κB signal transduction pathway. Nat Cell Biol 6(2):97. https://doi.org/10.1038/ncb1086
Brikos C, Wait R, Begum S, O’Neill LAJ, Saklatvala J (2007) Mass spectrometric analysis of the endogenous type I interleukin-1 (IL-1) receptor signaling complex formed after IL-1 binding identifies IL-1RAcP, MyD88, and IRAK-4 as the stable components. Mol Cell Proteomics 6(9):1551–1559. https://doi.org/10.1074/mcp.M600455-MCP200
Brusotti G, Calleri E, Colombo R, Massolini G, Rinaldi F, Temporini C (2018) Advances on size exclusion chromatography and applications on the analysis of protein biopharmaceuticals and protein aggregates: a mini review. Chromatographia 81(1):3–23. https://doi.org/10.1007/s10337-017-3380-5
Burckstummer T, Bennett KL, Preradovic A, Schutze G, Hantschel O, Superti-Furga G, Bauch A (2006) An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat Methods 3(12):1013–1019. https://doi.org/10.1038/nmeth968
Busby KN, Fulzele A, Zhang D, Bennett EJ, Devaraj NK (2020) Enzymatic RNA biotinylation for affinity purification and identification of RNA-protein interactions. ACS Chem Biol 15(8):2247–2258. https://doi.org/10.1021/acschembio.0c00445
Butter F, Scheibe M, Morl M, Mann M (2009) Unbiased RNA-protein interaction screen by quantitative proteomics. Proc Natl Acad Sci USA 106(26):10626–10631. https://doi.org/10.1073/pnas.0812099106
Chakrabarty JK, Bugarin A, Chowdhury SM (2020) Evaluating the performance of an ETD-cleavable cross-linking strategy for elucidating protein structures. J Proteomics 225:103846. https://doi.org/10.1016/j.jprot.2020.103846
Chavez JD, Bruce JE (2019) Chemical cross-linking with mass spectrometry: a tool for systems structural biology. Curr Opin Chem Biol 48:8–18. https://doi.org/10.1016/j.cbpa.2018.08.006
Childs D, Bach K, Franken H, Anders S, Kurzawa N, Bantscheff M, Savitski MM, Huber W (2019) Nonparametric analysis of thermal proteome profiles reveals novel drug-binding proteins. Mol Cell Proteomics 18(12):2506–2515. https://doi.org/10.1074/mcp.TIR119.001481
Cho KF, Branon TC, Rajeev S, Svinkina T, Udeshi ND, Thoudam T, Kwak C, Rhee HW, Lee IK, Carr SA, Ting AY (2020a) Split-TurboID enables contact-dependent proximity labeling in cells. Proc Natl Acad Sci USA 117(22):12143–12154. https://doi.org/10.1073/pnas.1919528117
Cho KF, Branon TC, Udeshi ND, Myers SA, Carr SA, Ting AY (2020b) Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat Protoc 15(12):3971–3999. https://doi.org/10.1038/s41596-020-0399-0
Ciuffa R, Uliana F, Mannion J, Mehnert M, Tenev T, Marulli C, Satanowski A, Keller LML, Rodilla Ramírez PN, Ori A (2022) Novel biochemical, structural, and systems insights into inflammatory signaling revealed by contextual interaction proteomics. Proc Natl Acad Sci 119(40):e2117175119. https://doi.org/10.1073/pnas.2117175119
Corleis B, Dorhoi A (2020) Early dynamics of innate immunity during pulmonary tuberculosis. Immunol Lett 221:56–60. https://doi.org/10.1016/j.imlet.2020.02.010
Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26(12):1367–1372. https://doi.org/10.1038/nbt.1511
Daniels CM, Kaplan PR, Bishof I, Bradfield C, Tucholski T, Nuccio AG, Manes NP, Katz S, Fraser IDC, Nita-Lazar A (2020) Dynamic ADP-ribosylome, phosphoproteome, and interactome in LPS-activated macrophages. J Proteome Res 19(9):3716–3731. https://doi.org/10.1021/acs.jproteome.0c00261
De Munter S, Gornemann J, Derua R, Lesage B, Qian J, Heroes E, Waelkens E, Van Eynde A, Beullens M, Bollen M (2017) Split-BioID: a proximity biotinylation assay for dimerization-dependent protein interactions. FEBS Lett 591(2):415–424. https://doi.org/10.1002/1873-3468.12548
DeBlasio SL, Wilson JR, Tamborindeguy C, Johnson RS, Pinheiro PV, MacCoss MJ, Gray SM, Heck M (2021) Affinity purification-mass spectrometry identifies a novel interaction between a polerovirus and a conserved innate immunity aphid protein that regulates transmission efficiency. J Proteome Res 20(6):3365–3387. https://doi.org/10.1021/acs.jproteome.1c00313
Del Toro N, Shrivastava A, Ragueneau E, Meldal B, Combe C, Barrera E, Perfetto L, How K, Ratan P, Shirodkar G, Lu O, Meszaros B, Watkins X, Pundir S, Licata L, Iannuccelli M, Pellegrini M, Martin MJ, Panni S, Hermjakob H (2022) The IntAct database: efficient access to fine-grained molecular interaction data. Nucleic Acids Res 50(D1):D648–D653. https://doi.org/10.1093/nar/gkab1006
Dhillon A, Persson BD, Volkov AN, Sülzen H, Kádek A, Pompach P, Kereïche S, Lepšík M, Danskog K, Uetrecht C (2024) Structural insights into the interaction between adenovirus C5 hexon and human lactoferrin. J Virol. https://doi.org/10.1128/jvi.01576-23
Doiron K, Goyon V, Coyaud E, Rajapakse S, Raught B, McBride HM (2017) The dynamic interacting landscape of MAPL reveals essential functions for SUMOylation in innate immunity. Sci Rep 7(1):107. https://doi.org/10.1038/s41598-017-00151-6
Duan Y, Zhang L, Angosto-Bazarra D, Pelegrin P, Nunez G, He Y (2020) RACK1 mediates NLRP3 inflammasome activation by promoting NLRP3 active conformation and inflammasome assembly. Cell Rep 33(7):108405. https://doi.org/10.1016/j.celrep.2020.108405
Dunham WH, Mullin M, Gingras AC (2012) Affinity-purification coupled to mass spectrometry: basic principles and strategies. Proteomics 12(10):1576–1590. https://doi.org/10.1002/pmic.201100523
Faoro C, Ataide SF (2014) Ribonomic approaches to study the RNA-binding proteome. FEBS Lett 588(20):3649–3664. https://doi.org/10.1016/j.febslet.2014.07.039
Fekete S, Beck A, Veuthey JL, Guillarme D (2014) Theory and practice of size exclusion chromatography for the analysis of protein aggregates. J Pharm Biomed Anal 101:161–173. https://doi.org/10.1016/j.jpba.2014.04.011
Franken H, Mathieson T, Childs D, Sweetman GM, Werner T, Togel I, Doce C, Gade S, Bantscheff M, Drewes G, Reinhard FB, Huber W, Savitski MM (2015) Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat Protoc 10(10):1567–1593. https://doi.org/10.1038/nprot.2015.101
Fujimoto S, Tashiro S, Tamura Y (2023) Complementation assay using fusion of split-GFP and TurboID (CsFiND) enables simultaneous visualization and proximity labeling of organelle contact sites in yeast. Contact (thousand Oaks) 6:25152564231153620. https://doi.org/10.1177/25152564231153621
George G, Valiya Parambath S, Lokappa SB, Varkey J (2019) Construction of Parkinson’s disease marker-based weighted protein-protein interaction network for prioritization of co-expressed genes. Gene 697:67–77. https://doi.org/10.1016/j.gene.2019.02.026
Gillen JG, Nita-Lazar A (2017) Composition of the myddosome during the innate immune response. J Immunol 198(1_Supplement):75.15. https://doi.org/10.4049/jimmunol.198.Supp.75.15
Gingras AC, Aebersold R, Raught B (2005) Advances in protein complex analysis using mass spectrometry. J Physiol 563(Pt 1):11–21. https://doi.org/10.1113/jphysiol.2004.080440
Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, Ruepp A (2019) CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res 47(D1):D559–D563. https://doi.org/10.1093/nar/gky973
Goncalves A, Burckstummer T, Dixit E, Scheicher R, Gorna MW, Karayel E, Sugar C, Stukalov A, Berg T, Kralovics R, Planyavsky M, Bennett KL, Colinge J, Superti-Furga G (2011) Functional dissection of the TBK1 molecular network. PLoS ONE 6(9):e23971. https://doi.org/10.1371/journal.pone.0023971
Goyon A, D’Atri V, Colas O, Fekete S, Beck A, Guillarme D (2017) Characterization of 30 therapeutic antibodies and related products by size exclusion chromatography: feasibility assessment for future mass spectrometry hyphenation. J Chromatogr B Analyt Technol Biomed Life Sci 1065–1066:35–43. https://doi.org/10.1016/j.jchromb.2017.09.027
Gregan J, Riedel CG, Petronczki M, Cipak L, Rumpf C, Poser I, Buchholz F, Mechtler K, Nasmyth K (2007) Tandem affinity purification of functional TAP-tagged proteins from human cells. Nat Protoc 2(5):1145–1151. https://doi.org/10.1038/nprot.2007.172
Guan C, Huang X, Yue J, Xiang H, Shaheen S, Jiang Z, Tao Y, Tu J, Liu Z, Yao Y, Yang W, Hou Z, Liu J, Yang XD, Zou Q, Su B, Liu Z, Ni J, Cheng J, Wu X (2021) SIRT3-mediated deacetylation of NLRC4 promotes inflammasome activation. Theranostics 11(8):3981–3995. https://doi.org/10.7150/thno.55573
Guo HT, Wang Q, Ghneim K, Wang L, Rampanelli E, Holley-Guthrie E, Cheng L, Garrido C, Margolis DM, Eller LA, Robb ML, Sekaly RP, Chen X, Su LS, Ting JPY (2021) Multi-omics analyses reveal that HIV-1 alters CD4 T cell immunometabolism to fuel virus replication. Nat Immunol 22(4):423. https://doi.org/10.1038/s41590-021-00898-1
Gutierrez CB, Yu C, Novitsky EJ, Huszagh AS, Rychnovsky SD, Huang L (2016) Developing an acidic residue reactive and sulfoxide-containing MS-cleavable homobifunctional cross-linker for probing protein-protein interactions. Anal Chem 88(16):8315–8322. https://doi.org/10.1021/acs.analchem.6b02240
Gutierrez CB, Block SA, Yu C, Soohoo SM, Huszagh AS, Rychnovsky SD, Huang L (2018) Development of a novel sulfoxide-containing MS-cleavable homobifunctional cysteine-reactive cross-linker for studying protein-protein interactions. Anal Chem 90(12):7600–7607. https://doi.org/10.1021/acs.analchem.8b01287
Haberger M, Leiss M, Heidenreich AK, Pester O, Hafenmair G, Hook M, Bonnington L, Wegele H, Haindl M, Reusch D (2016) Rapid characterization of biotherapeutic proteins by size-exclusion chromatography coupled to native mass spectrometry. MAbs 8(2):331–339
Han Y, Branon TC, Martell JD, Boassa D, Shechner D, Ellisman MH, Ting A (2019) Directed evolution of split APEX2 peroxidase. ACS Chem Biol 14(4):619–635. https://doi.org/10.1021/acschembio.8b00919
Hartmann WK, Saptharishi N, Yang XY, Mitra G, Soman G (2004) Characterization and analysis of thermal denaturation of antibodies by size exclusion high-performance liquid chromatography with quadruple detection. Anal Biochem 325(2):227–239. https://doi.org/10.1016/j.ab.2003.10.031
Hashimoto Y, Sheng X, Murray-Nerger LA, Cristea IM (2020) Temporal dynamics of protein complex formation and dissociation during human cytomegalovirus infection. Nat Commun 11(1):806. https://doi.org/10.1038/s41467-020-14586-5
Havugimana PC, Goel RK, Phanse S, Youssef A, Padhorny D, Kotelnikov S, Kozakov D, Emili A (2022) Scalable multiplex co-fractionation/mass spectrometry platform for accelerated protein interactome discovery. Nat Commun 13(1):4043. https://doi.org/10.1038/s41467-022-31809-z
Heo S, Baek J, Bae J, Seo BA, Kim S, Jeong S, Kim S, Ryu Y, Lee JJ (2023) Protein ligation-assisted reconstitution of split HRP fragments for facile production of HRP fusion proteins in E. coli. ChemBioChem 24(11):e202200700. https://doi.org/10.1002/cbic.202200700
Heusel M, Frank M, Kohler M, Amon S, Frommelt F, Rosenberger G, Bludau I, Aulakh S, Linder MI, Liu Y, Collins BC, Gstaiger M, Kutay U, Aebersold R (2020) A global screen for assembly state changes of the mitotic proteome by sEC-SWATH-MS. Cell Syst 10(2):133-155 e136. https://doi.org/10.1016/j.cels.2020.01.001
Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458(7237):514–518. https://doi.org/10.1038/nature07725
Hu J, Gao Q, Yang Y, Xia J, Zhang W, Chen Y, Zhou Z, Chang L, Hu Y, Zhou H, Liang L, Li X, Long Q, Wang K, Huang A, Tang N (2021) Hexosamine biosynthetic pathway promotes the antiviral activity of SAMHD1 by enhancing O-GlcNAc transferase-mediated protein O-GlcNAcylation. Theranostics 11(2):805–823. https://doi.org/10.7150/thno.50230
Hung V, Zou P, Rhee HW, Udeshi ND, Cracan V, Svinkina T, Carr SA, Mootha VK, Ting AY (2014) Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol Cell 55(2):332–341. https://doi.org/10.1016/j.molcel.2014.06.003
Jamilloux Y, Lagrange B, Di Micco A, Bourdonnay E, Provost A, Tallant R, Henry T, Martinon F (2018) A proximity-dependent biotinylation (BioID) approach flags the p62/sequestosome-1 protein as a caspase-1 substrate. J Biol Chem 293(32):12563–12575. https://doi.org/10.1074/jbc.RA117.000435
Jensen S, Thomsen AR (2012) Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol 86(6):2900–2910. https://doi.org/10.1128/JVI.05738-11
Jiang Y, Huang T, Chen L, Gao YF, Cai Y, Chou KC (2013) Signal propagation in protein interaction network during colorectal cancer progression. Biomed Res Int 2013:287019. https://doi.org/10.1155/2013/287019
Justice JL, Kennedy MA, Hutton JE, Liu D, Song B, Phelan B, Cristea IM (2021) Systematic profiling of protein complex dynamics reveals DNA-PK phosphorylation of IFI16 en route to herpesvirus immunity. Sci Adv 7(25):eabg6680. https://doi.org/10.1126/sciadv.abg6680
Kaake RM, Wang X, Burke A, Yu C, Kandur W, Yang Y, Novtisky EJ, Second T, Duan J, Kao A, Guan S, Vellucci D, Rychnovsky SD, Huang L (2014) A new in vivo cross-linking mass spectrometry platform to define protein-protein interactions in living cells. Mol Cell Proteomics 13(12):3533–3543. https://doi.org/10.1074/mcp.M114.042630
Kaja A, Barman P, Guha S, Bhaumik SR (2023) Tandem affinity purification and mass-spectrometric analysis of FACT and associated proteins. Base excision repair pathway: methods and protocols. Springer, New York, pp 209–227. https://doi.org/10.1007/978-1-0716-3373-1_14
Kanhaiya K, Czeizler E, Gratie C, Petre I (2017) Controlling directed protein interaction networks in cancer. Sci Rep 7(1):10327. https://doi.org/10.1038/s41598-017-10491-y
Kao A, Chiu CL, Vellucci D, Yang Y, Patel VR, Guan S, Randall A, Baldi P, Rychnovsky SD, Huang L (2011) Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol Cell Proteomics 10(1):M110 002212. https://doi.org/10.1074/mcp.M110.002212
Karbalaei R, Allahyari M, Rezaei-Tavirani M, Asadzadeh-Aghdaei H, Zali MR (2018) Protein-protein interaction analysis of Alzheimers disease and NAFLD based on systems biology methods unhide common ancestor pathways. Gastroenterol Hepatol Bed Bench 11(1):27
Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, Roux KJ (2016) An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell 27(8):1188–1196. https://doi.org/10.1091/mbc.E15-12-0844
Kim BR, Coyaud E, Laurent EM, St-Germain J, Van de Laar E, Tsao M-S, Raught B, Moghal N (2017) Identification of the SOX2 interactome by BioID reveals EP300 as a mediator of SOX2-dependent squamous differentiation and lung squamous cell carcinoma growth. Mol Cell Proteomics 16(10):1864–1888. https://doi.org/10.1074/mcp.M116.064451
Kim YC, Lee SE, Kim SK, Jang HD, Hwang I, Jin S, Hong EB, Jang KS, Kim HS (2019) Toll-like receptor mediated inflammation requires FASN-dependent MYD88 palmitoylation. Nat Chem Biol 15(9):907–916. https://doi.org/10.1038/s41589-019-0344-0
Kim M, Park J, Bouhaddou M, Kim K, Rojc A, Modak M, Soucheray M, McGregor MJ, O’Leary P, Wolf D, Stevenson E, Foo TK, Mitchell D, Herrington KA, Munoz DP, Tutuncuoglu B, Chen KH, Zheng F, Kreisberg JF, Krogan NJ (2021) A protein interaction landscape of breast cancer. Science 374(6563):eabf3066. https://doi.org/10.1126/science.abf3066
Kim D, Lee MS, Kim ND, Lee S, Lee HS (2023) Identification of alpha-amanitin effector proteins in hepatocytes by limited proteolysis-coupled mass spectrometry. Chem Biol Interact 386:110778. https://doi.org/10.1016/j.cbi.2023.110778
Kirkwood KJ, Ahmad Y, Larance M, Lamond AI (2013) Characterization of native protein complexes and protein isoform variation using size-fractionation-based quantitative proteomics. Mol Cell Proteomics 12(12):3851–3873. https://doi.org/10.1074/mcp.M113.032367
Kool J, Jonker N, Irth H, Niessen WM (2011) Studying protein-protein affinity and immobilized ligand-protein affinity interactions using MS-based methods. Anal Bioanal Chem 401(4):1109–1125. https://doi.org/10.1007/s00216-011-5207-9
Koyama S, Ishii KJ, Coban C, Akira S (2008) Innate immune response to viral infection. Cytokine 43(3):336–341. https://doi.org/10.1016/j.cyto.2008.07.009
Kumar S, Savane TS, Rathore AS (2023) Multiattribute monitoring of aggregates and charge variants of monoclonal antibody through native 2D-SEC-MS-WCX-MS. J Am Soc Mass Spectrom 34(7):1263–1271. https://doi.org/10.1021/jasms.2c00325
Kurzawa N, Mateus A, Savitski MM (2021) Rtpca: an R package for differential thermal proximity coaggregation analysis. Bioinformatics 37(3):431–433. https://doi.org/10.1093/bioinformatics/btaa682
Kwak C, Shin S, Park JS, Jung M, Nhung TTM, Kang MG, Lee C, Kwon TH, Park SK, Mun JY, Kim JS, Rhee HW (2020) Contact-ID, a tool for profiling organelle contact sites, reveals regulatory proteins of mitochondrial-associated membrane formation. Proc Natl Acad Sci USA 117(22):12109–12120. https://doi.org/10.1073/pnas.1916584117
Lee E, Redzic JS, Nemkov T, Saviola AJ, Dzieciatkowska M, Hansen KC, D’Alessandro A, Dinarello C, Eisenmesser EZ (2022) Human and bacterial toll-interleukin receptor domains exhibit distinct dynamic features and functions. Molecules 27(14):4494. https://doi.org/10.3390/molecules27144494
Lee YR, Kang W, Kim YM (2016) Detection of interaction between toll-like receptors and other transmembrane proteins by co-immunoprecipitation assay. Toll-like receptors, 2nd edn. Humana Press, New York, pp 107–120. https://doi.org/10.1007/978-1-4939-3335-8_7
Leitner A, Reischl R, Walzthoeni T, Herzog F, Bohn S, Forster F, Aebersold R (2012) Expanding the chemical cross-linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol Cell Proteomics 11(3):M111 014126. https://doi.org/10.1074/mcp.M111.014126
Li S, Wang L, Berman M, Kong YY, Dorf ME (2011) Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35(3):426–440. https://doi.org/10.1016/j.immuni.2011.06.014
Li BQ, Huang T, Liu L, Cai YD, Chou KC (2012) Identification of colorectal cancer related genes with mRMR and shortest path in protein-protein interaction network. PLoS ONE 7(4):e33393. https://doi.org/10.1371/journal.pone.0033393
Lin JS, Lai EM (2017) Protein-protein interactions: co-immunoprecipitation. Methods Mol Biol 1615:211–219. https://doi.org/10.1007/978-1-4939-7033-9_17
Liu F, Fitzgerald MC (2016) Large-scale analysis of breast cancer-related conformational changes in proteins using limited proteolysis. J Proteome Res 15(12):4666–4674. https://doi.org/10.1021/acs.jproteome.6b00755
Liu X, Zhang Y, Wen Z, Hao Y, Banks CAS, Lange JJ, Slaughter BD, Unruh JR, Florens L, Abmayr SM, Workman JL, Washburn MP (2020) Driving integrative structural modeling with serial capture affinity purification. Proc Natl Acad Sci USA 117(50):31861–31870. https://doi.org/10.1073/pnas.2007931117
Mackmull MT, Nagel L, Sesterhenn F, Muntel J, Grossbach J, Stalder P, Bruderer R, Reiter L, van de Berg WDJ, de Souza N, Beyer A, Picotti P (2022) Global, in situ analysis of the structural proteome in individuals with Parkinson’s disease to identify a new class of biomarker. Nat Struct Mol Biol 29(10):978–989. https://doi.org/10.1038/s41594-022-00837-0
Maere S, Heymans K, Kuiper M (2005) BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21(16):3448–3449. https://doi.org/10.1093/bioinformatics/bti551
Malinovska L, Cappelletti V, Kohler D, Piazza I, Tsai T-H, Pepelnjak M, Stalder P, Dörig C, Sesterhenn F, Elsässer F (2023) Proteome-wide structural changes measured with limited proteolysis-mass spectrometry: an advanced protocol for high-throughput applications. Nat Protoc 18(3):659–682. https://doi.org/10.1038/s41596-022-00771-x
Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, Ting AY (2012) Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol 30(11):1143–1148. https://doi.org/10.1038/nbt.2375
Martell JD, Yamagata M, Deerinck TJ, Phan S, Kwa CG, Ellisman MH, Sanes JR, Ting AY (2016) A split horseradish peroxidase for the detection of intercellular protein-protein interactions and sensitive visualization of synapses. Nat Biotechnol 34(7):774–780. https://doi.org/10.1038/nbt.3563
Mateus A, Kurzawa N, Becher I, Sridharan S, Helm D, Stein F, Typas A, Savitski MM (2020) Thermal proteome profiling for interrogating protein interactions. Mol Syst Biol 16(3):e9232. https://doi.org/10.15252/msb.20199232
May DG, Martin-Sancho L, Anschau V, Liu S, Chrisopulos RJ, Scott KL, Halfmann CT, Diaz Pena R, Pratt D, Campos AR, Roux KJ (2022) A BioID-derived proximity interactome for SARS-CoV-2 proteins. Viruses 14(3):611. https://doi.org/10.3390/v14030611
McGauran G, Dorris E, Borza R, Morgan N, Shields DC, Matallanas D, Wilson AG, O’Connell DJ (2020) Resolving the interactome of the human macrophage immunometabolism regulator (MACIR) with enhanced membrane protein preparation and affinity proteomics. Proteomics 20(19–20):e2000062. https://doi.org/10.1002/pmic.202000062
Meng Z, Xu R, Xie L, Wu Y, He Q, Gao P, He X, Chen Q, Xie Q, Zhang J (2021) A20/Nrdp1 interaction alters the inflammatory signaling profile by mediating K48-and K63-linked polyubiquitination of effectors MyD88 and TBK1. J Biol Chem. https://doi.org/10.1016/j.jbc.2021.100811
Milacic M, Beavers D, Conley P, Gong C, Gillespie M, Griss J, Haw R, Jassal B, Matthews L, May B, Petryszak R, Ragueneau E, Rothfels K, Sevilla C, Shamovsky V, Stephan R, Tiwari K, Varusai T, Weiser J, D’Eustachio P (2024) The reactome pathway knowledgebase 2024. Nucleic Acids Res 52(D1):D672–D678. https://doi.org/10.1093/nar/gkad1025
Moncrieffe MC, Bollschweiler D, Li B, Penczek PA, Hopkins L, Bryant CE, Klenerman D, Gay NJ (2020) MyD88 death-domain oligomerization determines myddosome architecture: implications for toll-like receptor signaling. Structure 28(3):281-289 e283. https://doi.org/10.1016/j.str.2020.01.003
Mortensen A, Skibsted LH (1997) Importance of carotenoid structure in radical-scavenging reactions. J Agric Food Chem 45(8):2970–2977. https://doi.org/10.1021/jf970010s
Morwitzer MJ, Tritsch SR, Cazares LH, Ward MD, Nuss JE, Bavari S, Reid SP (2019) Identification of RUVBL1 and RUVBL2 as novel cellular interactors of the Ebola virus nucleoprotein. Viruses 11(4):372. https://doi.org/10.3390/v11040372
Munoz A, Castellano MM (2018) Coimmunoprecipitation of interacting proteins in plants. Methods Mol Biol 1794:279–287. https://doi.org/10.1007/978-1-4939-7871-7_19
Murisier A, Andrie M, Fekete S, Lauber M, D’Atri V, Iwan K, Guillarme D (2022) Direct coupling of size exclusion chromatography and mass spectrometry for the characterization of complex monoclonal antibody products. J Sep Sci 45(12):1997–2007. https://doi.org/10.1002/jssc.202200075
Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J (2008) The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452(7183):103–107. https://doi.org/10.1038/nature06664
Niu T, De Rosny C, Chautard S, Rey A, Patoli D, Groslambert M, Cosson C, Lagrange B, Zhang Z, Visvikis O, Hacot S, Hologne M, Walker O, Wong J, Wang P, Ricci R, Henry T, Boyer L, Petrilli V, Py BF (2021) NLRP3 phosphorylation in its LRR domain critically regulates inflammasome assembly. Nat Commun 12(1):5862. https://doi.org/10.1038/s41467-021-26142-w
Oughtred R, Rust J, Chang C, Breitkreutz BJ, Stark C, Willems A, Boucher L, Leung G, Kolas N, Zhang F, Dolma S, Coulombe-Huntington J, Chatr-Aryamontri A, Dolinski K, Tyers M (2021) The BioGRID database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci 30(1):187–200. https://doi.org/10.1002/pro.3978
Pepelnjak M, de Souza N, Picotti P (2020) Detecting protein-small molecule interactions using limited proteolysis-mass spectrometry (LiP-MS). Trends Biochem Sci 45(10):919–920. https://doi.org/10.1016/j.tibs.2020.05.006
Pereira M, Gazzinelli RT (2023) Regulation of innate immune signaling by IRAK proteins. Front Immunol 14:1133354. https://doi.org/10.3389/fimmu.2023.1133354
Piazza I, Kochanowski K, Cappelletti V, Fuhrer T, Noor E, Sauer U, Picotti P (2018) A map of protein-metabolite interactions reveals principles of chemical communication. Cell 172(1–2):358-372 e323. https://doi.org/10.1016/j.cell.2017.12.006
Pleska M, Lang M, Refardt D, Levin BR, Guet CC (2018) Phage-host population dynamics promotes prophage acquisition in bacteria with innate immunity. Nat Ecol Evol 2(2):359–366. https://doi.org/10.1038/s41559-017-0424-z
Raida M (2011) Drug target deconvolution by chemical proteomics. Curr Opin Chem Biol 15(4):570–575. https://doi.org/10.1016/j.cbpa.2011.06.016
Rajpoot S, Kumar A, Zhang KYJ, Gan SH, Baig MS (2022) TIRAP-mediated activation of p38 MAPK in inflammatory signaling. Sci Rep 12(1):5601. https://doi.org/10.1038/s41598-022-09528-8
Ramirez CA, Egetemaier S, Bethune J (2021) Context-specific and proximity-dependent labeling for the proteomic analysis of spatiotemporally defined protein complexes with split-BioID. Methods Mol Biol 2247:303–318. https://doi.org/10.1007/978-1-0716-1126-5_17
Rebsamen M, Kandasamy RK, Superti-Furga G (2013) Protein interaction networks in innate immunity. Trends Immunol 34(12):610–619. https://doi.org/10.1016/j.it.2013.05.002
Reed TJ, Tyl MD, Tadych A, Troyanskaya OG, Cristea IM (2024) Tapioca: a platform for predicting de novo protein-protein interactions in dynamic contexts. Nat Methods 21(3):488–500. https://doi.org/10.1038/s41592-024-02179-9
Reinhard FB, Eberhard D, Werner T, Franken H, Childs D, Doce C, Savitski MF, Huber W, Bantscheff M, Savitski MM, Drewes G (2015) Thermal proteome profiling monitors ligand interactions with cellular membrane proteins. Nat Methods 12(12):1129–1131. https://doi.org/10.1038/nmeth.3652
Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY (2013) Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339(6125):1328–1331. https://doi.org/10.1126/science.1230593
Rinner O, Seebacher J, Walzthoeni T, Mueller LN, Beck M, Schmidt A, Mueller M, Aebersold R (2008) Identification of cross-linked peptides from large sequence databases. Nat Methods 5(4):315–318. https://doi.org/10.1038/nmeth.1192
Rix U, Superti-Furga G (2009) Target profiling of small molecules by chemical proteomics. Nat Chem Biol 5(9):616–624. https://doi.org/10.1038/nchembio.216
Rosenberger G, Heusel M, Bludau I, Collins BC, Martelli C, Williams EG, Xue P, Liu Y, Aebersold R, Califano A (2020) SECAT: quantifying protein complex dynamics across cell states by network-centric analysis of SEC-SWATH-MS profiles. Cell Syst 11(6):589-607 e588. https://doi.org/10.1016/j.cels.2020.11.006
Roux KJ, Kim DI, Burke B, May DG (2018) BioID: a screen for protein-protein interactions. Curr Protoc Protein Sci 91(1):19–23. https://doi.org/10.1002/cpps.51
Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Vidal M (2005) Towards a proteome-scale map of the human protein-protein interaction network. Nature 437(7062):1173–1178. https://doi.org/10.1038/nature04209
Savitski MM, Reinhard FB, Franken H, Werner T, Savitski MF, Eberhard D, Martinez Molina D, Jafari R, Dovega RB, Klaeger S, Kuster B, Nordlund P, Bantscheff M, Drewes G (2014) Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346(6205):1255784. https://doi.org/10.1126/science.1255784
Schmitt K, Kraft AA, Valerius O (2021) A multi-perspective proximity view on the dynamic head region of the ribosomal 40S subunit. Int J Mol Sci 22(21):11653. https://doi.org/10.3390/ijms222111653
Schopp IM, Amaya Ramirez CC, Debeljak J, Kreibich E, Skribbe M, Wild K, Bethune J (2017) Split-BioID a conditional proteomics approach to monitor the composition of spatiotemporally defined protein complexes. Nat Commun 8(1):15690. https://doi.org/10.1038/ncomms15690
Schopper S, Kahraman A, Leuenberger P, Feng Y, Piazza I, Muller O, Boersema PJ, Picotti P (2017) Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry. Nat Protoc 12(11):2391–2410. https://doi.org/10.1038/nprot.2017.100
Scott NE, Rogers LD, Prudova A, Brown NF, Fortelny N, Overall CM, Foster LJ (2017) Interactome disassembly during apoptosis occurs independent of caspase cleavage. Mol Syst Biol 13(1):906. https://doi.org/10.15252/msb.20167067
Shahinuzzaman ADA, Kamal AM, Chakrabarty JK, Rahman A, Chowdhury SM (2022) Identification of inflammatory proteomics networks of toll-like receptor 4 through immunoprecipitation-based chemical cross-linking proteomics. Proteomes 10(3):31. https://doi.org/10.3390/proteomes10030031
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504. https://doi.org/10.1101/gr.1239303
Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, Imamichi T, Chang W (2022) DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res 50(W1):W216–W221. https://doi.org/10.1093/nar/gkac194
Shi Y, Fernandez-Martinez J, Tjioe E, Pellarin R, Kim SJ, Williams R, Schneidman-Duhovny D, Sali A, Rout MP, Chait BT (2014) Structural characterization by cross-linking reveals the detailed architecture of a coatomer-related heptameric module from the nuclear pore complex. Mol Cell Proteomics 13(11):2927–2943. https://doi.org/10.1074/mcp.M114.041673
Shkel O, Kharkivska Y, Kim YK, Lee JS (2022) Proximity labeling techniques: a multi-omics toolbox. Chem Asian J 17(2):e202101240. https://doi.org/10.1002/asia.202101240
Shuken SR, Rutledge J, Iram T, Losada PM, Wilson EN, Andreasson KI, Leib RD, Wyss-Coray T (2022) Limited proteolysis–mass spectrometry reveals aging-associated changes in cerebrospinal fluid protein abundances and structures. Nat Aging 2(5):379–388. https://doi.org/10.1038/s43587-022-00196-x
Sinsky J, Pichlerova K, Hanes J (2021) Tau protein interaction partners and their roles in Alzheimer’s disease and other tauopathies. Int J Mol Sci 22(17):9207. https://doi.org/10.3390/ijms22179207
Skinnider MA, Foster LJ (2021) Meta-analysis defines principles for the design and analysis of co-fractionation mass spectrometry experiments. Nat Methods 18(7):806–815. https://doi.org/10.1038/s41592-021-01194-4
Slobodin B, Gerst JE (2011) RaPID: an aptamer-based mRNA affinity purification technique for the identification of RNA and protein factors present in ribonucleoprotein complexes. Methods Mol Biol 714:387–406. https://doi.org/10.1007/978-1-61779-005-8_24
Stutz A, Kolbe CC, Stahl R, Horvath GL, Franklin BS, van Ray O, Brinkschulte R, Geyer M, Meissner F, Latz E (2017) NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J Exp Med 214(6):1725–1736. https://doi.org/10.1084/jem.20160933
Szklarczyk D, Kirsch R, Koutrouli M, Nastou K, Mehryary F, Hachilif R, Gable AL, Fang T, Doncheva NT, Pyysalo S (2023) The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res 51(D1):D638–D646. https://doi.org/10.1093/nar/gkac1000
Sztacho M, Salovska B, Cervenka J, Balaban C, Hoboth P, Hozak P (2021) Limited proteolysis-coupled mass spectrometry identifies phosphatidylinositol 4,5-bisphosphate effectors in human nuclear proteome. Cells 10(1):68. https://doi.org/10.3390/cells10010068
Tampere M, Pettke A, Salata C, Wallner O, Koolmeister T, Cazares-Korner A, Visnes T, Hesselman MC, Kunold E, Wiita E, Kalderen C, Lightowler M, Jemth AS, Lehtio J, Rosenquist A, Warpman-Berglund U, Helleday T, Mirazimi A, Jafari R, Puumalainen MR (2020) Novel broad-spectrum antiviral inhibitors targeting host factors essential for replication of pathogenic RNA viruses. Viruses 12(12):1423. https://doi.org/10.3390/v12121423
Tang X, Munske GR, Siems WF, Bruce JE (2005) Mass spectrometry identifiable cross-linking strategy for studying protein-protein interactions. Anal Chem 77(1):311–318. https://doi.org/10.1021/ac0488762
Terracciano R, Preiano M, Fregola A, Pelaia C, Montalcini T, Savino R (2021) Mapping the SARS-CoV-2-host protein-protein interactome by affinity purification mass spectrometry and proximity-dependent biotin labeling: a rational and straightforward route to discover host-directed anti-SARS-CoV-2 therapeutics. Int J Mol Sci 22(2):532. https://doi.org/10.3390/ijms22020532
Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA (2011) Pattern recognition receptors and the innate immune response to viral infection. Viruses 3(6):920–940. https://doi.org/10.3390/v3060920
Tomkins JE, Manzoni C (2021) Advances in protein-protein interaction network analysis for Parkinson’s disease. Neurobiol Dis 155:105395. https://doi.org/10.1016/j.nbd.2021.105395
Tracy TE, Madero-Perez J, Swaney DL, Chang TS, Moritz M, Konrad C, Ward ME, Stevenson E, Huttenhain R, Kauwe G, Mercedes M, Sweetland-Martin L, Chen X, Mok SA, Wong MY, Telpoukhovskaia M, Min SW, Wang C, Sohn PD, Gan L (2022) Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell 185(4):712-728 e714. https://doi.org/10.1016/j.cell.2021.12.041
Trinkle-Mulcahy L (2019) Recent advances in proximity-based labeling methods for interactome mapping. F1000Res. https://doi.org/10.12688/f1000research.16903.1
Tsai BP, Wang X, Huang L, Waterman ML (2011) Quantitative profiling of in vivo-assembled RNA-protein complexes using a novel integrated proteomic approach. Mol Cell Proteomics. https://doi.org/10.1074/mcp.M110.007385
Van Quickelberghe E, De Sutter D, van Loo G, Eyckerman S, Gevaert K (2018) A protein-protein interaction map of the TNF-induced NF-kappaB signal transduction pathway. Sci Data 5(1):180289. https://doi.org/10.1038/sdata.2018.289
van Zuylen WJ, Doyon P, Clement JF, Khan KA, D’Ambrosio LM, Do F, St-Amant-Verret M, Wissanji T, Emery G, Gingras AC, Meloche S, Servant MJ (2012) Proteomic profiling of the TRAF3 interactome network reveals a new role for the ER-to-Golgi transport compartments in innate immunity. PLoS Pathog 8(7):e1002747. https://doi.org/10.1371/journal.ppat.1002747
Volkel P, Le Faou P, Angrand PO (2010) Interaction proteomics: characterization of protein complexes using tandem affinity purification-mass spectrometry. Biochem Soc Trans 38(4):883–887. https://doi.org/10.1042/BST0380883
Wehr T, Rodriguez-Diaz R (2005) Use of size exclusion chromatography in biopharmaceutical development. Analytical techniques for biopharmaceutical development, 1st edn. CRC Press, Boca Raton, pp 95–112
Weisbrod CR, Chavez JD, Eng JK, Yang L, Zheng C, Bruce JE (2013) In vivo protein interaction network identified with a novel real-time cross-linked peptide identification strategy. J Proteome Res 12(4):1569–1579. https://doi.org/10.1021/pr3011638
Wu G, Dawson E, Duong A, Haw R, Stein L (2014) ReactomeFIViz: a Cytoscape app for pathway and network-based data analysis. F1000Res 3:146. https://doi.org/10.12688/f1000research.4431.2
Xu Y, Fan X, Hu Y (2021) In vivo interactome profiling by enzyme-catalyzed proximity labeling. Cell Biosci 11(1):27. https://doi.org/10.1186/s13578-021-00542-3
Xu J, Coughlin JE, Szyjka M, Jabary S, Saluja S, Sosic Z, Chen Y, Xu C-F (2024) Evaluation of the impact of antibody fragments on aggregation of intact molecules via size exclusion chromatography coupled with native mass spectrometry. Mabs 16(1):2334783
Yang CS, Kim JJ, Kim TS, Lee PY, Kim SY, Lee HM, Shin DM, Nguyen LT, Lee MS, Jin HS, Kim KK, Lee CH, Kim MH, Park SG, Kim JM, Choi HS, Jo EK (2015) Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat Commun 6(1):6115. https://doi.org/10.1038/ncomms7115
Yu C, Huang L (2018) Cross-linking mass spectrometry (XL-MS): an emerging technology for interactomics and structural biology. Anal Chem 90(1):144
Zhang C, Wang J, Hanspers K, Xu D, Chen L, Pico AR (2013) NOA: a cytoscape plugin for network ontology analysis. Bioinformatics 29(16):2066–2067. https://doi.org/10.1093/bioinformatics/btt334
Zhang Y, Song G, Lal NK, Nagalakshmi U, Li Y, Zheng W, Huang P-J, Branon TC, Ting AY, Walley JW (2019) TurboID-based proximity labeling reveals that UBR7 is a regulator of N NLR immune receptor-mediated immunity. Nat Commun 10(1):3252. https://doi.org/10.1038/s41467-019-11202-z
Zhang W, Wei Y, Zhang H, Liu J, Zong Z, Liu Z, Zhu S, Hou W, Chen Y, Deng H (2021) Structural alternation in heat shock proteins of activated macrophages. Cells 10(12):3507. https://doi.org/10.3390/cells10123507
Zhang Y, Shang L, Zhang J, Liu Y, Jin C, Zhao Y, Lei X, Wang W, Xiao X, Zhang X, Liu Y, Liu L, Zhuang MW, Mi Q, Tian C, Wang J, He F, Wang PH, Wang J (2022) An antibody-based proximity labeling map reveals mechanisms of SARS-CoV-2 inhibition of antiviral immunity. Cell Chem Biol 29(1):5-18 e16. https://doi.org/10.1016/j.chembiol.2021.10.008
Acknowledgements
This research was supported by the Division of Intramural Research of NIAID, NIH.
Funding
Open access funding provided by the National Institutes of Health. This article is funded by Division of Intramural Research.
Author information
Authors and Affiliations
Contributions
D. K. and A. N.-L. conceptualized and wrote the manuscript. D. K. prepared the figures and Table 1. Both authors edited and reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, D., Nita-Lazar, A. Progress in mass spectrometry approaches to profiling protein–protein interactions in the studies of the innate immune system. J Proteins Proteom (2024). https://doi.org/10.1007/s42485-024-00156-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42485-024-00156-6
Keywords
- Protein–protein interactions
- Affinity purification mass spectrometry (AP-MS)
- Proximity labeling mass spectrometry (PL-MS)
- Cross-linking mass spectrometry (XL-MS)
- Size exclusion chromatography coupled with mass spectrometry (SEC-MS)
- Limited proteolysis-coupled mass spectrometry (LiP-MS)
- Thermal proteome profiling (TPP)