
Abstract
Pyrite is the most common among the group of sulfide minerals in the Earth and abundant in most geological settings. This gangue mineral in association with garnet, hematite, magnetite, and other sulfide minerals acts as an indicator mineral in the Kubi concession of the Asante Gold corporation in Ghana. X-ray diffraction (XRD), air annealing in a furnace, energy-dispersive x-ray spectroscopy (EDX), and X-ray photoelectron spectroscopy (XPS) were applied to investigate the crystal structure, identify individual elements, permanence, transformation, and chemical/electronic properties of such pyrite. The study aims to identify individual elements and to gain an understanding of the surface reaction mechanisms, as well as the properties of precipitated pyrite particles observed during the hydrothermal formation of the ore deposit. XRD shows that pristine and annealed samples contain some hematite and quartz besides pyrite. Results from air annealing indicate that the relationship between pyrite and hematite-magnetite is controlled by temperature. EDX reveals that the sample has O and C as contaminants, while XPS in addition reveals Ba, Au, P, Al, and N. These elements are attributed to pyrite that bonds metallically or covalently to neighboring ligands/impurity minerals such as oxides, chalcogenide sulfides, as well as the gangue alteration minerals of magnetite and hematite in the pyrite sample.
These findings suggest that during the hydrothermal flow regime, pyrite, pathfinder elements, and impurity minerals/metals were in contact with quartz minerals before undergoing hematite transformation, which thus becomes an indicator mineral in the Kubi gold concession.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Pyrite (FeS2), and other sulfide groups associated with gold (arsenopyrite, chalcopyrite, pyrrhotite, covellite, sphalerite, and galena) form tiny particles of gold attached to the sulfide compound grains [1,2,3]. Pyrite is one of the most common minerals hosting gold in most ore deposits and part of the gold-bearing minerals in the Kubi concession area alongside garnet and the aforementioned sulfide groups of minerals that are of economic importance. It is one of the most common sulfide minerals that belong to the Fe-S system which forms the basis for understanding phase interactions and thermochemistry of other key systems, e.g., Zn-Fe-S, Cu-Fe-S, Fe–Ni-S, and Fe-As-S [4]. Under the oxidation process in the presence of air, pyrite causes acid mine drainage to release sulfuric acid and heavy metals, which is a serious environmental problem and a concern in the mineral industry. As a semiconductor material with redox behavior, pyrite can cause a galvanic effect when in contact with other minerals [5, 6]. It is therefore important to consider how other sulfide minerals affect the chemical reactivity of pyrite minerals towards oxidation when in contact with each other.
X-ray photoelectron spectroscopy (XPS) and other techniques have previously been used in the study of gold deposition from aqueous solutions in pyrite [7] and other indicator minerals of gold [8] and have been a contributory technique in the study of products of oxidation formed on sulfide-containing minerals. Murphy and Strongin [9] conducted surface reactivity studies on pyrite (FeS2) and pyrrhotite (FexS1-x) using XPS and X-ray absorption spectroscopy techniques to examine the structure of the original pyrite and sulfide surfaces [9]. XPS and low-energy electron diffraction were used to analyze iron sulfide surface reactions by depositing cystine on pyrite (100) to reveal how the orientation of the chemical functionalities of the cystine molecules compensates for the iron surface coordination [10]. Frost et al., used XPS to study mineral, synthetic, and coal-associated pyrites, to detect, monitor, and clarify pyrite surface-oxidative changes. These changes can influence surface-dependent coal cleaning methods, such as flotation of froth (separation of hydrophobic materials from hydrophilic materials), and provide a direct means of analyzing coal sulfur, by determining the existence of oxidizing conditions to remove oxidized surface pyrite of the same XPS binding energy (Eb) as the organic sulfur peak of coal [11].
The main advantage of XPS in mineral studies is the ability to define complete information from high-resolution spectra for samples of macroscopic sizes of different mineral phases. In view of this, it is of interest to reveal the individual elements using XPS and assign them to the original host minerals (indicator minerals) [12,13,14].
In the present study, we combine XRD, EDX, and XPS to investigate pyrite samples from the alluvial deposit as part of our continuous research on the characterization of pathfinders and indicators of Gold on the Kubi concession [15].
The objectives of this study are to identify individual elements and to gain insight into surface reaction mechanisms, as well as the properties of precipitated pyrite particles observed during the hydrothermal formation of the ore deposit. The samples were investigated in their pristine state and after air annealing in a furnace.
1.1 Geology and Mineralization of the Kubi Gold Deposit
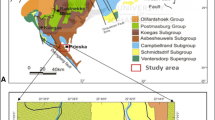
Figure 1 [16] is a simplified geological map of Ghana, the southwestern part of the country is underlain by the Paleoproterozoic Birimian Supergroup comprising thick sequences of northeasterly-southwesterly trending belts of steeply dipping metavolcanic rocks (chiefly metamorphosed, basic, and intermediate lavas and pyroclastic rocks) that alternate with metasedimentary rock units (phyllites, schist and greywackes) all of which have been intruded by syn- and post-tectonic granitoids [17]. The Tarkwaian group represents metamorphosed erosional deposits of Birimian [17,18,19] and occupy the troughs within sections of the Birimian metavolcanic sequence. All the major gold deposits in Ghana are hosted in the Birimian and the Tarkwaian rocks and usually associated with shearing, deformation alteration along major contact zones and granite intrusions.

Generalized Geological Map of Ghana (After Peterson et al. [16]) showing Kubi concession area
The Kubi gold deposit is situated at the intersection of the main NE-SW trending Birimian-Tarkwaian contact and a major north–south trending basement fault that strikes at 020°, dips 85° to the west [19, 20], and a conjugate E-W set which strikes 275° and dips 79° to the north. The gold mineralization at Kubi is located within garnetiferous horizons of variable thickness within Birimian metasediments where fine-grained gold associated with minor (10–20%) pyrite, pyrrhotite are found with coarser gold associated with relatively narrow quartz veins.
The Kubi Gold deposit was once an old open-pit-mine which has been refilled and currently under-consideration for deep mining after series of exploration activities on the concession, hence the location is ideal for pathfinders, alterations, and transformation investigations. The Offin River is the major drainage of the area and has numerous dendritic tributaries.
2 Experimental Details
2.1 Sample Collection and Preparation
A pyrite nugget of approximately 37.50 g, 4.8 cm × 2.2 cm (Fig. 2a) was collected from the alluvial Au deposit of 12 m depth at a small-scale mining site at the Kubi Gold concession near Dunkwa-on-Offin. The sample was washed with clean water to remove dirt and other contaminations and then put into a sample bag. With the aid of a geological hammer, a smaller piece was broken off and filed/polished into a dimension of 0.6 cm × 0.6 cm (Fig. 2b) using a metallographic polisher (Struers polishing machine) for XRD, EDX, and XPS analysis. The remaining nugget was then crushed and sieved into fine- and coarse-grained samples. The fine-grained sample was annealed at a temperature of 1100 °C using an air annealing furnace. This air annealing oxidation process was performed to examine the effect on mineral grains, to (1) analyze the effect of temperature on the mineral grains, facilitation of water-heat transfer of ions between and within minerals and any growth of new minerals and (2) to promote chemical reactions such as oxidation and hydrolysis with oxygen and moisture that may lead to mineralogical alteration. This nugget sample is part of a bulk pyrite sample from the 12 m depth. Before this study, pyrite in quartz and pyrite close to the orebody had been identified from this Kubi concession as cubic \(P\overline{a }3\) space group (lattice constant a = 5.41 Å) and distinguished from marcasite. Therefore, the one pyrite sample in this study is a representative of the Kubi pyrite and a continuation of our research on the characterization of pathfinders and indicators of Gold on the Kubi concession [15].

a Pyrite nugget with a chipped-off piece, b pristine pyrite nugget, c crushed powder pyrite sample before annealing, and (d) fused pyrite sample after annealing
2.2 Measurements
Initially, the mineralogical compositions of the samples (pristine and annealed) were investigated by XRD. The measurements were performed using a PANAnalyical X’pert powder diffractometer with a θ-2θ configuration of Cu-Kα radiation with a wavelength of 1.5406 Å. A Cu long-fine-focus X-ray tube was set to 45 kV and 40 mA with a scan step size of 0.0084; a counting time of 19.68 s per step, and a scan range between 10 and 100 degrees in 2-θ scans during data collection. The data were quantitatively analyzed with the aid of the MAUD software [21] using Rietveld refinement, where analytical functions of pseudo-Voigt and Pearson VII and Marquardt Least Squares algorithm [22] were employed to model the experimental XRD patterns. The fitting parameters (background, peak shape, crystal structure, microstructure, strain, and texture) were used for the refinement while considering the least squares for the iteration [21] to minimize the residual and to fit the anisotropic size-strain broadening for the crystallite size and possible micro-strain [23].
The pyrite powder sample was subjected to annealing using a compact 2" tube furnace OTF-1200X-S-DVD type from MTI corporation. The sample was manually placed in an inner sample sliding glass tube of the furnace for rapid thermal processing (RTP) under room atmosphere conditions for two hours and at a furnace temperature of 1100 °C. XRD measurement was again performed on the annealed sample.
The pyrite sample was analyzed using SEM/EDX operated at 20 kV. The sample was prepared by mounting the crystalline sample into an aluminum stub with adhesive carbon tape. The Zeiss Supra 35 VP field emission SEM type EDX was used in this study with field emission scanning electron microscopy (FESEM) equipped with an EDX detector, which was employed for elemental mapping of the sample to understand the elemental distribution.
X-ray photoelectron spectroscopy (XPS) was used to analyze the elemental distribution and chemistry of the pyrite sample. The measurement was performed in an Axis Ultra DLD instrument from Kratos Analytical (UK) that uses monochromatic Al Kα radiation (hν = 1486.6 eV) and operates with a base pressure lower than 1.1 × 10–9 Torr (1.5 × 10–7 Pa) during data acquisition. XPS depth profiles were acquired by sputter etching with 0.5-keV Ar+ ions incident at an angle of 70° relative to the sample surface normal. The low Ar+ energy and shallow incidence angle were chosen to minimize the effect of sputtering damage on the sample. The spot size on the sample analyzed by XPS was 0.3 × 0.7 mm2 and was located in the center of 3 × 3 mm2 ion-etched regions. The binding energy scale was calibrated using the standard ISO-certified procedure [24]. The measurements were made with the oxygen signal monitored and the highest intensity height adjusted. The recorded spectra include core-levels (Fe 2p, S 2p, O 1 s, and C 1 s) and valence band, with a total acquisition time of 348 s with the charge neutralizer off.
3 Results
Figure 3 (a and b) shows XRD patterns from pristine and annealed pyrite. The diffractograms of pyrite exhibit pronounced high-intensity reflection peaks that reveal the presence of pyrite, hematite, and quartz consistent with powder diffraction data for pyrite, hematite, and quartz, that crystallize in the cubic \(P\overline{a }3\) space group (lattice constant a = 5.41 Å), rhombohedral in the \(R\overline{3 }c\) space group (lattice constant a = b = 5.03 Å, c = 13.77 Å), and hexagonal in the P3221 (154) space group (lattice constant a = b = 4.91 Å, c = 5.40 Å), respectively. The lattice parameters are in good agreement with the values in the literature [25,26,27]. The polished pristine pyrite (Fig. 3a) shows negligible levels of impurity minerals, while the untreated and annealed pyrite contains quartz and a transformed pyrite mineral phase of hematite. During the annealing process, there is a restructuring of mineral grains as dislocations are removed, allowing grain growth of SiO2 to occur since the annealing temperature is above the recrystallization temperature of quartz. In the cubic Pa̅3 space group, the Fe4+ atoms are bonded to six equivalent S2− atoms and form corner-sharing FeS6 octahedra while the S2− atoms are bonded in a 3-coordinate geometry to three equivalent Fe4+ atoms.

Pyrite X-ray diffractograms (a) before annealing and (b) after annealing
Figure 4 shows EDX spectra of polished pristine pyrite that is dominated by Fe and S with weaker peaks from C and O as impurity species, accounting for segregations and incorporation of these species as contamination on the FeS2 surface. Table 1 shows a list and quantitative analysis of the identified elemental species in the polished pristine pyrite (FeS2), where the 0.01 detection limits of atomic percentages of the EDX are normalized to their nearest percentage units.

EDX spectra of FeS2 with the elemental distribution of polished pyrite dominated by S and Fe sample
Figure 5 shows a survey XPS spectrum of a well-ordered pyrite surface after sputter etching with assigned peaks of identified elements. After the sputtering process, the sulfur peaks were identified as a spectroscopic signature to confirm the presence or absence of the pyrite surface/material. Table 2 lists the Ebs of the observed elements and their corresponding assigned compounds; consistent with the reference data [28]. The survey XPS data of the investigated pristine pyrite in Fig. 5 shows Fe 2p3/2,1/2 peaks located at 707.3 and 720.1 eV respectively, Fe 3 s at 92 eV, Fe 3p at 54 eV, Al 2 s at 116 eV, Al 2p at 76 eV, S 2 s at 226 eV, S 2p 162 eV, Ba 4 s at 251 eV, Au 4f5/2 at 91.2 eV, and P 2 s at 187 eV. Also, a Ba 5p3/2 peak located at the same Eb of 14.50 eV as the N 2 s. In addition, there are C 1 s and O 1 s peaks at 285 and 532 eV, respectively [28]. The Fe 3 s peak at 92 eV occurs close to an atomic position of the Au 4f5/2 peak belonging to the alkali-chloride class of caesium gold (III) tetrachloride [29]. The XPS observations of N 2 s, O 1 s, and C 1 s in Fig. 5 provide information on surface cleanliness and carbonic/graphitic contamination as the sample from the alluvial environment may contain discharged mined fluids, dissolution of carbonate minerals of different pH, and dissolved oxides [30].

XPS survey spectrum of polished FeS2 recorded after sputtering plotted on a binding energy scale. The broken vertical lines indicate the core levels as well as the spin–orbit splitting of Fe. VB denotes valence band
Figure 6 shows high-resolution XPS spectra of Fe 2p, S 2p, O 1 s, and C 1 s before and after sputtering, where the signals from O and C before sputtering are attributed to oxide and carbonate contaminations. From the high-resolution XPS spectra in Fig. 6 (a-d), the Fe and the signal from oxygen could be attributed to Fe2O3/Fe3O4 (Fig. 6a, c), while the Fe and S elements observed in the polished pristine pyrite sample are attributed to FeS2 in the sample (Fig. 6a, b) [31]. In Fig. 6a, the peaks located at 707.48 eV (Fe (III)-S) and 720.58 eV (Fe (II)-S) after sputtering, are the characteristic signals of the Fe 2p3/2 and 2p1/2 subshells, respectively. The peak at 735.40 eV is a satellite peak of Fe [32]. The Fe 2p peaks in the spectrum correspond to different chemical states of Fe oxide with the 2p3/2 peak at 707.48 eV and the spin–orbit splitting of 13.1 eV to the 2p1/2 peaks. The Fe 2p peaks in Fig. 6a show an asymmetric shape characteristic for metals due to screening with a well-resolved Fe 2p3/2 peak that exhibits multiplet splitting. There is a high Eb shift because of charge transfer from metal atoms to S ligands.

Core level XPS spectra of polished FeS2 recorded before and after sputtering (a) Fe 2p, (b) S 2p, c O 1 s, and (d) C 1 s plotted on a binding energy scale
Figure 7 shows experimental (Fig. 7a) and calculated (Fig. 7b) spectra of the valence band of pyrite. The calculated spectra in Fig. 7b are the density of states (DOS with space group Pa̅3 in Fig. 8a) weighted by the photoionization cross sections et al. Kα X-rays (1486.6 eV) [33] and an energy-dependent broadening relative to the Fermi level (EF). Generally, there is good agreement between the experimental and calculated valence band spectra although the cross-section of the Fe 3d states close to the EF is somewhat overestimated in the calculation. The data are also consistent with previous measurements of the valence band of pyrite [34] (Fig. 7).

Valence band XPS spectra of the polished FeS2 (a) before and after sputtering on a binding energy scale, b calculated spectra of the pyrite valence band with weighted cross sections and increasing energy-dependent broadening from the EF

a Calculated total and partial density of states (DOS) of FeS2 (cubic Pa̅3 space group) with the main hybridization regions indicated. b calculated t2g-eg crystal-field splitting of the Fe 3d states in pyrite
Figure 8a shows the results of calculated total and partial density of states (DOS) for the cubic Pa̅3 space group of pyrite (nonmagnetic). The geometry relaxation was performed using the Perdue-Burke-Ernzerhof (PBE) functional [35] including the Grimme–van der Waals density-functional theory (DFT)-D2 scheme [36]. The first-principal calculations were carried out with a 25 × 25x25 k grid using DFT implemented in the Vienna ab initio simulation package (VASP) [37] with an exchange-potential functional using the general gradient approximation (GGA). As observed in Fig. 8a, the states are dominated by Fe 3d states (0–1.5 eV), S 2p (2–8 eV), and S 2 s (11–17 eV) with different hybridization regions. The main hybridization regions are the Fe 3d-t2g—S 3pxy (π-bonding, 0–1 eV) at the top of the valence band close to the EF, Fe 3d-eg—S 3pz (σ-bonding, 2–4 eV), and a broad double peak at the bottom of the valence band that consists of S 3 s- σ—S 3 s- σ bonding and S 3 s- σ*—S 3 s- σ* anti-bonding (S2 dimer) states, respectively [38]. Figure 8b shows the calculated Fe 3d t2g-eg splitting in pyrite. As observed, the Fe 3d-eg bonding states are located 1.5–8 eV below the EF while the Fe 3d states closest to the EF mainly consist of non-bonding and anti-bonding Fe 3d-t2g states. Thus, the Fe 3d—S 3p hybridization and bonding occur 1.5–5 eV below EF while the S 3 s—S 3 s dimer bonding occurs at the bottom of the valence band, 11–17 eV below EF.
4 Discussion
Upon annealing the pristine pyrite, it transforms from pyrite to hematite with quartz as an impurity as observed in the XRD data in Fig. 3. This signifies that the pyrite mineral was in contact with quartz or intruded from a quartz vein. The annealed powder pyrite sample first transforms into iron (III) sulfide and then dissociates into hematite between 100 ℃ to 1100 ℃ [39]. The light purple, brown to black color in Fig. 2d signifies hematite alteration, influenced by oxidation and temperature. The thermal reaction process of pyrite produces different intermediate products such as pyrrhotite, maghemite, magnetite, and iron oxides, and the final by-products of hematite and sulfur dioxide [40] are formed above 1000° C. Above 300 °C, the pyrite sample attains a hexagonal pyrrhotite phase where iron and vacancies are created and distributed in the cation sites. In this air-annealed experiment, where oxygen and little water (moist) are present, oxidation and reduction processes may occur with O2/H2O gas mixtures with H2O being adsorbed through a Fe–O interaction in a reduction process. The O2 reacts with Fe to form Fe–O bonds leaving the oxidation of S2− as a free electron transfer. The Oxygen from H2O is then incorporated into the sulfate ions (SO42−) which are formed when pyrite undergoes oxidation [9]. Here, oxygen bonding with sulfur is easier to form sulfur oxides and to be adsorbed on the pyrite surface to form stable S–O bonds at elevated temperatures than Fe − O bonds [41, 42]. Therefore, pyrite can be oxidized into hematite or magnetite when Fe in the pyrite experience cathodic reduction of O2 leading to electron transfer between Fe(II) and Fe(III) species [42] according to the reaction Fe3+ + e → Fe+2. Therefore, in the hydrothermal system, different mineral phases (sulfides and oxides) occur when pyrite undergoes oxidation and reduction processes at elevated temperatures. At these higher temperatures and pressures, hematite undergoes reduction to form magnetite in the presence of water; represented by the Eq. 3Fe2O3 + H2 = 2Fe3O4 + H2O. Secondly, the reduction of hematite to magnetite in contact with Fe metal can be influenced by water at elevated temperatures and pressure by the process Fe + 4Fe2O3 = 3Fe3O4 [42]. However, in the alluvial environment, the reduction process of sulfates involves contributions from organic matter and bacteria according to the equation SO42− + 8H+ + 8e → S− + 4H2O and it is influenced by pH. The dissolution of pyrite is influenced by the pH conditions of the environment; increases with increasing alkalinity. Under acidic conditions, microbial activities help facilitate the oxidation of Fe (Fe2+ to Fe3+) and S, such that the alteration products of pyrite containing ferric minerals, Fe2(SO4)3, FeOH(SO4), FeOOH, and Fe(OH)3 are formed at higher pH and iron oxides at elevated temperatures and pressure [42]. The reaction of iron sulfide with air and water produces sulfuric acid and dissolved iron. This iron can undergo precipitation to form coloured (red–orange-yellowish) sediments containing mine drainage to settle at the bottom of the rivers and streams around the Kubi village. The overflow acid can dissolve heavy metals such as mercury, lead, and copper into the nearby water bodies, contaminating and disrupting growth and reproduction of aquatic plants and animals as well as corroding parts of infrastructures like bridges. These coloured sediments are also seen to be constituents of the alteration minerals of hematite and magnetite. Thus, the different minerals and alterations associated with iron oxides and sulfides, e.g., pyrrhotite, magnetite, hematite, marcasite, and goethite in the Kubi concession, are intermediate and by-products of pyrite.
The EDX spectra in Fig. 4 and quantitative results in Table 1 reveal that the sample is of a pyrite nature and consists mainly of Fe and S, with Si, O, and C as contaminants. As shown in Table 3, the quantitative XPS analysis gives the Fe, S, O, and C contents in the pyrite sample to be 20at.%, 68at.%, 7at.%, and 5at.%, respectively, indicating the presence of natural FeS2. By combing EDX and XPS results, the impurity elements in the pristine sample are Ba, P, Al, C, O, and Si. The chemical shift of 0.28 eV towards higher Eb compared to metallic Fe observed in Fig. 6a indicates that the sample originated from the oxide zone; ascribed to FeO, Fe2O3, and Fe3O4 as indicated in Table 3. The XPS spectra lack multiplet splitting and are attributed to the pyrite group of compounds [31]. Therefore, it can be inferred that the deficient main Fe 2p3/2 peak in the sample under investigation contains natural FeS2, which is an indicator mineral in the Kubi study area.
The sulfur peak shows two pronounced S 2p1/2,3/2 peaks at 163.92 eV and 162.71 respectively, in Fig. 6b. This is consistent with sulfur atoms bonded to thiolate species (gold-associated species) that correspond to S–S bonds present in natural pyrite and in agreement with studies by Zhang et al. (2017) and Jana et al. (2021) [43, 44]. The S 2p peak has closely spaced spin–orbit components (Δ = 1.2 eV). The observed splitting of S 2p between components has a chemical shift of 0.20 eV compared to pure sulfur with an asymmetric peak shape due to an unresolved doublet that depends on the chemical state. The shift towards lower Eb is an indication that the sulfur atom is surrounded by more electropositive elements, e.g., Fe and other metals that form part of the pathfinder elements of Au.
The weak impurity signal of the O 1 s spectra in Fig. 6c could not be well fitted as it consists of two peaks positioned at 535.0 and 530.4 eV and ascribed to COO−, (C–O–C), (C-O-Fe) and oxides respectively (Fig. 6c and in Table 3) and in agreement with literature [32]. For metal oxides, the bonding occurs at a considerably distinct BE compared to other oxygen species that show much narrower O 1 s peaks (e.g., carbonates) [32]. The observed O 1 s spectra in Fig. 6c show a broad feature with multiple overlapping components (carbonates, water, and organic contaminations) [45].
The C 1 s peaks in Fig. 6d are obtained from two contributions and identified at an Eb of 285.0 eV, assigned to the C–N groups [44], whereas the second component observed at 288.75 eV can be assigned to the COOH groups [46], indicative of metal carbonates, e.g., CaCO3, as this sample is collected from an alluvial environment containing metal oxides [47].
In the pyrite valence band spectra in Fig. 7, the Fe 3d states dominate close to the EF with significant influence from S at the bottom of the valence band. The 3d orbitals of Fe split into two states, i.e., t2g and eg in a pseudo-octahedral field with empty eg orbitals and completely filled t2g orbitals (6 electrons). The eg states interact with the sp3 hybrid orbital of S2 (dimers) to bridge with the two iron centers to form σ-bonds of 12–18 eV below EF. Furthermore, there is a charge transfer excitation from the total filled t2g orbitals to the empty eg antibonding states due to the hybridization between Fe 3d and S 2p states. The eg states move towards higher Eb along the axis where they experience strong repulsive forces from ligands due to the high involvement of the 3d orbitals of the eg states in metal–ligand σ interaction. Hence, the eg states attain higher energy than the t2g states to favor covalent bonding [48]. Consequently, due to σ interactions, a shift in Eb along the eg states of empty orbitals will covalently bond with oxides within the alluvial environment. At atmospheric pressure, Fe has two crystal structures: bcc α-phase which is stable at the ground state, and fcc γ-Fe, which is stable up to 1665 K, while above this temperature it transforms back into bcc phase (σ-Fe) [49]. Thus, in the hydrothermal system, the fcc-Fe structure bonds metallically with neighboring ligands due to the lower energy of the t2g orbitals which favors the contribution of π interactions with ligands by virtue of their weaker interactions in metal complexes [48, 50].
In terms of electrical conductivity, sulfur vacancies act as dopants, and the conductivity is determined by the Fe 3d and S 3p states. Above the valence band, the pyrite consists mainly of Fe 3d t2g states while the bottom of the conduction band is occupied by Fe 3d eg states. The sulfur S 3p states are located close to both the valence and conduction bands. This implies that the conductivity of pyrite is composed mainly of eg orbitals from the Fe surface atoms with less contribution from the S 3p orbitals.
The pyrite sample from the alluvial sediments is associated with other sulfide minerals and oxides in gangue quartz veins, sedimentary rocks, or as a product of contact metamorphism and can be formed as a high-temperature hydrothermal mineral. The mineral undergoes different stages of transformation, forming mineral phases and alterations by oxidation and reduction processes. The mineral bonds metallically to other iron oxides (FeO, Fe2O3, and Fe3O4) and covalently to carbonates minerals. The reactivity of pyrite depends on the oxidation and reduction of Fe and S states by electronic charge transfer; influenced by O2 and H2O at elevated temperatures. Finally, we note that the conductivity of pyrite is mostly influenced by the S 3p states of sulfur, in addition to Fe 3d t2g and eg states located close to the EF in the valence and conduction bands, respectively; and that the conductivity may be improved by impurity elements.
The results suggest that the identified elemental species in the pyrite mineral and the impurity phases acting as alteration minerals in the pyrite, as well as any possible Au inclusions, were formed at the same time from the same fluid [51]. Furthermore, it suggests that other minerals or elemental species acting as pathfinder elements were originally incorporated into the sulfides (pyrite) through cracks, fractures, and voids during the hydrothermal flow regime before being solidified.
5 Conclusions
By combining EDX with XRD, air-annealed furnace, core-level, and valence-band XPS electronic structure calculations, the electronic structure and chemical bonding in pyrite has been investigated. The complementary results show that a polished pyrite (FeS2) sample transforms to hematite during annealing. The sample has O, C, Ba, P, Al, and N as contaminants as well as Au. These are attributed to impurity minerals such as oxides of barium and aluminum, chalcogenide sulfides, and phosphorus, as well as the gangue alteration minerals such as magnetite and hematite in the sample. A chemical shift of 0.28 eV for metal Fe toward higher energies due to screening effect and charge transfer, is attributed to the bonding between Fe atoms and the neighboring ligands in the indicator or rock-forming minerals in the Kubi concession. The charge transfer contributes to the surface reactivity and conductivity of pyrite. Thus, the pyrite mineral bonds metallically to metallic oxides and covalently to non-metallic components due to hybridization, σ, and π, interactions in metal complexes.
From the air-annealing results, we conclude that the relationship between pyrite, hematite, and magnetite is controlled by temperature and pressure with magnetite being formed at high temperatures of about 1100 °C. These transformation processes (pyrite-hematite-magnetite) occur under oxidation and reduction conditions and at certain pH levels. Additionally, the conversion of pyrite-hematite with intermediate phases in the gangue quartz mineral in the Kubi gold concession might be caused by an increase in temperature and pressure associated with regional metamorphism and facilitated by the presence of oxidizing and reduction conditions.
These combined techniques can provide pertinent information to understand surface chemistry and to characterize chemical changes in pyrite mineral surfaces.
Data and Code Availability
A brief description what data or code is available to readers and how it can be accessed, for example, a DOI to an electronic repository.
Data is available on reasonable request from the corresponding author.
Change history
03 April 2024
A Correction to this paper has been published: https://doi.org/10.1007/s42461-024-00960-0
References
Cook NJ, Chryssoulis SL (1990) Concentrations of ‘“invisible gold”’ in the common sulfides. Can Mineral 28:1–16
Fleet ME, Mumin AH (1997) Gold-bearing arsenian pyrite and marcasite and arsenopyrite from Carlin trend gold deposits and laboratory synthesis. Am Mineral 82:182–193
Friedl J, Wagner FE, Wang N (1995) On the chemical state of combined gold in sulfidic ores: conclusions from Mössbauer source experiments. Neues Jahrb Mineral-Abh 169:279–290
Craig JR, Scott SD (2018) Sulfide phase equilibria. In: Ribbe PH (ed) Sulfide mineralogy, 1976. De Gruyter, Berlin, Boston, pp 124–233. https://doi.org/10.1515/9781501508332-008
Dos-Santos EC, Lourenç MPO, Pettersson LGM, Duarte HA (2017) Stability, structure, and electronic properties of the pyrite/arsenopyrite solid-solid interface-A DFT study. J Phys Chem C 121:8042–805
Abraitis PK, Pattrick RAD, Kelsall GH, Vaughan DJ (2004) Acid leaching and dissolution of major sulphide ore minerals: processes and galvanic effects in complex systems. Mineral Mag 68:343–351
Scaini MJ, Bancroft GM, Knipe SW (1997) An XPS, AES, and SEM study of the interactions of gold and silver chloride species with PbS and FeS2: comparison to natural samples. Geochim Cosmochim Acta 61:1223–1231
Velasquez P, Leinen D, Pascual J, Ramos-Barrado JR, Grez P, Gomez H, Schrebler R, Del-Rio R, Cordova R (2005) A chemical, morphological, and electrochemical (XPS, SEM/EDX, CV, and EIS) analysis of electrochemically modified electrode surfaces of natural chalcopyrite (CuFeS2) and pyrite (FeS2) in alkaline solutions. J Phys Chem B 109:4977–4988
Murphy R, Strongin DR (2009) Surface reactivity of pyrite and related sulfides. Surf Sci Rep 64:1–45 (Elsevier)
Sanchez-Arenillas M, Mateo-Marti E (2016) Pyrite surface environment drives molecular adsorption: cystine on pyrite (100) investigated by X-ray photoemission spectroscopy and lowenergy electron diffraction. Phys Chem Chem Phys 18:27219–27225
Frost DC, Leeder WR, Tapping RL, Wallbank B (1977) An XPS study of the oxidation of pyrite and pyrites in coal. Fuel 56(3):277–280
Buckley AN, Skinner WM, Harmer SL, Pring A, Lamb RN, Fan LJ, Yang YW (2007) Examination of the proposition that Cu (II) can be required for charge neutrality in a sulfide lattice-Cu in tetrahedrites and sphalerite. Can J Chem 85:767–781
Harmer SL, Pratt AR, Nesbitt WH, Fleet ME (2004) Sulfur species at chalcopyrite (CuFeS2) fracture surfaces. Am Miner 89:1026–1032
Smart RSC, Amarantidis J, Skinner WM, Vanier LL, Grano SR (2003) Topics in applied physics. Wandelt K, Thurgate S (eds) Solid–liquid interfaces, vol 85. Springer, New York, NY, USA, pp 3–60
Nzulu G, Eklund P, Magnusson M (2021) Characterization and identification of au pathfinder minerals from an artisanal mine site using X-ray diffraction. J Mater Sci 56:7659–7669
Petersson A, Scherstén A, Gerdes A (2018) Zircon extensive reworking of Archaean crust within the Birimian terrane in Ghana as revealed by combined zircon U-Pb and Lu-Hf isotopes. Geosci Front 9:173–189
Kesse GO (1985) The mineral and rock resources of Ghana. A.A. Balkema, Rotterdam/Bostom, 610
Leube A, Hirdes W, Mauer R (1986) The Birimian supergroup of Ghana: depositional environment, structural development and conceptual model of an early proterozoic suite. Technical Cooperation Ghanaian-German Mineral Prospecting Project, 80, 2040, 6
Marfo E, Darko E, Faanu A, Mayin S (2016) Study of the radiological parameters associated with small-scale mining activities at Dunkwa-on-Offin in the central region of Ghana. Radiat Prot Environ 39:83
Kesse GO, Foster RP (1984) The occurrence of gold in Ghana. In: Balkema AA (ed) Gold 82: the geology, geochemistry and genesis of gold deposits. Geological Society of Zimbabwe: Rotterdam, The Netherlands, pp 648–650
Lutterotti L (2010) Total pattern fitting for the combined size-strain-stress-texture determination in thin film diffraction. Nucl Inst Methods Phys Res B 268:334–340
Marquardt D (1963) An algorithm for least-squares estimation of nonlinear parameters. SIAM J Appl Math 11(2):431–441
Popa NC (1998) The (hkl) dependence of diffraction-line broadening caused by strain and size for all Laue groups in Rietveld refinement [J]. J Appl Cryst 31(2):176–180
Kratos Analytical Ltd.: library filename: “casaXPS_KratosAxis-F1s.lib”
Bayliss P (1977) Crystal structure refinement of a weakly anisotropic pyrite cubic model. Am Miner 62:1168–1172
Blake RL, Hessevick RE, Zoltai T, Finger LW (1966) Refinement of the hematite structure. Am Mineral 51:123–129
Kihara K (1990) An X-ray study of the temperature dependence of the quartz structure sample: at T = 298 K. Eur J Mineral 2:63–77
Fuggle JC, Mårtensson N (1980) Core-level binding energies in metals. J Electron Spectrosc Relat Phenom 21:275
Kitagawa H, Kojima N, Nakajima TJ (1991) Studies of mixed-valence states in three-dimensional halogenbridged gold compounds, Cs2AulAulllX6, (X = Cl, Br or I). Chem Soc Dalton Trans 3121
Dochartaigh BÉÓ, Smedley PL, MacDonald AM, Darling WG, Homoncik S (2011) Groundwater chemistry of the Carboniferous sedimentary aquifers of the Midland Valley, British Geological Survey, Groundwater program
Biesinger MC, Payne BP, Andrew P, Grosvenor AP, Laua LWM, Gerson AR, Smart RStC (2011) Resolving surface chemical states in XPS analysis of first row transition metals, oxides, and hydroxides: Cr Mn, Fe, Co and Ni. Appl Surf Sci 257:2717–2730
Yang S, Song X, Zhang P, Sun J, Gao L (2014) Small, self-assembled α-Fe2O3 mesocrystals/graphene nanohybrid for enhanced electrochemical capacitors. Nano Micro Small Wiley 10:2270–2279
Jackson AJ, Ganose AM, Regoutz A, Egdell RG, Scanlon DO (2018) Galore: broadening and weighting for simulation of photoelectron spectroscopy. J Open-Source Softw 3(26):773. https://doi.org/10.21105/joss.007733
Nesbitt HW, Berlich AG, Harmer SL, Uhlig I, Bancroft GM, Szargan R (2004) Identification of pyrite valence band contributions using synchrotron-excited X-ray photoelectron spectroscopy. Am Miner 89:382–389
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865
Grimme S (2006) Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J Comput Chem 27:1787
Kresse G, Furthmüller J (1996) Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B 54:11169
Heide VDH, Hemmel R, Bruggen CFV, Haas C (1980) X-ray photoelectron spectra of 3d transition metal pyrite. J Solid-State Chem 33:17–25
Leskovar B, Vrabec M, Dolenec MM, Naglič I, Dolenec T, Dervarič E, Markoli B (2017) Temperature-initiated structural changes in FeS2 pyrite from Pohorje Eastern Alps, north-eastern Slovenia. MTAEC9 51(2):259
Kopp OC, Kerr PF (1958) Differential thermal analysis of pyrite and marcasite. Am Mineral 43(11–12):1079–1097
Kendelewicz T, Doyle CS, Bostick BC, Brown GE (2004) Initial oxidation of fractured surfaces of FeS2(100) by molecular oxygen, water vapor, and air. Surf Sci 558:80–88
Santos ECD, Silva JCd-M, Duarte HA (2016) Pyrite oxidation mechanism by oxygen in aqueous medium. J Phys Chem 120(5):2760–2768. https://doi.org/10.1021/acs.jpcc.5b10949
Zhang CL, Deng LB, Zhang PX, Ren XZ, Li YL, He TS (2017) Electrochim Acta 229:229–238
Jana SK, Gucchait A, Susmita P, Tultul S, Acharya S, Hoque KM, Misra AK, Chatterjee BK, Chatterjee T, Chakrabarti P (2021) Virstatin-conjugated gold nanoparticle with enhanced antimicrobial activity against the vibrio cholerae El Tor biotype. ACS Appl Biomater 4(4):3089–3100. https://doi.org/10.1021/acsabm.0c01483
Nohira H, Tsai W, Besling W, Young E, Petry J, Conard T, Vandervorst W, Gendt SDe, Heyns M, Maes J, Tuominen M (2002) Characterization of ALCVD-Al2O3 and ZrO2 layer using X-ray photoelectron spectroscopy. J Non-Cryst Solids 303:83–87
Fischer S, Papageorgiou AC, Marschall M, Reichert J, Diller K, Klappenberger F, Allegretti F, Nefedov A, Wöll C, Barth JV (2012) l-Cysteine on Ag (111): a combined STM and X-ray spectroscopy study of anchorage and deprotonation. J Phys Chem C 116:20356–20362
Shchukarev A, Korolkov D (2004) XPS Study of group IA carbonates. J Open Chem:347–362
Zhang J-X, Sheong FK, Zhenyang L (2020) Superatomic ligand-field splitting in ligated gold nanoclusters. Inorg Chem 59(13):8864–8870
Pepperhoff W, Acet M (2001) The structure of iron. In: Constitution and magnetism of iron and its alloys. Engineering materials. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-04345-5_1
Nzulu GK, Bakhit B, Högberg H, Hultman L, Magnuson M (2021) Elucidating pathfinding elements for gold from the alluvia small-scale mining site. Minerals 11:912–933
Tuba G, Kontak DJ, Zajacz Z, Petrus JA (2019) Bulk microanalysis of assemblages of small fluid inclusions by LA-ICP-MS: methodology and application to orogenic gold systems. Chem Geol 529:119326
Acknowledgements
We thank the Swedish Government Strategic Research Area in Materials Science on Functional Materials at Linköping University (Faculty Grant SFO-Mat-LiU No. 2009 00971). M.M. acknowledges financial support from the Swedish Energy Research (Grant No. 43606-1) and the Carl Tryggers Foundation (CTS23:2746, CTS20:272, CTS16:303, CTS14:310).
Funding
Open access funding provided by Linköping University.
Author information
Authors and Affiliations
Contributions
Gabriel K. Nzulu:—Conceptualization, resources, methodology, data curation, formal analysis, investigation, software, validation, visualization, writing – original draft, and writing – review & editing.
Hans Högberg:—Supervision, project administration, validation, and writing – review & editing.
Per Eklund:—Supervision, project administration, validation, visualization, and writing – review & editing.
Lars Hultman:—Funding acquisition, project administration, validation, and writing – review & editing.
Prosper M. Nude:—Validation, and writing – review & editing.
Abu Yaya:—Validation, and writing – review & editing.
Martin Magnuson:—Methodology, supervision, software, funding acquisition, project administration, and writing – review & editing.
Corresponding author
Ethics declarations
Ethical Approval
A statement that approval was granted to carry out experiments involving human tissue by an institutional review board or equivalent ethics committee.
Not applicable.
Conflicts of Interest or Competing Interests
A statement that declares all relationships or interests of the manuscript’s authors that could potentially influence or bias the submitted work. If no conflicts of interest exist, the authors must include a statement stating this. Details on potential conflicts of interest are included under Publishing Ethics. Manuscripts that do not include a conflict-of-interest statement will be returned to the authors for amendment before any editorial consideration.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nzulu, G.K., Högberg, H., Eklund, P. et al. Chemical Reactivity and Alteration of Pyrite Mineral in the Kubi Gold Concession in Ghana. Mining, Metallurgy & Exploration 41, 1013–1023 (2024). https://doi.org/10.1007/s42461-024-00932-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42461-024-00932-4