
Abstract
Forming complexes with β-cyclodextrin can enhance stability, dissolution rate, solubility, and bioavailability of an active pharmaceutical ingredient. In this study, the inclusion behavior between β-cyclodextrin (β-CD) and diphenhydramine, clonidine, and tolperisone in DMSO-d6 was investigated using NMR spectroscopy. 1H, 13C, COSY, HMQC, and ROESY data were applied to determine the structure of inclusion complexes, and molecular docking analysis was engaged to identify the most favorable host–guest interactions in the inclusion complexes. Complexation of β-CD with diphenhydramine, clonidine, and tolperisone is accompanied by the insertion of a molecular fragment of the guest molecule, one molecule of diphenhydramine and tolperisone, and two molecules of clonidine, into the inner sphere of one host molecule. The reported study provides useful information for the potential application of the complexation of β-CD with diphenhydramine, clonidine, and tolperisone. This may be a good strategy for the development of solid pharmaceutical dosage forms based on β-CDs as a drug delivery system.
Article highlights
-
The inclusion complexes of β-CD and diphenhydramine, clonidine, and tolperisone were synthesized and analyzed using 1Н, 13С, COSY, HMQC, and ROESY spectroscopy.
-
Diphenhydramine, clonidine, and tolperisone interact with β-CD with the formation of stable 1:1 stoichiometric complexes for β-CD:diphenhydramine and β-CD:tolperisone, and 1:2 stoichiometric complex for β-CD:clonidine.
-
Possible structures of the inclusion complexes between β-CD and diphenhydramine, clonidine, and tolperisone were determined using molecular docking in the software AutoDock 4.0.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Currently, due to the intensive development of the pharmaceutical industry, the search for new forms of drugs has great significance [1]. In the modern pharmaceutical industry, great prospects are associated with the encapsulation of drugs with effective receptors, which make it possible to obtain solid dosage forms from liquid ones, help to stabilize active pharmaceutical ingredients (APIs) toward the action of light and heat, increase the solubility of the drug, improve its bioavailability, and mask unwanted odors and taste [2]. The encapsulation of pharmaceuticals allows obtaining drugs with prolonged action and increases the possibility of targeted drug transport in the body directly to the site of its action. In this regard, the development of supramolecular forms of APIs diphenhydramine 1, clonidine 2, and tolperisone 3 with β-cyclodextrin (β-CD) and the investigation of their structure is an urgent task of modern chemistry and medicine.
The choice of APIs diphenhydramine, clonidine, and tolperisone can be explained by their high pharmaceutical activity and the importance of searching for long-term action forms of these APIs [3,4,5]. Diphenhydramine, an antihystamine with a bitter taste, is mainly used to relieve symptoms of allergy, insomnia, and fever, to treat tremor and nausea. This drug has a bitter taste due to the amino groups. Studies [6] have shown that the inclusion of diphenhydramine inside the cavity of β-CD masks the bitter taste of the drug due to the interaction between the amino groups of diphenhydramine and the hydrogen atom of β-CD. Clonidine is known as an adjuvant to local anesthetics, which prolongs their action, reducing the dosage required for anesthesia. In vitro studies have shown that complexation of clonidine with β-CD increases clonidine’s adjuvant effect of clonidine without changing the intrinsic toxicity of clonidine [7]. Tolperisone is a centrally acting muscle relaxant, which is used to treat increased muscle tone caused by neurological diseases. Side effects include body weakness, nausea, dizziness, increase in liver enzymes and muscle pain. According to the studies [6, 7], the inclusion of the APIs into the β-CD cavity may result in reduced side effects of the APIs. Presumably, the same effect may be obtained in the complex between tolperisone and β-CD (Fig. 1).

Structures of (1) diphenhydramine, (2) clonidine and (3) tolperisone. Drawn in ChemDraw
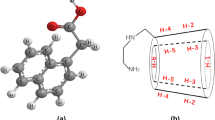
As for the host molecule, β-CD has been chosen because among the currently known encapsulating receptors for APIs, it stands out in several remarkable properties due to its structure [8]. β-CD is a cyclic oligosaccharide composed of seven D-glucopyranose units. The β-CD molecule has a shape of a truncated cone. Protons H-3 and H-5 are located in the inner hydrophobic bonding surface, and H-2 and H-4 are located in the outer one. The most important feature of β-CD is its ability to bind the guest molecule in its cavity in the aqueous environment (Fig. 2).

Structure of β-CD. Drawn in ChemDraw
One of the main methods of studying supramolecular inclusion complexes is NMR spectroscopy [8, 9]. We used this method to study new complexes of APIs diphenhydramine, clonidine, and tolperisone with β-CD.
According to [9], the 1H NMR spectrum of β-CD, obtained in DMSO-d6, consists of six groups of signals in the range 3.23–3.32; 3.45–3.53; 3.56–3.60; 4.47–4.49; 4.77–4.78; 5.66–5.73 ppm. Herein, we study the formation of inclusion complexes of β-CD with APIs 1–3 by determining the difference in the values of 1H and 13C chemical shifts of substrates (1–3) and the receptor (β-CD) in the free state and as a part of complexes due to the intermolecular interaction. By the magnitude of the chemical shifts of the internal or external protons of β-CD, it is possible to reveal the formation of, respectively, internal, external, or mixed complexes. Changes in the 1H and 13C chemical shifts in the spectra of the substrate make it possible to determine the direction of entry of the latter into the β-CD cavity or interaction with the outer segment of the cavity [12, 13].
Firstly, we explored the NMR data for compounds 1–3.
In the 1H NMR spectrum of 1 (Table 1), methyl protons H-12 and H-19 appeared as a six-proton singlet signal at 2.29 ppm. The alicyclic protons H-10 and H-9 resonated with two-proton quadruplets at 2.63 and 3.59 ppm., respectively, with a spin–spin coupling constant of 3 J 6.0 Hz. The tertiary proton H-7 appeared as a single-proton singlet at 5.39 ppm. In the aromatic region, one two-proton (7.22–7.27 ppm) and two four-proton multiplets (7.30–7.34 and 7.37–7.39 ppm) reflect resonation of aromatic protons H-4,16, H-2,6,14,18 and H-3,5,15,17, respectively.
In the 13C NMR spectrum (Table 1), aliphatic carbon atoms appeared at 46.13 (C-12, 19), 59.07 (C-10), 67.60 (C-9) and 84.14 (C-7). Aromatic carbon atoms resonated at 127.12 (C-2,6,14,18), 127.54 (C-4.16), 128.49 (C-3,5,15,17) and 142.40 (C-1,13) ppm.
The structure of 1 was also confirmed by two-dimensional NMR spectroscopy COZY (1H–1H) and HMQC (1H–13C), which make it possible to establish spin–spin interactions of homo- and heteronuclear nature. The observed correlations in the molecule are shown in Fig. 3. In the COZY 1H–1H spectra of the compound, spin–spin correlations are observed for the neighboring protons H9–H10 of the methylene groups of the aliphatic fragment through three bonds (cross-peaks at 3.59, 2.63 and 2.63, 3.59 ppm). Heteronuclear interactions of protons with carbon atoms through one bond were established using HMQC 1H–13C spectroscopy for the following pairs present in the compound: H12,19–C12,19 (2.29, 46.13), H10-C10 (2.63, 59.08), H9-C9 (2.63, 59.08), H7–C7 (5.38, 84.00) and H2-6.14–18–C2-6.14–18 (7.33, 128.52) ppm.

Correlations in the COZY (a) and HMQC (b) spectra of 1. Drawn in ACD/ChemSketch
Analysis of the 1H NMR spectra of compound 1 and supramolecular complex 4 showed (Table 1) that most of the protons change chemical shifts during complexation. Moreover, the greatest screening (− 0.18 − (− 0.20) ppm) is observed for two methyl-group protons H-12, H-19, and aliphatic protons H-10. Due to the superposition of the chemical shifts of the H-9 protons with more intense signals from the cyclodextrin molecule, aliphatic protons could not be detected in the supramolecular complex. All the aromatic protons undergo a significant shift to the upfield region (− 0.06 − (− 0.08) ppm) as a result of supramolecular interaction. The smallest change in the chemical shifts of the spectra is recorded for the tertiary proton H-7.
The supramolecular interaction of β-CD with 1 was accompanied by a shift of 5 of 6 considered cyclodextrin signals to the weak field region (0.01–0.03 ppm). The greatest difference (− 0.06 ppm) in the values of chemical shifts of protons was observed for the intracavitary proton H-3, located in the middle of the cyclodextrin cone. A significant change in the chemical shifts of the intracavitary H-5 proton located in the narrow side of the cyclodextrin rim, as well as the position of adjacent H-6 protons, confirms the formation of inclusion complexes. It can be assumed that the formation of inclusion complexes is accompanied by the entry of two hydrophobic phenyl radicals into the β-CD cavity. Changes in the chemical shifts of the intracavity protons of β-CD can also occur when methyl and methylene aliphatic protons of dimethylaminoethoxy fragment enter its cavity. The supramolecular interaction of 1 with external protons of β-CD is accompanied by a change in the chemical shifts of the latter and the formation of external complexes. Comparison of the integral intensities of the signals of protons 1 and β-CD in the composition of supramolecular complex 4 showed that supramolecular ensembles are mainly formed with the composition of one molecule 1 per one molecule of β-CD.
The study of the 1H NMR spectrum of 2 showed (Table 2) the presence of an intense four-proton singlet signal at 3.48 ppm (protons of the five-membered heterocycle H-2 and H-3). Heterocyclic and bridging amine protons H-4 and H-6 appeared as single-proton singlets at 5.33 and 7.32 ppm, respectively. The protons of the aromatics resonated with a one-proton triplet at 6.79 (H-10, 3 J 8.0 Hz) and a two-proton doublet at 7.23 (H-11 and H-9, 3 J 7.6 Hz), respectively.
In the carbon NMR spectrum of 2 (Table 2), the carbon atoms of the heterocyclic fragment appeared at 42.57 (C-2, C-3) and 158.08 (C-5) ppm. Aromatic carbon atoms appeared at 122.74 (C-10), 128.34 (C-9, C-11), 129.76 (C-8, C-12) and 145.16 (C-7) ppm.
The study of two-dimensional NMR spectra COZY (1H-1H) and HMQC (1H-13C) shows correlations in molecule 2 shown in Fig. 2. In the 1H-1H COZY spectra of the compound, spin–spin correlations are observed through three bonds of protons of neighboring methine groups H9 and H11 with H10 of the benzene ring by cross-peaks with coordinates at 7.23, 6.80 and 6.80, 7.23 ppm. Heteronuclear interactions of protons with carbon atoms through one bond were established using 1H-13C HMQC spectroscopy for the pairs present in the compound: H2 and H3 with C2 and C3 (3.48, 42.58) and H9 and H11 with C9 and C11 (7.22, 128.41) ppm (Fig. 4).

Correlations in the COZY (a) and HMQC (b) spectra of 2. Drawn in ACD/ChemSketch
The supramolecular interaction of β-CD with 2 was accompanied by a shift of all considered cyclodextrin 6 signals to the weak field region (0.01–0.03 ppm), and the greatest shift of proton signals is observed in both external (H-4, H-6) and internal (H-3, H-5) cyclodextrin protons. Significant changes in chemical shifts in complex 5 compared to molecule 2 occurred for the amine protons H-4 and H-6. The hydrophilic nature of the pyrazole fragment of molecule 2 allows us to conclude that this fragment cannot enter the inner cavity of β-CD. Therefore, there is a high probability of the formation of external supramolecular complexes of molecules 2 with β-CD. Changes in chemical shifts in the process of supramolecular complexation of aromatic protons H-9 and H-11 of 2 are much less noticeable than those of the analogous aromatic protons of molecule 1 in complex 4. This may indicate a lower complexation activity of the dichlorophenyl fragment during the formation of an internal complex. Comparison of the integral intensities of the signals of protons 2 and β-CD in the composition of supramolecular complex 5 showed that predominantly supramolecular ensembles with the composition of two molecules of 2 per one molecule of β-CD are formed.
The study of the 1H NMR spectrum of 3 showed (Table 3) the presence of a multiplet signal at 1.10–1.13 ppm with an integrated intensity of 3H of protons of the methyl group H-9. Piperidine protons appeared as multiplet signals at 1.30–1.31 (2H, H-3ax, H-5ax), 1.40–1.44 (4H, H-3 eq, H-4ax, H-4 eq, H-5 eq), 2.31–2.35 (4H, H-2ax, H-2 eq, H-6ax, H-6 eq) ppm. Aliphatic methylene protons H-7ax and H-7 eq appeared as single-proton multiplets at 2.31–2.35 and 2.75–2.81 ppm. The tertiary proton H-8 resonated with a single-proton multiplet at 3.62–3.70 ppm. Methyl protons H-18 resonated as a multiplet at 2.31–2.35 ppm. The equivalent aromatic protons H-14 and H-16 and H-13 and H-17 appeared as two-proton doublets with 3J 7.6 Hz at 7.19 and 7.81 ppm, respectively.
In the 13C NMR spectrum of 3 (Table 3), the carbon atoms of the methyl groups appeared at 17.00 (C-9) and 21.68 (C-18) ppm. The carbon atoms of the piperidine fragment appeared at 24.28 (C-4), 25.97 (C-3, C-5), and 54.94 (C-2, C-6) ppm. Alicyclic carbon atoms C-7 and C-8 appeared at 62.30 and 38.69 ppm, respectively. Aromatic carbon atoms appeared at 128.51 (C-13, C-17), 129.33 (C-14, C-16), 134.29 (C-12) and 143.66 (C-15) ppm. Carbonyl carbon atoms C-10 appeared in the lowest-field part of the spectrum at 203.63 ppm.
The structure of compound 3 was also confirmed by two-dimensional NMR spectroscopy COZY (1H–1H) and HMQC (1H–13C). The observed correlations in the molecule are shown in Fig. 5. In the 1H–1H COZY spectra, spin–spin correlations are observed through three bonds of protons of neighboring methylene groups H-3ax, H-5ax with H-4ax, H-4eq (1.31, 1.44 and 1.44, 1.31), H-3eq, H-5eq with H-2eq, H-6eq (1.43, 2.31 and 2.31, 1.43), H-2eq, H-6eq with H-7eq (2.34, 2.75 and 2.75, 2.34) of the piperidine and methylene fragments, methyl-methine groups H9–H8 (1.12, 3.68 and 3.68, 1.12) and methine groups H-14, H-16 with H-13, H-17 (7.20, 7.84 and 7.84, 7.20) of the aromatic substituent. Heteronuclear interactions of protons with carbon atoms through one bond were established using 1H-13C HSQC spectroscopy for the following pairs present in the compound: H-9 with C-9 (1.12, 17.44), H-3ax and H-5ax with C-3 and C-5 (1.31, 25.81), H-3 eq and H-5 eq with C-3 and C-5 (1.44, 25.81), H-18 with C-18 (2.36, 22.09), H-2ax and H-6ax with C-2 and C-6 (2.32, 55.22), H-7ax with C-7 (2.35, 62.11), H-7 eq with C-7 (2.79, 62.11), H-8 with C-8 (3.67, 38.65), H-13 and H-17 with C-13 and C-17 (7.83, 128.56), H-14 and H-16 with C-14 and C-16 (7.21, 129.49) ppm.

Correlations in the COZY (a) and HMQC (b) spectra of 3. Drawn in ACD/ChemSketch
The supramolecular interaction of β-CD with 3 was accompanied by a shift of all the considered six cyclodextrin signals to the weak field region (0.01–0.03 ppm), and the greatest shift of proton signals is observed in both external (H-4, H-6) and internal (H-3, H-5) cyclodextrin protons. Significant changes in chemical shifts in molecule 3 of complex 6 occurred in the piperidine protons H-2, H-6, and H-3, H-5. The equatorial components exhibit a more significant shift in comparison with the axial ones. In the aromatic fragment, the greatest change in chemical shifts is observed for the H-14, H-16 protons. Large chemical shifts also occur in the methyl protons H-9 and protons H-7ax and H-8. These results may indicate the formation of both internal and external complexes of β-CD with 3. There is a high probability of the formation of internal complexes with the entry of aromatic and piperidine fragments into the internal cavity of β-CD. In this case, aliphatic protons of molecule 3 can interact with external protons of cyclodextrin. Comparison of the integrated intensities of the signals of protons 3 and β-CD in the composition of supramolecular complex 6 showed that supramolecular assemblies are mainly formed with a composition of slightly less than one molecule 3 per one molecule of β-CD.
In order to construct the inclusion complexes, molecular docking was used. The binding mode of a guest to its host was investigated by the software AutoDock 4.0 [14]. The 3D structure of the host (β-CD molecule) was downloaded from the Protein Data Bank (PDB ID: 2V8L) [15]. In the downloaded.pdb file β-CD molecule is located in the “A” chain, the chain “B” was deleted. 3D structures of guest molecules (ligands)—diphenhydramine [16], clonidine [17], and tolperisone [18] were downloaded from PubChem databases in.sdf format and then converted into.pdbqt format in Open Babel software [19]. Gasteiger method was used to assign partial charges of host and guests molecules. The grid spacing was set at 0.0375 nm in each dimension, and each grid map consisted of 40^40^40 grid points. To find a globally optimized conformation and model the interaction between docked molecules (guest and host), the Lamarckian genetic algorithm (LGA) was implied. Fifteen runs were performed during each docking experiment. As for the inclusion complex between β-CD and clonidine (1:2), two docking processes were run consequently. Cluster analysis to docking results was performed to select a complex as a representative binding mode. Complexes were selected by the lowest docking energy.
Possible structures of the inclusion complexes between β-CD and diphenhydramine (1a), clonidine (1b), and tolperisone (1c) are presented below in Fig. 6.

Possible structures with lowest docked energies of the inclusion complex between β-CD and (a) diphenhydramine (1:1); (b) clonidine (1:2); (c) tolperisone (1:1). Drawn in AutoDock 4.0
2 Experimental part
99% purity β-CD (manufactured by Fluka) was used in the experiment. 1H and 13C NMR spectra of substrates (1–3) and β-CD and their supramolecular complexes (4–6) were recorded in DMSO-d6 and d-chloroform on a JNM-ECA 400 spectrometer (400 MHz for 1H and 100 MHz for 13C nuclei) by Jeol (manufactured in Japan). Chemical shifts were measured relative to residual protons or carbons of deuterated dimethyl sulfoxide. In all experiments, the temperature was maintained at 298 K, and standard 5 mm NMR tubes were used.
To obtain inclusion complexes of 1–3 with β-CD, the method of coprecipitation from a water–ethanol solution was implied. An ethanol solution of API (0.5 g) was added dropwise to an aqueous solution of β-CD (1:1 mol ratio) at 40–50 °C, at a dropping rate of 1 drop per minute, the mixture was stirred for 20–30 min and then left to evaporate naturally to produce inclusion complexes.
3 Conclusion
In this study, we have shown that diphenhydramine, clonidine, and tolperisone interact with β-CD with the formation of stable 1:1 stoichiometric complexes for β-CD:diphenhydramine and β-CD:tolperisone, and 1:2 stoichiometric complex for β-CD:clonidine. The chemical shifts in the 1H and 13C NMR spectra of the inclusion complexes showed similar characteristics with slight differences compared to their parent molecules. This study provides the idea to support the application of β-CD as a key tool to enhance the pharmaceutical and pharmacological aspects of diphenhydramine, clonidine, and tolperisone and to turn these widely available APIs into more effective drug forms.
Data availability
All data generated or analysed during this study are included in this published article.
References
Domingos S, André V, Quaresma S, Martins ICB, Fátima M, Piedade M, Duarte MT (2015) New forms of old drugs: improving without changing. J Pharmacy Pharmacol. https://doi.org/10.1111/jphp.12384
Uekama K, Hirayama F, Irie T (1998) Cyclodextrin drug carrier systems. Chem Rev. https://doi.org/10.1021/cr970025p
Jain R, Segal S, Kollins SH, Khayrallah M (2011) Clonidine extended-release tablets for pediatric patients with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. https://doi.org/10.1016/j.jaac.2010.11.005
Opota DO, Joachim G, Kalantzis G, Piccerelle P, Reynier JP, Joachim J (1999) Controlled-release behavior of diphenhydramine hydrochloride loaded neutral microgranules and coated using ethylcellulose water dispersion. Drug Dev Ind Pharm. https://doi.org/10.1081/DDC-100102145
Antal I, Kiss D, Orgovan G, Stiedl B, Zelko R, Klebovich I, Noszal B (2012) EU Patent EP2175839B1. https://patents.google.com/patent/EP2175839B1/zh-CN
Adhikari S, Daftardar S, Fratev F, Rivera M, Sirimulla S, Alexander K, Boddu SHS (2018) Elucidation of the orientation of selected drugs with 2-hydroxylpropylβ-cyclodextrin using 2D-NMR spectroscopy and molecular modeling. Int J Pharm 545(2018):357–365. https://doi.org/10.1016/j.ijpharm.2018.05.016
Braga MA, Martini MF, Pickholz M, Yokaichiya F, Franco MKD, Cabeca LF, Guilherme VA, Silva CMG, Limia CEG, de Paula E (2015) Clonidine complexation with hydroxypropyl-beta-cyclodextrin: from physico-chemical characterization to in vivo adjuvant effect in local anesthesia. J Pharma Biomed Anal. https://doi.org/10.1016/j.jpba.2015.11.015
Nowakowski M, Ejchart A (2014) Complex formation of fenchone with α-cyclodextrin: NMR titrations. J Incl Phenom Macrocycl Chem. https://doi.org/10.1007/s10847-013-0356-4
Maheshwari A, Sharma M, Sharma D (2013) Complexation of sodium picosulphate with beta cyclodextrin: NMR spectroscopic study in solution. J Incl Phenom Macrocycl Chem. https://doi.org/10.1007/s10847-012-0251-4
Rasheed A, Kumar ASK, Sravanthi VV (2008) Cyclodextrins as drug carrier molecule: a review. Sci Pharm. https://doi.org/10.3797/scipharm.0808-05
Schneider HJ, Hacket F, Rudiger V, Ikeda H (1998) NMR studies of cyclodextrins and cyclodextrin complexes. Chem Rev. https://doi.org/10.1021/cr970019t
Pirnau A, Floare CG, Bogdan M (2014) The complexation of flurbiprofen with β-cyclodextrin: a NMR study in aqueous solution. J Incl Phenom Macrocycl Chem. https://doi.org/10.1007/s10847-012-0277-7
Kemelbekov U, Saipov A, Abdildanova A, Ospanov I, Luo Y, Guskov W, Saenger W, Imachova Sh, Nasyrova S, Pichkhadze G (2013) Structure and pharmacological studies of the anaesthetic 1-(3-n-butoxypropyl)-4-benzoyloxypiperidin hydrochloride and its complex with β-cyclodextrin in solution. J Incl Phenom Macrocycl Chem. https://doi.org/10.1007/s10847-012-0239-0
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem. https://doi.org/10.1002/jcc.21256
Tung JY, Liu YY, Sun YJ (2008) Carbohydrate-binding of the starch binding domain of Rhizopus oryzae glucoamylase in complex with beta-cyclodextrin and maltoheptaose. Biochem J. https://doi.org/10.1042/BJ20080580
National Center for Biotechnology Information (2021) PubChem Compound Summary for CID 3100, Diphenhydramine. https://pubchem.ncbi.nlm.nih.gov/compound/Diphenhydramine. Accessed 2 Aug 2021
National Center for Biotechnology Information (2021) PubChem Compound Summary for CID 2803, Clonidine. https://pubchem.ncbi.nlm.nih.gov/compound/Clonidine. Accessed 2 August 2021
National Center for Biotechnology Information (2021) PubChem Compound Summary for CID 5511, Tolperisone. https://pubchem.ncbi.nlm.nih.gov/compound/Tolperisone. Accessed 2 August 2021
O’Boyle N, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open babel: an open chemical toolbox. J Cheminf. https://doi.org/10.1186/1758-2946-3-33
Funding
This research is funded by the Ministry of Education and Science of the Republic of Kazakhstan (Grant No. AP08857345).
Author information
Authors and Affiliations
Contributions
SZ, AT, TZ, VY, TS, AB, SB, MA and AZ have contributed equally.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhumakova, S., Ten, A., Zharkynbek, T. et al. NMR study of the inclusion complexes of β-cyclodextrin with diphenhydramine, clonidine and tolperisone. SN Appl. Sci. 4, 75 (2022). https://doi.org/10.1007/s42452-022-04958-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-022-04958-5