
Abstract
The present application wish to seem at the event of validation of bio analytical method and pharmacokinetic study of selexipag and its related impurities in rat plasma using LC–MS/MS. The optimized method contains gradient elution of selexipag with a flow rate of 1 ml/min and X-Bridge phenyl column (150 × 4.6 mm, 3.5 µ). A buffer of 1 mL formic acid in l liter water and acetonitrile mixture is used as mobile phase. 30 min run time was used for separation of selexipag and its related impurities with Ambrisentan as internal standard and impurity-D as active metabolite. The linearity curves are linear in between the percentages of 10 to 200% of rat plasma and R2 value of each analyte was observed as 0.999. This application denotes all the parameters like precision, accuracy, recovery and stability were got the results within the limit of USFDA guidelines. This method applies effectively for the investigation of pharmacokinetic studies using rat plasma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
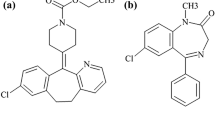
Selexipag is obtained from Actelion [1] is employed pulmonary arterial hypertension (PAH) [2, 3] to slow down disease development and decrease the possibility of hospitalization. PAH may be a comparatively rare disorder with normally a poor forecast needing more caring options to extend long term out comes. Retailed by Actelion drug under name Uptravi. Selexipag and its active metabolite are agonists [4, 5] of the professional stacycline receptor, which results in vasodilation [6] within the circulation [7, 8] and reduce elevated pressure within the blood cells carrying blood to the lungs. Selexipag has been related to a coffee rate of anti-body adjuvant elevations during treatment, but has yet to be indicated in cases of scientific evident acute liver wound [9]. Europe country selexipag used as strong inhibitors in liver enzyme in gemfibrozil [10, 11] is contraindicated because it increases concentrations of selexipag two fold and its working metabolite 11 fold, potentially resulting in more adverse effects. The adverse effects of selexipag are almost like those of intravenous prostacyclin[12] used for pulmonary arterial hypertension. General side effects are head ache, and jaw pain, hyperthyroidism [13] has also been noted in people taking selexipag. Bio analysis is area of analytical chemistry [14] in quantitative measurement of xenobiotics [15, 16] and biotics in biological systems. The chemical structure of Selexipag as shown in Fig. 1. Bio analytical common techniques are, hyphenated techniques: LCMS, GCMS, LC-DAD, CE-MS. Chromatographic methods: HPLC, GC, UPLC super critical fluid chromatography. It is deals with organictestscarrying the substance alongside a various range of chemicals which will have an unfavorable effect on the exact and specific amount of the analyte. As such a good range of techniques are applied to extract the analytes from its matrix. It is having protein precipitation [17], liquid–liquid extraction [18], solid phase extraction [19]. Selexipag yields it active metabolite [20] by hydrolysis of the acylsulfonamide by the enzyme hepatic carboxylesterase [21]. Oxidative metabolism catalyzed by CYP3A4 and CYP2C8 results in hydroxylated [22] and dealkylated products. UGT1A3 and UGT2B7 are involved in the glucuronidation [23] of the active metabolite, other metabolites in circulation do not exceed 3% of the total drug related material. In fact, last few years, we are working on formulations, validation and degradation study of various drugs [24,25,26]. In this study, we focused on method development and validation on new a LCMS/MS method for the quantification of slexipag and related impurities along with their pharamcokintics studies.

Structural representation of selexipag
2 Materials and method
2.1 Chemicals and reagents
The reference standards of selexipag (purity-99.9%) and its related impurities (purity-99.9%) were got from Candila health care ltd, Ahmedabad, India. HPLC marked acetonitrile, formic acid were got from Merck (India), Worli, Mumbai, India. HPLC grade water was gained from Milli Q (Milli Q system, USA) water purification. Rat plasma was obtained from Bharat biotech Ltd, Hyderabad.
2.2 Equipment
Waters, Alliance e2695 model HPLC provided with column oven, auto sampler and degasser was operated for analysis. The HPLC system was coupled to SCIEX QTRAP 5500 mass spectrometer equipped with electro spray ionization interface. SCIEX software was utilized for the interpretation of the data of chromatograms. In the separation and validation of selexipag a column of X-Bridge phenyl is used.
2.3 Chromatographic conditions
2.3.1 HPLC conditions
A mixture of buffer (1 ml of formic acid in 1 L of water) and acetonitrile in a gradient mode was operated as mobile phase with a movement of 1 ml/min. In this validation gradient elution with an injection volume 10 µl is used.
2.3.2 LC/MS/MS conditions
Multiple reaction monitoring mode of mass spectrometer with positive ion mode electro spray ionization was used in the separation of selexipag and its impurities. Collision energy of 15 and 14 V, source temperature of 550 °C, ion spray voltage 5500 V, drying gas temperature of 120–250 °C, collision gas of nitrogen, entrance and exit potentials of 10 V, 7 V and dwell time of 1 s was used in mass spectrometer.
2.4 Preparation of standard and quality control samples
The standard solution of selexipag (50 ng/ml), impurity-1 (0.5 ng/ml), impurity-A (50 pg/ml), impurity-B (0.26 ng/ml), impurity-C (20 pg/ml), impurity-D (active metabolite) (0.56 pg/ml), impurity-E (1 ng/ml), n-oxide (10 pg/ml), and SE-60 (20 pg/ml) was made by diluting with diluents. Prepared standard solutions were stored at 4 °C and bring back to room temperature before using them.
2.5 Preparation of sample solution
The sample solution was prepared by mixing 200 µl of plasma, 800 µl of acetonitrile, 500 µl of internal standard and 500 µl of standard stock. To precipitate all the proteins, mix in vortex cyclomixture. Centrifuge at 400 rpm for 20 min. Collect the supernatant solution in HPLC vial and inject into the chromatogram.
2.6 Method validation
2.6.1 Selectivity
The optimized LC–MS/MS technique was studied by examining six lots of individual plasma samples. The chromatograms of spiked plasma samples at LLOQC amount were differentiate to above mentioned blank plasma samples.
2.7 Matrix effect
The matrix effect of selexipag along with its impurities and IS was assessed by comparing the area of the peak ratio in the extracted plasma sample taken from six different rats. It was studied at LQC and HQC levels in three replicates.
2.7.1 Dilution integrity
Dilution integrity should be exhibited by doping the matrix with substance accumulation exceeding the ULOQC and mixing this selected sample with blank matrix.
2.7.2 Precision and accuracy
Same day accuracy and precision were tested in six replicates in a single set using samples from HQC, MQC, LQC and LLOQC levels. The inter-day precision and accuracy were tested by analyzing HQC, MQC, LQC and LLOQC concentration samples on three successive separate batches. The precision was expressed by percent CV and accuracy by percent recovery.
2.7.3 Carry over
The analyte retained by the chromatographic system during the injection of a sample that appears in subsequent blank or unknown samples.
2.7.4 Recovery
The withdrawal efficiencies of selexipag and its impurities were calculated by examination of six duplicates at each quality control concentration. The percentage recovery was evaluated by peak responses of extracted standards to the peak responses of non-extracted standards.
2.7.5 Stability
Stability is done by area response and internal standard in the stability sample with the area response of typical prepared from new solution. Stability studies were carryout in low and high quantity accumulation levels using six preparations in every level. Samples are stable below 15% as per USFDA guidelines. Bench top stability samples were stored at 2–8 °C in auto sampler tested up to 24 h. Freeze thaw stability samples are frozen at −30 °C and thawed three times compared to freshly prepared control sample. Six different samples of LQC and HQC levels are used for freeze thaw stability. Wet extract stability samples are stored temperature below 10 °C for 24 h.
3 Results and discussion
The optimization of instrument to offer sensitivity and signal stability throughout in fusion of the analyte within the continuous flow of movable phase to electro spray ion source operated at both polarities at flow of 10 µl/min. Selexipag and its impurities give more response in positive ion mode in comparison with negative ion mode.
To obtain the simplest chromatographic condition, different columns like C18, C8 and CN-propyl and mobile phases composed of 0.1% OPA and acetonitrile were tested. The simplest chromatographic separation occurred on X-Bridge phenyl column with a movable phase of acetonitrile and 0.1% formic acid with a gradient elution rate of 1 ml/min. The mass spectras of Selexipag and their internal standard Ambrisentan was shown in Fig. 2a and b.

a Mass spectra of Selexipag b Mass spectra of Ambrisentan (IS)
3.1 Method validation
3.1.1 Selectivity and sensitivity
Blank plasma and spiked plasma with LOQ sample in of selexipag and its impurities. The % interference of retention time of analytes between six different lots of rat plasma, including hemolyzed and lipedemic plasma containing K2EDTA as an anti-coagulant for selexipag respectively, it is within the acceptable criteria. Six replicates of extracted samples at LLOQC level in one of the plasma sample having least interference at the retention time of selexipag was prepared and analyzed. The % CV of the area ratios of these six replicates of samples was 1.54% of selexipag. The blank and LLOQC chromatogram was shown in Figs. 3 and 4.

Blank plasma chromatogram of selexipag

LLOQC chromatogram for selexipag and its related impurities
3.1.2 Matrix effect
The ion suppression/enhancement % CV in the signal was observed to be 1.0% at MQC level for selexipag and its impurities. It indicates that the matrix effect on the ionization of analyte is within the allowable limit.
3.1.3 Linearity
From the calibration curve it was clear that the peak response ratios were proportional to the concentration. The regression coefficient of selexipag and its impurities were observed to be ≥ 0.999. Linearity of Selexipag and their impurities concentration vs area ratio values are shown in Table 1 and their calibration curves are shown in Fig. 5.

Calibration Curves for a selexipag; b Selexipag Imp-1; c Selexipag Imp-A; d Selexipag Imp-B; e Selexipag Imp-C; f Selexipag Imp-D (active metabolite); g Selexipag Imp-E; h Selexipag Imp n-oxide; i Selexipag Imp SE-60
3.1.4 Prediction linearity
From the calibration curve it was clear that the peak response ratios were proportional to the concentration. In this prediction linearity it was observed that the regression coefficient of selexipag and its impurities were observed to be within the allowable limit. (R2 = 0.999). Prediction linearity of Selexipag and their impurities concentration vs area ratio values are shown in Table 2 and their calibration curves are shown in Fig. 6.

Calibration Plot for a Selexipag; b Impurity-1; c Impurity-A; d Impurity-B; e Impurity-C; f Impurity-D; g Impurity-E; h Impurity-h; i Impurity SE-60 prediction linearity
3.1.5 Precision and accuracy
The inter-run and accuracy were calculated by merging all separate results of reproduce quality control over five individual batch runs examined on four dissimilar days. The % CV of inter-run precision was < 5% and the value of inter-run accuracy was in the range of 85–115% for selexipag and its impurities. Precision and accuracy results are tabulated in Table 3.
3.1.6 Recovery
For recovery determination LQC, HQC and MQC concentration levels for selexipag and its impurities were prepared and obtained areas for extracted samples of the same accumulation levels from a precision and accuracy batch run on a same day.
3.1.7 Carry over
System error that may affect the measured value of the sample is called carry over. Samples carry over on a LC–MS/MS system configured with waters alliance was evaluated using the following procedure. A system blank injection of 10 µl 0.1% formic acid and acetonitrile in a gradient mode was made on to a waters Zspray triple quadrupole mass detector using flow injection analysis. From this we can say that it does not affect the accuracy and precision of the proposed method. Sample carry over was intimated as both % carry over and nL carry over.
3.1.8 Re-injection and reproducibility
Throughout real subject sample examination, re injection re-reducibility was performed to check the instrument after hard ware deactivation due to any instrument failure. At LQC and HQC levels the changes were less than 2.0, hence throughout real subject test examination the batch was re injected in the case of instrument miscarriage. Samples were prepared and re injected after 24 h, shows % change should be less than 2.0 at LQC and HQC levels. Hence inside the case of instrument failure the sample can be re injected after 24 h.
3.1.9 Stability
In solution stability analysis, selexipag and its impurity solutions were prepared with diluents and put in storage at 2–8 °C in a refrigerator. Fresh stock solutions were related to aged stock solutions prepared earlier 24 h. From the values of % changes of selexipag and its impurities, it is clear that the stock solutions are stable up to 24 h when stored in 2–8 °C. Bench top and auto sampler stabilities were observed at LQC, MQC and HQC levels.
At room temperature the selexipag and its impurities were steady in plasma for 24 h, and in auto sampler at 20 °C for 24 h. From this it was confirmed that, at LQC and HQC levels repeated freezing and thawing of plasma samples spiked with selexipag and its impurities didn’t influence their stabilities. From long term stability it was clear that selexipag and its impurities were stable at 24 h at a storage temperature of −30 °C. The stability data are shown in Table 4.
3.1.10 Pharmacokinetic study
The method has been validated successfully to quantify the concentration of selexipag and its impurities in 6 different groups of rats were administered of selexipag and its impurities sample as an oral dose, under fasting condition. After injecting the drug samples into a rat body, collect the samples at selected intervals of time like 0, 1, 2, 2.5, 3, 4 and 6 h from the rat body. After that as per test method sample is prepared and injected into the chromatographic system and record the values. The evaluated pharmacokinetic parameters were Cmax, tmax, and t1/2 were calculated. Pharmacokinetic results were shown in Table 5 and there plots are shown in Fig. 7.

Recovery plot of selexipag (a) and its impurities, active metabolite (b, c)
4 Conclusion
For the first time higher sensitive HPLC-LC–MS/MS method was evolved and validated for the determination of selexipag and its impurities in rat plasma. Here the described method is fast, rugged and reproducible bio analytical method. Easy and systematic method was developed and can be used in pharmacokinetic studies and to check the investigated analyte in body fluids.
References
US FDA (2019) Seeks additional data from Actelion's NDA for Opsumit to treat CTEPH (NDA 204410/S-020). PharmaBiz. Athena information solutions PVT. Ltd, South San Francisco, California, USA
Hadinnapola C, Bleda M, Haimel M, Screaton N, Swift A, Dorfmuller P et al (2017) EIf2AK4 mutation carriers in a large cohort of patients diagnosed clinically with pulmonary arterial hypertension. Circulation 136(21):2022–2033
George MP, Champion HC, Pilewski JM (2011) Lung transplantation for pulmonary hypertension. Pulm Circ 1(2):182–191
Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, Devree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M et al (2013) Structure and function of an irreversible agonist-β (2) adrenoceptor complex. Nature 469(7329):236–40
De Min A, Matera C, Bock A et al (2017) A new molecular mechanism to engineer protean agonism at a G-protein coupled receptor. Mol Pharm 91(4):348–356
Nisha Charkoudian (2010) Mechanisms and modifiers of reflex induced cutaneous vasodilation and vasoconstriction in humans Journal of applied physiology. Am Physiol Soc 109(4):1221–1228
Elmaghawry M, Zanatta A (2014) Zampieri F (2014) The discovery of pulmonary circulation from imhotep to William Harvey. Glob Cardiol Sci Pract 2:103–116
Akmal M, Zulkifle M, Ansari A (2010) Ibn nafis, a forgotten genius in the discovery of pulmonary blood circulation. Heart Views Off J Gulf Heart Assoc 11(1):26–30
Segura Sampedro JJ, Pinenoflores C et al (2019) New hemostatic device for grade IV, V liver injury in porcine model a proof of concept. World J Emerg Surg 14(1):58
Rodney G, Uhlendorf P, Max well RE (1976) The hypolipidaemic effect of gemfibrozil in laboratory animals. Proceedings of the royal society of medicine 69 suppl 2:6-10
Doummar J, Aoun M (2018) Assessment of origin and transport of four selected emerging micro pollutants sucralose, Acesulfame K, Zemfibrozil and iohexol in a karst spring during a multi event spring response. J Contam Hydrol 215:11–20
Hache M, Denault A, Belisle S, Robitaille D, Couture P et al (2003) Inhaled epoprostenol (prostacycline) and pulmonary hypertension before cardiac surgery. J Thorac Cardiovasc Surg 125(3):642–649
Deveruaex D, Tewelde SZ (2014) Hyperthyroidism and thyrotoxicosis. Emerg med clin North Am 32(2):277–292
Skoog Douglas A, West, Donald M, Holler F, James, Crouch SR (2014) Fundamentals of analytical chemistry, 9th edn. Brooks/Cole, Cengage Learning, Belmont, CA
Mansuy D (2013) Metabolism of xenobiotics, beneficial and adverse effects. BiolAujourdhui 207(1):33–37
Kun Lu, Mahbub R, Fox JG (2015) Xenobiotics interaction with the intestinal microflora. ILAR J 56(2):218–227
Zellner M, Hayden H, Diestinger M, Eliasen M et al (2005) Quantitative validation of different protein precipitation methods in proteome analysis of blood platelets. Electrophoresis 26(12):2481–2489
Marcilla A, Reyers L, Juan A, Olaya M (2017) Should be trust all published LLE correlation parameters in phase equilibria?. Necessity of their assessment prior to publication. Fluid Phase Equilib 433:243–252
Augusto F, Hantao LW, Mogollon NGS, Braga SCGN (2013) New materials and trends in sorbents for solid phase extraction. TrAC, Trends Anal Chem 43:14–23
Obach SR, Esbenshade TA (2013) Pharmacologically active drug metabolites, impact on drug discovery and Pharmacotherapy. Pharmacol Rev 65(2):578–640
Aranda J, Cerqueira NMFSA, Fernandes PA, Roca M, Tunon I, Ramos MJ (2014) The catalytic mechanism of carboxylesterases A computational study. Biochemistry 53(36):5820–5829
Huang X, Groves JT (2017) Beyond Ferryl mediated hydroxylation 40 years of the rebound mechanism and C-H activation. JBIC J Biol Inorg Chem 22(3):185–207
Liston H, Marcowitz J, Devane C (2001) Drug glucuronidation in clinical psychopharmacology. J Clin Psychopharmacol 21(5):500–515
Amara Babu NLA, Srinivas B, Sreeram V, Rao KP et al (2020) Development and validation for simultaneous estimation of perindopril and Indapamide by UPLC–UV in tablet dosage form. Res J Pharm and Tech 13(1):287–292
Amara Babu NLA, Koganti K, Palakeeti B, Srinivas KS, Rao KP et al (2020) Development and validation of UPLC method for separation and determination of rivaroxaban and its related substances in bulk drugs. Drug Invent Today 13(4):611–618
Amara Babu NLA, Koganti K, Palakeeti B, Srinivas KS, Rao KP et al (2020) Bioanalytical LC-MS/MS method for determination and comparison of Selexipag Assay in various Biological materials and its application to Pharmacokinetics studies in Rat plasma. Int. J. Res. Pharm. Sci 11(2):2210–2220
Acknowledgements
Namburi. L. A. Amara babu thanks VFSTR for the research avenues and facilities. Dr. K. P. Rao Thanks DST-SERB, for financial support under project for early career, project no. EMR/2014/001114.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Authors declare that there was no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rao, K.P., babu, N.L., Koganti, K. et al. Related substances method development and validation of an LCMS/MS method for quantification of selexipag and its related impurities in rat plasma and its application to pharmacokinetic studies. SN Appl. Sci. 3, 321 (2021). https://doi.org/10.1007/s42452-021-04219-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-021-04219-x