
Abstract
This study was conducted to understand the environmental behavior of mercury released by artisanal and small-scale gold mining (ASGM) activities. For this purpose, we attempted to assess the effect of diffused mercury on mercury concentrations in soil, demonstrate the presence of methylmercury in soil affected by the deposited mercury and determine the reactions associated with methylmercury production. The vertical profiles of mercury were obtained from two sites in the forest of the ASGM village in Pongkor (West Java, Indonesia) and from two sites in Mount Halimun-Salak National Park, which is approximately 12 km from the ASGM village. The highest total mercury concentration, 8.9 mg kg−1, was observed for soil samples collected at the ASGM village. The mercury was concentrated at the surface or in the subsurface layers, and the concentrations were several times to more than ten times higher than the lowest values observed in the deeper layers at each site. Even in the national park, the highest concentration of 1.9 mg kg−1 was observed in the upper soil layer. These results suggest that the primary source of mercury in the forest soil is atmospheric deposition; fallen plant leaves also deliver accumulated mercury to the soil surface. The organic mercury percentages of the total mercury were 0.2 ± 0.1% for the national park and 0.3 ± 0.2% for ASGM sites. The vertical variation in organic mercury concentration did not always match that in total mercury concentration, which suggested that the formation of methylmercury in soil was closely related to the decomposition of organic matter near the surface. The soil surface is an important reaction field for methylmercury production in forested areas.
Similar content being viewed by others


1 Introduction
The behavior of mercury discharged into the environment from both natural and anthropogenic sources has become a global concern. A volatile gaseous mercury form, presumably elemental mercury, Hg0, has an atmospheric residence time of at least a few months, perhaps even 1 or 2 years [1]. Mercury emitted into the atmosphere can disperse over a wide area and settle on soil surfaces through wet and dry deposition; oxidized mercury and particulate mercury are more readily deposited, with relative rapidity after their formation [1, 2].
The deposited mercury may be taken up by microorganisms and converted to methylmercury (MeHg). MeHg is one of the most toxic chemical forms of mercury, and MeHg can accumulate in the ecosystem at levels that constitute a health hazard [3, 4]. Thus, the soil is an important reservoir of discharged mercury and a critical medium for understanding the dynamics of mercury that is released into the environment [5].
In recent years, the use of mercury in artisanal small-scale gold mining (ASGM) has increased in many developing countries; consequently, ASGM has become a major source for emissions and releases of mercury worldwide [2]. Emissions from ASGM are estimated to comprise 37% of anthropogenic emissions. There are many reports on the effects of mercury released from ASGM on workers and local residents [6,7,8,9]. ASGM workers and their families are exposed to mercury vapor, and workers, workers’ families and residents of nearby and downstream communities consume fish heavily contaminated by MeHg [6]. Currently, mercury exposure appears to be the inhalation of volatile mercury from the atmosphere rather than the consumption of MeHg-contaminated food (rice and fish) [8]. On the other hand, although the inhalation of volatile mercury can be drastically decreased by stopping the use of mercury in gold mining, the mercury dispersed into the environment will not disappear. The distribution of mercury in the surrounding environment is important. The mercury emitted into the atmosphere may deposit on surfaces around the ASGM site, and soil is an important receiver of such mercury; the deposited mercury can be subjected to methylmercury formation in soil. Contaminated soil may become one of the continuous sources of mercury for the ecosystem, and the relative importance of exposure via the consumption of contaminated food may increase in the future. Some studies have reported the distribution of mercury in soil around ASGM regions [10, 11]. As described in these studies, the soil surface receives mercury from the air and serves as the field of contact between mercury and the plants and animals in the respective area. On the other hand, there are few reports on the detailed vertical variation in mercury and soil chemistry, and the reports on MeHg concentration in soil around ASGM sites are still limited [12,13,14]. The soil surface is a dynamic field where material supply and degradation proceed, and vertical variation in mercury species may provide important information for tracking the effects of such variation.
The purpose of this study is to assess the impact of mercury emitted by ASGM activity on mercury concentrations in soil and to follow MeHg formation in soil. For this purpose, soil samples were collected around an ASGM site in West Java, Indonesia, at surface depths of 0–20 cm, and the vertical variation in total mercury (T-Hg) and organic mercury (org-Hg) concentrations in soil was measured. The relation between MeHg production and the decomposition of organic matter is discussed based on the variation in nitrogen, organic carbon content and soil chemical composition. The results of this study offer both basic and significant information that will enable an understanding of the dynamics of mercury released by ASGM activity.
2 Methods and materials
2.1 Sampling
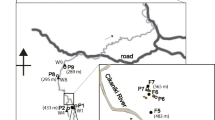
The study area is located in the Nanggung subdistrict of Bogor Regency, West Java, Indonesia. Since the Pongkor gold–silver deposits were discovered in 1981, there has been much ASGM activity in this area. The sampling points are shown in Fig. 1. The mining work sites in this area are mainly located in the villages. Gold is extracted from ore by traditional methods. Approximately 20 to 40 kg of crushed gold ore is added to an iron ball mill together with 0.3 to 0.5 kg of mercury for whole-ore amalgamation. A series of small mills runs for 4 to 8 h, and then, the product is discharged. The amalgam and gold–unbound mercury are separated from the tailings by panning. After removing the excess mercury by squeezing in a piece of cloth, the amalgam is burned in an open pan using a gasoline–air torch and the mercury is emitted into the atmosphere, contaminating miners, their families and neighbors. The mercury-contaminated tailings are either dumped into rivers or sold to processing plants that leach the residual gold with cyanide. The whole-ore amalgamation process loses an average of 50% of the initial Hg and recovers less than 30% of the gold in the ore [8, 15].

Map of the study area
Surveys about the total amount of mercury used in this region were not possible because there were places that we could not enter. From time to time, police enter the village, and workers are detained, so workers are wary of visitors. Therefore, it is unknown how much mercury has been emitted into the environment. The amount of mercury dumped with tailings into the river by the local miners was estimated to be approximately 2 kg h−1 in the daytime [16], which leads one to believe that 10–20 tonnes/a of mercury has been released and emitted into the region where the artisanal miners are active. Contamination of the river water, sediments [17,18,19,20,21] and atmosphere [22, 23] around this area has been reported.
The soil samples were collected from two sites in the forest of the ASGM village (approximately 0.2–0.5 km from the nearest workplace) and from two sites in Mount Halimun-Salak National Park, which is approximately 12 km from the ASGM site. The altitude difference between the ASGM village and the national park is approximately 500 m. The samplings were conducted in March and August of 2014. At each site, after collecting the fallen leaves from a 20 × 20 cm sample area, soil samples were collected at 2-cm intervals from the surface to 20 cm below the surface.
The samples were placed in plastic bags with seals and were imported to Japan with the permission of the Minister of Agriculture, Forestry and Fisheries in accordance with plant protection law. The samples were freeze-dried, ground in an agate mortar, and preserved for determination of their T-Hg and org-Hg concentrations, total organic carbon (TOC) and total nitrogen (TN) contents and soil chemical compositions.
2.2 Mercury measurements
The T-Hg concentrations were determined using the method described by Akagi and Nishimura [24] with modifications by Akagi et al. [25]. The precision and accuracy of the method have been repeatedly verified by interlaboratory calibration exercises [26, 27], including the analysis of reference standards (e.g., IAEA 085 and 086). In the present study, the accuracy of the T-Hg measurement was also verified on the certified reference material CRM 7302-a (marine sediment) prepared by the National Institute of Advanced Industrial Science and Technology (AIST), Japan. The values determined by this method of 0.51 ± 0.02 mg kg−1 (n = 8) agreed well with the reference value of 0.52 ± 0.03 mg kg−1.
The procedure for the T-Hg measurement is as follows. A known amount of sample (0.1–0.5 g) was placed in a 50-mL volumetric flask. After adding 1 mL of pure water, 2 mL of a 1:1 nitric acid–perchloric acid solution and 5 mL of concentrated sulfuric acid, the flask was left to stand for a few minutes and then heated on a hot plate at 230 °C for 20 min. After cooling, the volume of the digested sample was adjusted to 50 mL with water, and a suitable aliquot of the resulting solution (< 10 mL) was analyzed for its mercury content by cold vapor atomic absorption spectrometry (CVAAS) using a semiautomated mercury analyzer (Model Hg-201, Sanso Seisakusho Co., Ltd., Japan). Separately, 1.0 mL of water and the standard mercury solution were transferred into three sample digestion flasks and treated by the above-mentioned procedure, to obtain blank and standard test solutions for measuring the total mercury concentration. The absolute limit of detection, calculated as triple the standard deviation of the blank reading (n = 10), was 0.02 ng; on 0.2 g of sample, the detection limit was 0.1 μg kg−1. The repeatability was confirmed with duplicate measurements of the soil samples, and the variation ranged from 0.3 to 11.5% (mean, 3.9%).
The org-Hg concentrations were determined by the method of Tomiyasu et al. [28] with some modifications [29]. This method does not require special equipment; mercury species were separated by solvent extraction and measured by CVAAS. In natural soil, not only methylmercury but also ethylmercury can be present [30]. Thus, by this method, the total amount of these organic mercury species will be determined. The procedure for org-Hg measurement is as follows. A 1.0-g sample of each soil was placed in a centrifugation tube for org-Hg determination. After adding 0.4 g of CuCl and 10 mL of 1 M HCl (3% NaCl) solution, the tube was shaken for 20 min and then centrifuged for 10 min at 2000 rpm. The resulting supernatant was poured into another 50-mL centrifugation tube. The extraction was repeated in another 10 mL of 1 M HCl (3% NaCl) solution, and the second supernatant was combined with the first. To the combined HCl solutions in the centrifugation tube, 10 mL of toluene was added, and the tube was shaken for 10 min. After centrifugation for 2 min at 2000 rpm, the HCl solution was discarded and the remaining toluene was cleaned by adding 5 mL of 1 M HCl (3% NaCl) solution to the tube and shaking for 5 min. The mixture was centrifuged for 2 min at 2000 rpm, and 7 mL of the toluene was collected into a 10-mL tube with a cap. After adding 2 mL of 0.1% cysteine solution, this tube was shaken for 2 min and centrifuged for 2 min at 1200 rpm. The toluene layer was discarded and 1.5 mL of the cysteine layer was digested with HNO3–HClO4–H2SO4 and the organic mercury was measured by the same procedure as that was employed for the total Hg determination. To prepare the calibration curves, aliquots of MeHg standard solutions were prepared in 50-mL centrifugation tubes as described for the soil samples. The repeatability was checked by duplicating measurements of the real soil samples. The duplicate measurements were deviated by 1.11 ± 0.69 μg kg−1 (n = 9). The accuracy of the org-Hg measurement was verified on the certified reference material ERM-CC580 (an estuarine sediment). The result of the current methods (0.077 ± 0.004 mg kgˉ1; n = 7) was consistent with the reference value for MeHg (0.075 ± 0.004 mg kgˉ1).
Because MeHg is predominant (> 90%) in the natural soil, we can discuss the behavior of MeHg based on the org-Hg concentration obtained by this method. Indeed, the org-Hg concentration obtained by the method agreed well with the reference value for MeHg. Thus, basically, “org-Hg” was used for the measurement value and “MeHg” was used for the discussion of environmental behavior of mercury in this study. A schematic diagram of the procedures for T-Hg and org-Hg measurements is shown in Fig. S1.
2.3 TOC and TN in the soil samples and chemical composition analysis
The TOC was determined as the difference between the total carbon (TC) and inorganic carbon (IC) using a total carbon analyzer (TOC-V SCN, Shimadzu Co., Ltd., Japan) attached to a solid-sample module (SSM-5000A, Shimadzu Co., Ltd., Japan). The TC was determined by combusting a portion of the dried sample (50 mg) at 980 °C. The IC was determined by adding aqueous H3PO4 (1:1 v/v H3PO4/H2O) to a 50-mg dried soil sample, followed by heating at 240 °C. The TN was measured by organic elemental analysis (Flash 2000, Thermo Scientific, Tokyo, Japan). The chemical compositions of the soil samples were analyzed by the wavelength-dispersive X-ray diffraction fluorescence technique (ZSX-mini II, Rigaku Co., Ltd., Japan).
2.4 Data analysis and statistics
The data and graphical analyses were performed by the statistical analysis functions of Microsoft Excel 2010 (Microsoft Corporation, USA).
3 Results
3.1 Vertical distributions of T-Hg, org-Hg, TOC and TN in the soil samples
The analytical results are listed in Table 1, and the vertical variation in T-Hg, TOC, TN and org-Hg in the forest soil collected in August 2014 is shown in Fig. 2. The highest concentrations of T-Hg observed in the national park and the ASGM village were 1.9 mg kg−1 and 8.9 mg kg−1, respectively. The T-Hg was most concentrated near the soil surface and decreased with increasing depth, suggesting that mercury had been deposited from the atmosphere. The T-Hg was lower in the national park than at the ASGM site, but the vertical variation showed similar trends at all sites, suggesting that the atmospherically deposited mercury was discharged from ASGM activities. In 2010, 0–6-cm-depth soil samples were collected at 3 points in the same forest in the national park [31]. The average of these samples was 0.34 mg kg−1, and the Hg concentration in the 0–2 cm surface soil layer at each point was 0.25, 0.40 or 0.47 mg kg−1. These values were the highest at each point. Thus, at that time, the soil surface had received mercury emitted by the mining activity, but greater than 1 mg kg−1 T-Hg was not observed. In 2014, the soil sample of NP2 was obtained at the same place as in the sampling campaign in 2010. Therefore, it can be considered that the ASGM activity in this region increased during the following 4 years, leading to an increase in the Hg concentration at the soil surface at those sites.

Vertical variations of T-Hg, TOC, TN and org-Hg in the forest soils collected in August 2014; the values at 0 cm depth were taken from fallen leaves collected on the soil surface
The T-Hg concentrations in the national park soils were maximized just below the surface, whereas the TOC levels were maximized at the soil surface. Therefore, near the surface, the mercury concentrations decreased with increasing TOC content. As mentioned above, the mercury concentration near the soil surface may have been increased by wet or dry deposition of mercury from the atmosphere. The oxidized and deposited mercury may have been stabilized by adsorbing to organic matter in the soil [32]. Fallen leaves are a primary source of organic matter in the soil and can contribute mercury that has accumulated on them from the atmosphere. Decomposition decreases the weight of the fallen leaves, concentrating the mercury in the organic matter. Additional mercury can be deposited in organic matter from the atmosphere. Therefore, the mercury concentration should be higher in a mature soil layer than in a soil layer topped with fresh fallen leaves. The highest concentrations of org-Hg observed at the national park and ASGM village were 5.0 μg kg−1 and 11.4 μg kg−1, respectively. The same tendency as shown by T-Hg was observed in the vertical variation in org-Hg, and a higher concentration was observed near the surface. As described above, the increase in the T-Hg concentration near the surface might be caused by the deposition of mercury from the atmosphere. The increase in the org-Hg concentration in the surface soil around the ASGM site indicates the progress of methylation of deposited mercury in the soil. The org-Hg percentages of the T-Hg were 0.2 ± 0.1% for the national park and 0.3 ± 0.2% for ASGM sites.
3.2 TOC-to-TN ratio and organic matter decomposition
When fresh organic matter, such as fallen leaves, is decomposed by microbial metabolism, carbon is released into the atmosphere as CO2, so that the ratio of TOC to TN gradually decreases and approaches 10:1. As shown in Fig. 3a, when TN was plotted against TOC, a linear relationship was observed in the lower range of TOC (< 25%). In the range of TOC higher than 25%, the plots deviated from a linear relationship. The soil layer containing more than 25% organic carbon may include fresh and/or insufficiently decomposed fallen leaves; since the TOC-to-TN ratio approached 10:1 in the lower range of TOC, it can be considered that the fallen leaves were decomposed aerobically by organisms. At lower than 20% TOC, the CaO content showed no significant change with low values, and at higher than 20% TOC, the CaO content was positively related to TOC. Calcium can be considered to be derived from plant debris. The chemical compositions were similar in the soils of both sites, and a significantly high Ca content in the fallen leaf layer was common to both sites (Fig. S2), supporting the conclusion that the samples containing high percentages of Ca were topped by insufficiently decomposed fallen leaves. As shown in Fig. S3, the vertical variation in CaO and that in N/C were consistent with each other. It can be said again that in the upper layer containing fresh fallen leaves, the decomposition of the organic matter proceeds aerobically.

Relationships between TOC and TN (a), and TOC and CaO (b)
4 Discussion
There are only a few reports about the MeHg concentration in the soil around ASGM sites. Among the reports, the highest concentration value of MeHg, < 1–162 μg kg−1, with a median of 13 μg kg−1 (28 ± 19% of T-Hg), was reported for an ASGM in Ghana in the wet season [13]. The concentration values of 0.40–16 μg kg−1, with a median of 5.0 μg kg−1 (0.14 ± 0.16% of T-Hg), were reported for ASGM sites in China [14], and 0.052–48 μg kg−1, with a median of 0.78 μg kg−1, were reported for ASGM sites in Senegal [12]. In the case of Senegal, although the raw data are not applicable, the range and median of T-Hg were 0.050–130 mg kg−1 and 0.68 mg kg−1, respectively. The percentage of org-Hg may be similar to the value reported for China. These studies suggest that a portion of the mercury released by ASGM activity is converted to MeHg in the surrounding environment via several routes. The MeHg % reported in these studies was similar to the values found in our study. In Ghana, the highest concentration of MeHg was observed in the rainy season; however, MeHg was not detected in the dry season. This suggests that in Ghana, the water content of the soil is an important factor for MeHg formation. On the other hand, in West Java, Indonesia, precipitation is approximately 50 mm/month, even in the dry season, which is several times greater than in Ghana. Therefore, water cannot be considered the factor limiting MeHg formation in this area, and methylation factors other than water should be considered. Therefore, the relationships between the MeHg formation process and chemical components in soil were studied in detail.
Figure 4 plots the T-Hg and org-Hg concentrations as functions of TOC content. The T-Hg and org-Hg were positively linearly related to TOC contents, except in the uppermost layer topped by insufficiently decomposed fallen leaves, in which the TOC values exceeded 25%. Within the linear range of the T-Hg versus TOC plots, the slopes were an order of magnitude steeper at the ASGM sites than at the national park sites. The steeper slopes suggest a higher depositional rate of Hg at the ASGM sites than in the national park [31]. As the fallen leaves decompose, they discharge carbon as CO2, and the mass of organic matter decreases, concentrating mercury in the organic matter. During this process, the organic matter also traps and accumulates deposited mercury. Hg(II) bound with the organic matter generally has low mobility in soil, but if bound to soluble organic complexes, it can migrate to deeper soil layers [33]. The linear range of TOC versus T-Hg plots may show the migration of mercury with organic compounds. A linear relationship was observed between TOC and org-Hg in the same range of TOC % (Fig. 4b). On the other hand, the increase in the concentration of org-Hg observed in the surface and subsurface soils (Fig. 2) suggests that MeHg production occurs in these layers. The decomposition of organic matter near the surface can be considered to occur under aerobic conditions. It is well known that MeHg is produced under anaerobic conditions by microorganismal activity. However, the observation suggests that MeHg can be formed in the soil layer where organic matter is aerobically decomposed. To follow the relation between aerobic decomposition of organic matter and MeHg formation, further investigations will be required. Very similar relations between the decomposition process of organic matter and the increase in org-Hg species in soil have been reported around the Idrija mercury mine in Slovenia [30]. The climates of Indonesia and Slovenia are very different: The annual average temperatures (precipitation) in 2014 are 25.3 °C (1628 mm) and 10.3 °C (1027 mm) in Bogor and Idrija, respectively [34]. These facts suggest that the soil surface is an important reaction field for mercury dynamics in forests under wide environmental conditions.

Dependencies of T-Hg (a) and org-Hg (b) concentrations on TOC contents at (1) ASGM sites and (2) national park sites. The samples including > 30% of TOC were excluded from the linear regressions
Palmieri et al. [5] reported that mercury occurs not only as Hg(II) predominantly bound to organic components in most of the samples but also in the form of cinnabar in some. Since HgS is stable, Hg(II) will be subject to MeHg formation [5]. Even when amalgam burning is not performed, the concentration of mercury in the atmosphere in the workplace is much higher than that outside [23], suggesting that the generated mercury vapor is adsorbed on the indoor walls and floor. That is, in the immediate vicinity of the workplace, there is a possibility that elemental mercury will be adsorbed onto the surrounding surfaces. In this study, a glass filter, 2.4 M KCl solution and 0.1% KMnO4 solution were connected in a series to a pump, and ambient air in the workplace was introduced into the solutions. During amalgam burning, mercury trapped in the KCl solution used as a washing solution increased and accounted for 0.7–5.2% (median: 1.3%) of the total Hg. The mercury trapped in the KCl solution may have included Hg(II). Although further investigation is needed to quantitatively discuss the species trapped in the solution, the possibility of deposition of Hg0 and Hg(II) can be considered in the ASGM village. However, the formation of MeHg from the deposited Hg0 is unlikely to proceed because re-evaporation of deposited Hg0 should occur, and Hg0 needs to be oxidized to Hg(II) to be converted into MeHg [35]. Although it is difficult to estimate the degree of oxidation of Hg0 to Hg(II) in soil, in a study about mercury speciation and the mobility of Hg in soils affected by Hg emissions from three chlor-alkali plants, the Hg0 emitted from the plants could not be detected in any of the investigated soils, which indicated quantitative re-emission or oxidation of Hg0 in the atmosphere or soils [33]. Oxidation of Hg0 to Hg(II) in water in the presence of a complexing agent has also been reported [36,37,38]. In the presence of water and a complexing agent such as humic acid, oxidation of deposited Hg0 to Hg(II) at the soil surface can presumably occur. Since we have no adequate data concerning the relation of chemical forms of Hg deposited on the surfaces and the distance from the workplace, a quantitative discussion is not possible here. This relation should be investigated in the future.
The T-Hg and org-Hg concentrations observed in this study were in the range of 0.35–1.94 mg kg−1 (av. ± s.d. = 1.04 ± 0.43 mg kg−1) and 0.20–7.76 μg kg−1 (av. ± s.d. = 2.4 ± 1.8 μg kg−1) in the national park, and 0.33–8.94 mg kg−1 (av. ± s.d. = 1.90 ± 1.92 mg kg−1) and 1.36–11.40 μg kg−1 (av. ± s.d. = 3.76 ± 3.60 μg kg−1) in the ASGM village, respectively. The ratio of org-Hg to T-Hg was 0.23 ± 0.14% and 0.34 ± 0.20% for the national park and ASGM village, respectively. As it was shown that only a small portion of mercury emitted into the environment was converted to MeHg, it can be said that a direct and serious health impact in this region is due to exposure to mercury emitted into the surrounding atmosphere. However, according to the Guidelines for Recovery of Contaminated Land Waste Hazardous and Toxic Materials (Regulation of the Minister of Environment and Forestry of the Republic of Indonesia), if the mercury concentration in leaching solution of soil is 0.02 mg L−1 < Hg ≦ 0.05 mg L−1 and/or the mercury concentration in the soil is 0.3 mg kg−1 < Hg ≦ 75 mg kg−1, the said land must be managed according to non-B3 waste management [39]. Although simple comparisons may not be possible, the observed T-Hg value in this study was significantly higher than 0.3 mg kg−1. Although only 0.2–0.3% of the emitted mercury was converted to MeHg, its concentration may be higher than the background level. The surface soil can be transported by heavy rain into the water system, and the MeHg formed in the surface soil can have an impact on aquatic ecosystems [40]. The methylmercury contamination of ecosystems in this region should be studied continuously.
5 Conclusion
An increase in the T-Hg concentration in the upper layer of forest soil around the ASGM site was observed, which suggested that the deposition of discharged mercury from ASGM caused an increase in the mercury concentration at the soil surface. The T-Hg concentration observed in this study was 1.04 ± 0.43 mg kg−1 in the national park and 1.90 ± 1.92 mg kg−1 in the ASGM village. The percentage of org-Hg to T-Hg were 0.23 ± 0.14% for the national park and 0.34 ± 0.20% for the ASGM village. The vertical variation in the org-Hg concentration was consistent with the decomposition of organic matter estimated by variations in TN, TOC and soil chemical composition, which suggests that MeHg is formed in the soil layer where organic matter is aerobically decomposed. Even though MeHg accounts for only a small portion of the total mercury, the mercury concentration in the soil surface layer around the ASGM site is increasing. To follow up on changes in the mercury concentration, and to estimate the impact on ecosystems, a continuous survey is required.
References
Lindqvist O, Rodhe H (1985) Atmospheric mercury—a review. Tellus 37B:136–159
UNEP (2013) Global Mercury Assessment 2013: sources, emissions, releases and environmental transport. UNEP Chemicals Branch, Geneva, Switzerland, pp 1–32
Harris HH, Pickering IJ, George GN (2003) The chemical form of mercury in fish. Science 301:1203
Qiu G, Feng X, Li P, Wang S, Li G, Shang L, Fu X (2008) Methylmercury accumulation in rice (Oryza sativa L.) grown at abandoned mercury mines in Guizhou, China. J Agric Food Chem 56:2465–2468
Palmieri ELH, Nalini AH Jr, Leonel VL, Windmöller CC, Santos CR, de Brito W (2006) Quantification and speciation of mercury in soils from the Tripuí Ecological Station, Minas Gerais, Brazil. Sci Total Environ 368:69–78
Gibb H, O’Leary KG (2014) Mercury exposure and health impacts among individuals in the artisanal and small-scale gold mining community: a comprehensive review. Environ Health Perspect 122:667–672
Arifin YI, Sakakibara M, Sera K (2015) Impacts of artisanal and small-scale gold mining (ASGM) on environment and human health of Gorontalo Utara Regency, Gorontalo Province, Indonesia. Geosciences 5:160–176
Obiri S, Yeboah PO, OsaeS Adu-kumiS, Cobbina SJ, Armah FA, Ason B, Antwi E, Quansah R (2016) Human health risk assessment of artisanal miners exposed to toxic chemicals in water and sediments in the Prestea Huni Valley District of Ghana. Int J Environ Res Public Health 13:139
Krisnayanti BD (2018) ASGM status in West Nusa Tenggara Province, Indonesia. J Degrad Min Lands Manag 5:1077–1084
Elias Nyanza C, Deborah Dewey, Deborah Thomas S K, Mark Davey, Sospatro Ngallaba E (2014) Spatial distribution of mercury and arsenic levels in water, soil and cassava plants in a community with long history of gold mining in Tanzania. Bull Environ Contam Toxicol 93:716–721
Tania Terán-Mita A, Angel Faz, Flor Salvador, Joselito Arocena M, Jose Acosta A (2013) High altitude artisanal small-scale gold mines are hot spots for Mercury in soils and plants. Environ Pollut 173:103–109
Gerson JR, Driscoll CT, Hsu-Kim H, Bernhardt ES (2018) Senegalese artisanal gold mining leads to elevated total mercury and methylmercury concentrations in soils, sediments, and rivers. Elem Sci Anth 6:11–24
Donkor AK, Bonzongo JC, Nartey VK, Adotey DK (2006) Mercury in different environmental compartments of the Pra River Basin, Ghana. Sci Total Environ 368:164–176
Feng X, Dai Q, Qiu G, Li G, He L, Wang D (2006) Gold mining related mercury contamination in Tongguan, Shaanxi Province, PR China. Appl Geochem 21:1955–1968
Veiga MM, Angeloci G, Hitch M, Velasquez-López PC (2014) Processing centers in artisanal gold mining. J Clean Prod 64:535–544
Tomiyasu T, Hamada YK, Baransano C, Hidayati N, Rahajoe JS (2019) Time variation in transfer amounts of mercury by a river system near an artisanal and small-scale gold mining area in West Java, Indonesia. Environ Earth Sci 78:686
Yustiawati Syawal MS, Terashima M, Tanaka S (2006) Speciation analysis of mercury in river water in West Java, Indonesia. Tropics 15:425–428
Hidayati N, Juhaeti T, Syarif F (2009) Mercury and cyanide contaminations in gold mine environment and possible solution of cleaning up by using phytoextraction. J Biosci 16:88–94
Kido M, Yustiwati Syawal MS, Sulastri Hosokawa T, Tanaka S, Saito T, Iwakuma M, Kurasaki M (2009) Comparison of general water quality of rivers in Indonesia and Japan. Environ Monit Assess 156:317–329
Yasuda M, Yustiawati Syawal MS, Sikder MdT, Hosokawa T, Saito T, Tanaka S, Kurasaki M (2011) Metal concentrations of river water and sediments in West Java Indonesia. Bull Environ Contam Toxicol 87:669–673
Tomiyasu T, Hamada YK, Kodamatani H, Hidayati N, Rahajoe JS (2019) Transport of mercury species by river from artisanal and small-scale gold mining in West Java, Indonesia. Environ Sci Pollut Res 26:25262–25274
Kono Y, Rahajoe JS, Hidayati N, Kodamatani H, Tomiyasu T (2012) Using native epiphytic ferns to estimate the atmospheric mercury levels in a small-scale gold mining area of West Java, Indonesia. Chemosphere 89:241–248
Kono Y, Tomiyasu T (2013) Quantitative evaluation of real-time measurements of atmospheric mercury in a mercury-contaminated area. Environ Saf 4:153–157
Akagi H, Nishimura H (1991) Speciation of mercury in the environment. In: Suzuki T, Imura N, Clarkson TW (eds) Advances in mercury toxicology. Plenum Press, New York, pp 53–76
Akagi H, Malm O, Branches FJP, Kinjo Y, Kashima Y, Guimaraes JRD, Oliveira RB, Haraguchi K, Pfeiffer WC, Takizawa Y, Kato H (1995) Human exposure to mercury due to gold mining in the Tapajos River basin, Amazon, Brazil: speciation of mercury in human hair, blood and urine. Water Air Soil Pollut 80:85–94
Matsuo N, Suzuki T, Akagi H (1989) Mercury concentration in organs of contemporary Japanese. Arch Environ Health 44:293–303
Malm O, Branches FJP, Akagi H, Castroa MB, Pfeiffer WC, Harada M, Bastos WR, Kato H (1995) Mercury and methylmercury in fish and human hair from the Tapajós river basin, Brazil. Sci Total Environ 175:141–150
Tomiyasu T, Nagano A, Sakamoto H, Yonehara N (1996) Differential determination of organic mercury and inorganic mercury in sediment, soil and aquatic organisms by cold-vapor atomic absorption spectrometry. Anal Sci 12:477–481
Tomiyasu T, Hamada YK, Baransano C, Kodamatani H, Matsuyama A, Imura R, Hidayati N, Rahajoe JS (2020) Mercury concentrations in paddy field soil and freshwater snails around a small-scale gold mining area, West Java, Indonesia. Toxicol Environ Health Sci 12:23–29
Tomiyasu T, Kodamatani H, Imura R, Matsuyama A, Miyamoto J, Akagi H, Kocman D, Kotnik J, Fajon V, Horvat M (2017) The dynamics of mercury near Idrija mercury mine, Slovenia: horizontal and vertical distributions of total, methyl, and ethyl mercury concentrations in soils. Chemosphere 184:244–252
Tomiyasu T, Kono Y, Kodamatani H, Hidayati N, Rahajoe JS (2013) The distribution of mercury around the small-scale gold mining area along the Cikaniki river, Bogor, Indonesia. Environ Res 125:12–19
Tomiyasu T, Okada M, Imura R, Sakamoto H (2003) Vertical variations in the concentration of mercury in soils around Sakurajima Volcano, Southern Kyushu, Japan. Sci Total Environ 304:221–230
Biester H, Müller G, Schöler HF (2002) Binding and mobility of mercury in soils contaminated by emissions from chlor-alkali plants. Sci Total Environ 284:191–203
World Weather Online. com (2020). https://www.worldweatheronline.com/. Accessed 14 June 2020
Hu H, Lin H, Zheng W, Tomanicek SJ, Johs A, Feng X, Elias DA, Liang L, Gu B (2013) Oxidation and methylation of dissolved elemental mercury by anaerobic bacteria. Nat Geosci 6:751–754
Yamamoto M (1995) Possible mechanism of elemental mercury oxidation in the presence of SH compounds in aqueous solution. Chemosphere 31:2791–2798
Meech JA, Veiga MM, Tromans D (1998) Reactivity of mercury from gold mining activities in darkwater ecosystems. Ambio 27:92–98
Ravichandran M (2014) Interactions between mercury and dissolved organic matter—a review. Chemosphere 55:319–331
State Gazette of the Republic of Indonesia (2018) Berita Negara Republik Indonesia, Pedoman Pemulihan Lahan Terkontaminasi Limbah Bahan Berbahaya Dan Beracun, Peraturan Menteri Lingkungan Hidup Dan Kehutanan Nomor P.101/MENLHK/SETJEN/KUM.1/11/2018, https://app.box.com/s/qtq4nc45geyl56evojtoo5r8qey9qrx4. Accessed 14 June 2020
Bravo AG, Bouchet S, Tolu J, Björn E, Mateos-Rivera A, Bertilsson S (2017) Molecular composition of organic matter controls methylmercury formation in boreal lakes. Nat Commun 8:14255
Acknowledgements
This work was supported by Grants-in-Aid (No. 22404002 and 25257302) for Scientific Research from the Japan Society for the Promotion of Science (JSPS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tomiyasu, T., Baransano, C., Hamada, Y.K. et al. Distribution of total and organic mercury in soils around an artisanal and small-scale gold mining area in West Java, Indonesia. SN Appl. Sci. 2, 1213 (2020). https://doi.org/10.1007/s42452-020-3008-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-3008-5