
Abstract
An erodible polyamide 6,10 based drug carrier was fabricated for continuous, extended release of a model hydrophilic drug, amitriptyline hydrochloride. The developed polyamide-based drug loaded compact disc was stable, semi-crystalline, mechanically robust and demonstrated the potential to prolong drug release while displaying controlled in vitro matrix erosion that was quantified as gravimetric matrix loss and changes in conductivity potential. 14.06%w/w of amitriptyline hydrochloride content was released over a 30-day period. A zero-order, linear mathematical fitting of the drug release profile predicted a 100% release in approximately 240 days. Drug release kinetics was predicted to be zero order and regulated more by matrix relaxation than Fickian diffusion. The synthesized polyamide compact disc was identified as being physicochemically stable and physicomechanically robust. In conclusion, the preliminary in vitro data generated serves as proof-of-concept that the polyamide-based disc can potentially function as a useful polymeric biomaterial for designing extended release, erodible drug carriers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The primary purpose of administering drugs in vivo is usually to enable the desired pharmacological action. Generally, conventional dosage forms/delivery systems are commonly known to utilize high drug doses, most of which is often excreted without exerting any preferred biological activity except for undesired adverse reactions. Hence, scientific advancements continually detail the concept of polymer-based, modified drug delivery approaches that function as facilitators for timed release and site-specific administration, favouring therapeutic performance and patient convenience, not frequently offered by conventional medicines [1,2,3,4]. Drug delivery encompasses the use of novel technologies suitable for presenting bioactive molecules to preferred in vivo administration sites to facilitate absorption or transport across biological membranes, bioavailability and pharmacological efficacy [1, 5]. The synthesis, optimization and development of new drug candidates is expensive and time consuming. Consequently, improving the safety and pharmacological performance of conventional medicines/dosage form by reformulating them into improve delivery systems are often quicker, cheaper and well-known to aid anticipated treatment outcomes [6]. This led to the invention of novel drug carriers that are capable of facilitating modified release, absorption, bioavailability, distribution, metabolism and elimination with the end goal of optimizing effectiveness, patient adherence and safety [1, 4, 6,7,8,9].
Polymer-based extended release drug carriers have received increased attention in recent times and this is predominantly because of their numerous clinical and biomedical advantages, which include reduced dosing frequency, administration site-specificity, minimized adverse effects, effective management of chronic conditions, improved bioavailability, fewer medical visits and better patient compliance [2, 10,11,12,13]. They are widely applied as invasive or non-invasive platforms typified by their unique functionalities which are based on novel delivery technologies such as reservoir systems, monolithic matrices, pumps (osmotic, peristaltic, infusion) and nano/micro structures [13]. Together with the continuous attempts to increase human life expectancy and key advances in polymer-based biomaterials, devices and fabrication methods; different kinds of extended release carriers have been optimized and engineered for disease management [14]. They permit targeted and localized release mechanisms which usually results in the achievement of therapeutic effect with lower drug concentrations [10, 11, 15, 16]. When employed as implantable systems, they are capable of increasing the bioavailability of poorly absorbed oral drugs and bypasses first pass metabolism and/or chemical degradation within the gastrointestinal tract. They are also flexible and offer the opportunity for quick, earlier than planned removal if elicited side effects outweigh benefits and needs treatment cessation [10, 17, 18]
Semi-crystalline thermoplastic polymers form an important group of polymeric biomaterials with many biomedical applications as drug carriers [19]. Poly [Imino-1,6-Hexanediylimino (1,10–Dioxo-1,10-Decanediyl)] otherwise known as polyamide 6,10 or nylon 6,10 is an example of a semi-crystalline thermoplastic polymers characterized with an even-even configured linear chain, poly-condensed synthetic aliphatic polyamide [20,21,22]. Aliphatic polyamides are very useful and versatile polymeric materials that are valuable because of their superior physicochemical and physicomechanical properties such as chemical inertness, good level of purity after production, abrasion resistance, relatively high mechanical strength, ease of processing, thermoplasticity, controlled matrix dissolution, hydrophilic, slow degradation, high melting points and thermal resistance [20,21,22,23,24]. These physical properties and widespread clinical uses of synthetic aliphatic polyamides, as both absorbable and non-absorbable surgical sutures, demonstrate their biocompatibility and non-toxicity making them attractive for the design of extended release drug carriers [25,26,27,28,29].
As already reiterated above, polymers play a major role in the engineering of multifaceted extended release drug delivery systems where they function as sole carriers or external coats. They are constantly in high demand and scientist are seeking additional polymeric carriers for this application [2]. Researchers have documented the attractive clinical application, biocompatibility and potent drug release modulating properties of different aliphatic polyamides [29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44]. Yet, there is still not much information regarding their use as extended release drug carriers. To execute the study objective, polyamide 6,10 was formulated into a monolithic matrix loaded with amitriptyline hydrochloride to form a cylindrical compact disc. According to the Biopharmaceutics Classification System (BCS), amitriptyline hydrochloride is a BCS class I drug meaning that it is highly soluble and permeable in nature [45] leaving it with higher tendencies of eliciting burst effect, irregular and rapid release kinetics. These properties make it a suitable specimen for evaluating the in vitro extended release and bioerodible capabilities of the polyamide-based delivery system. This proof-of-concept study therefore aims to investigate the extended in vitro drug release behaviour, bioerosion, physicochemical and physicomechanical characteristics of a typical semi-crystalline aliphatic polyamide (i.e. polyamide 6,10), formulated as a drug loaded compact disc.
2 Materials and methods
2.1 Materials
1,6-Diaminohexane, decanedioyldichloride, anhydrous n-hexane and anhydrous potassium bromide, amitriptyline hydrochloride, cyclohexane and anhydrous sodium hydroxide were purchased from Sigma Chemical Company (St. Louis, MO, USA). All other reagents were of analytical grade and used as received.
2.2 Chemical synthesis of polyamide 6,10 by interfacial polymerization
Polyamide 6,10 was synthesized by interfacial polymerization employing decanedioyldichloride and 1,6-diaminohexane as monomers, cyclohexane/hexane and deionized water as non-polar and polar dispersants respectively and sodium hydroxide as a neutralizing agent (Fig. 1) [34,35,36]. Two solutions were prepared with the aqueous phase containing 1.75 g of 1,6-diaminohexane plus 0.4 g of sodium hydroxide dissolved in 10 mL deionized water while the non-polar phase was composed of 0.2 g decanedioyldichloride evenly dispersed in hexane in cyclohexane (1:4). The two homogenous solutions were gradually added together to form two immiscible phases which presented with the formation of the polyamide film at the liquid–liquid interface. Formed polyamide film was collected as a mass by rotating a glass rod at the interface of the two immiscible liquids phases. The isolated polyamide film was then thoroughly rinsed multiple time with deionized water, blotted on a 110 mm filter paper to eliminate any excess solvent and dried to constant weight at 40 ± 0.5 °C over 4 days in a Memmert 854 well-insulated thermal oven (Schwabach, Germany). Dried polyamide 6,10 were stored away in airtight, opaque glass jar containing desiccant bags for further testing.

Schematic showing the interfacial polymerization reaction
2.3 Calculation of yield
The amount of polyamide 6,10 produced for each synthesis round was calculated as percentage yield (%w/w) based on the mathematical relationship between the actual and theoretical yields. The theoretical yield was derived from the reaction stoichiometry while the actual yield was measured as the real mass of the polyamide 6,10 produced at the end of each synthesis.
2.4 Formulation of the polyamide-based drug loaded implantable delivery system
Dehydrated polyamide 6,10 was subjected to dry milling using a laboratory scale blending machine (Kinematica GMBH, Eschbach, Germany) until a free-flowing powered was formed. Each drug loaded delivery system contained 300 mg pulverized polyamide and 50 mg amitriptyline hydrochloride (based on pilot experiments performed by changing one-variable-at-a-time), dry milled in the laboratory blender and sieved through a 1000 mm aperture size Endecotts sieve (Endecotts Limited, London, UK) resulting in particle size range of 711–1000 μm. Thorough mixing of the respective solids was carried out for about 15 min using a laboratory scale blender (CG 100, Kenwood Ltd., Cambridge, UK). Final homogenous dry blends were compressed into flat-surfaced cylinder-shaped matrices with an average diameter of 13 ± 0.5 mm and thickness of 4 ± 0.4 mm using a Beckman hydraulic press (Beckman Instruments, Inc., Fullerton, USA) under a pressure of 1 ton for 1 min. The amitriptyline-polyamide delivery matrices were stored away in opaque, airtight jars for subsequent testing.
2.5 Evaluation of extended amitriptyline hydrochloride release kinetics
In vitro drug release studies was performed on the drug loaded polyamide delivery matrices as six replicates. Each formulation was immersed in 100 mL simulated plasma contained in closed glass vessels and subjected to a 30-day release study under biorelevant conditions of 37 ± 0.5 °C and 50 rpm in a thermoregulated shaking water bath (Stuart SBS 40, Staffordshire, UK). Simulated plasma (pH 7.4.) was prepared by dissolving 0.144 g potassium dihydrogen phosphate (KH2PO4), 0.795 g anhydrous disodium phosphate (Na2HPO4) and 9.000 g sodium chloride (NaCl) in 1L of deionized water [46]. At specific time intervals over the planned 30-day period, 5 mL sample was withdrawn and collected volume of fluid replaced with freshly prepared, simulated plasma that was preserved at 37 ± 0.5 °C. Collected dissolution samples were appropriately reconstituted and filtered through a 0.20 μm pore size Cameo Acetate membrane filter (Milipore Co., Bedford, Mass). A correction factor was appropriately applied in all cases where dilutions of samples were required. Drug release was quantified by ultraviolet spectrophotometry (Specord 40, Analytik Jena, AG, Germany) at a lambda maximum of 240 nm using the isolated filtrate. Background readings were performed on blank simulated plasma and placebo solutions and actual amitriptyline hydrochloride release per unit time was computed using fitted linear calculation (y = 53.01x; R2 = 0.98). Additionally, release kinetic models that best-fit the formulation dissolution profile was simulated using model dependent strategies such as zero-order, first order, second order, Higuchi, Peppas and Sahlin, Korsmeyer-Peppas and Michaelis–Menten [46] based on the KinetDS, version 3.0 open source software. The selection of the mathematical model of best-fit that optimally describes the mechanisms governing amitriptyline hydrochloride release from these matrices was based on the correlation coefficient (r) with a value closest to one.
2.6 Investigating in vitro matrix erosion
In vitro matrix weight loss referred to as erosion was measured to evaluate the bio-erodible properties of polyamide-based discs. The biorelevant testing conditions already described for the dissolution studies in Sect. 2.5 above was utilized. At specific time intervals up to 30 days, each drug loaded formulation was removed, blotted using lint-free filter paper and dried to constant weight at 40 ± 0.5 °C in an oven (Thermo Scientific™ PR305220G, Fisher Scientific, Waltham, MA, USA). All measurements were performed in triplicate and matrix erosion was quantified as percentage weight loss per time-point.
2.7 Evaluation of polymeric matrix solvation using electrolyte conductivity testing
In this case, in vitro electrolyte conductivity studies were carried out on the drug-free polyamide matrices placed in 100 mL deionized water maintained at 37 ± 0.5 °C and 50 rpm in a shaking water bath to exclude ionic interferences between the electrolytes present in the simulated plasma solution, amitriptyline hydrochloride and the intrinsic polyamide ions. Measurements were targeted towards assessing the potential bio-erodible properties of the polyamide discs. A calibrated, conductivity tester (TDS Testr 40 with ATC, Oakton, USA) with a dual measurement range (0.00–199.90 μS/cm and 200.00–1999.00 μS/cm) and an automatic temperature compensation system ranging from 5–50 °C was used to detect the conductivity transitions that occurred as a result of the polyamide matrix dissolution. During a typical test, the basal conductivity reading of the deionized water was subtracted from the subsequent actual conductivity values obtained from immersing tester into 10 mL of each replicate sample for 60 s and a test duration of 24 h. The tester was thoroughly cleaned after each measurement and three replicate samples tested per time.
2.8 Peripheral swelling and water uptake analyses
Peripheral swelling and water uptake characteristics were studied employing the same set up as earlier described for the dissolution studies in Sect. 2.5. above. In addition, the hydrated drug loaded matrices were removed from the dissolution medium (simulated plasma) at scheduled time intervals, gently patted using lint free laboratory towels to remove excess fluids and then weighed on an analytical balance (ME204TE/00, Mettler Toledo, LLC, OH, USA) to assess any swelling related weight changes (%Sw). The quantity of fluid absorbed was indicative of the matrix swelling capacity and was calculated as the difference between the anhydrous and hydrated weights of each formulation. Besides, matrix swelling was also expressed as volumetric measures in which case each formulation was treated as having a cylindrical geometry. Thus, volumetric dimensions were computed using mathematical expressions (volume = Πr2h; the radius (r) and height were measured using a digital micrometer screw gauge at different time-points). Percentage volumetric swelling (%Vsw) was then calculated based on the dimensional volumetric transitions between the wet and dry matrices.
2.9 Physicomechanical texture of the drug loaded polyamide-based compact discs
The physicomechanical properties were quantified as: (i) resilience (Res)—a measure of the disc’s matrix resistance to deformation; (ii) energy of deformation (Edef)—degree of work performed during matrix disruption; (iii) hardness (H)—a measure of resistance against localized deformation by an external indenter and (iv) Brinell hardness number (BH#)—a number that quantifies hardness obtained by pressing a hard steel ball/sphere into the surface of a material, all of which are measures of matrix rigidity and robustness [47,48,49]. A calibrated textural analyzer (TA.XTplus, Stable Micro Systems, Surrey, England) fitted with either a cylindrical steel probe (50 mm diameter for Res) or flat-tipped steel probe (2 mm diameter for H and Edef) or ball probe indenter (diameter 0.5 mm for BH#). Data was captured with the texture exponent software. The parameter settings were pretest, test and post-test speeds of 1 mm/sec, 0.5 mm/sec, 1 mm/sec respectively; compression force of 40 N; an automatic trigger type setting, trigger force of 0.5 N, 50 kg load cell, 50% compression strain for resilience and indentation depth set at 0.25 mm for the BH# determinations. Furthermore, the maximum force generated as a result matrix indentation using the 0.5 mm diameter ball probe was integrated into Eq. 1 and employed in the computation of the BH#. All tests were carried out as three replicates.
where F = maximum force generated as a result of matrix indentation (N), D = diameter of ball probe indenter (0.5 mm) and d = indentation depth (0.25 mm).
2.10 Physicochemical characterization of the polyamide-based delivery matrices
2.10.1 Elucidation of structural configuration
Fourier Transform Infra-Red (FTIR) spectrum of the polyamide-based matrices were generated using a Perkin Elmer Spectrum 100 FTIR Spectrophotometer equipped with the Spectrum V 6.2.0 software (Beaconsfield, Buckinghamshire, UK). The sample receptacle and machine stage were carefully cleaned with ethanol, allowed to dry before background scans were captured. Thereafter, approximately 5 mg of test samples were applied to the machine stage and scanned for spectral patterns. Measurements were made three times and each recorded spectrum was an average of 32 scans within the wavenumber range of 4000–650 cm−1 and a resolution of 4 cm−1.
2.10.2 Determination of thermal transitions
The thermal behavior of the polyamide-based matrices was analyzed using a differential scanning calorimeter equipped with 50-position autosampler and the Platinum™ software (Q2000, TA Instruments, New Castle, DE, USA). Three powdered test samples (10 mg each) were placed in separate aluminum pans with perforated lids plus an empty pan which served as the reference point. Each sample was subjected to a three cycle heating process at temperature ranges between 25–400 °C at a rate of 10 °C/min under an inert nitrogen flow of 25 mL/minute.
2.10.3 Molecular mass quantification
High resolution electron ionization mass spectrometry analyses were performed on a Kratos MS 9/50, VG 70 SEQ mass spectrophotometer (VG Analytical, Manchester, England) under high and low resolutions using powdered samples. The instrumental settings employed for the measurement include electron ionization, resolution = 7500, mass range = 3.00 amu (8\kv), scan rate 5 s/decade (external). Measurements were made using 2 mg sample dissolved in 2 mL of meta-nitrobenzylalcohol.
2.10.4 Crystallinity assessment
X-ray diffraction patterns of the pulverized polyamide matrices were examined using the Bruker D8 Advance Diffractometer (Bruker, Karlsruhe, Germany). Test sample (200 mg) was placed in the aluminium holder tested using the X-ray diffractometer programmed at a generator voltage of 40 kV, current of 30 mA, scanning speed of 2 degrees per minute, step width 0.025 degrees, divergence and anti-scatter slits of 2 mm each and detector slit of 0.2 mm.
2.10.5 Surface topography visualization
The surface structural morphology was identified using micrographs generated from a JSM-840 Scanning Electron Microscope (JEOL 840, Tokyo, Japan) at a voltage of 20 keV and a magnification of 1000 × . Test samples with dimensions of 5 mm × 2.5 mm were sputter coated with gold–palladium and viewed accordingly.
2.11 Preliminary stability investigations
The drug loaded polyamide were subjected to a preliminary, short-term stability testing which was executed according to recommendations in compliance with the World Health Organization’s guidelines for stability testing intended for the global market [50]. Test was conducted on samples stored at a temperature of 30 ± 2 °C and relative humidity of 65 ± 5% in a laboratory scale incubator (Model FSIH4, Labcon, Johannesburg, South Africa) for 60 days. The drug content was selected as the stability indicator and measurements were made at intervals of 0, 30 and 60 days. For each determination, samples were withdrawn and retested for amitriptyline hydrochloride content in triplicate. Results were presented as the average reading ± standard deviation and statistical significance was assessed with the GraphPad Prism 7 software (GraphPad, San Diego, CA, USA) using a two-way Analysis of Variance (ANOVA).
3 Results and discussion
3.1 Synthesis, physical appearance and yield
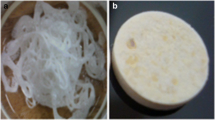
Physically, the synthesized polyamide appeared as off-white, crystalline and densely packed solid (Fig. 2a) which when subjected to dry milling produced free-flowing powder with a continuous consistency. The calculated yield amounted to 95.89 ± 1.32%w/w which is good indication that the selected reaction stoichiometry that was based on the molar ratios of the monomeric components, partitioning efficiency explained as the capability of the solute particles to segregate into the solvents, volume ratios and polarity of the solvent systems employed for the synthesis had a positive influence on the perceived quality and yield of the polymer. The formulated amitriptyline hydrochloride containing polyamide matrices were round, flat-surfaced, cylindrical compacts with a diameter of 1.3 ± 0.2 cm, average thickness of 0.4 cm and total weight of 350 ± 3.45 mg (Fig. 2b).

Digital photographs of: a polyamide 6, 10 dry milled powder and b the polyamide-based drug loaded compact discs
3.2 Prolonged in vitro drug release kinetics and matrix erosion trends over time
The drug loaded polyamide-based matrices constantly released 14.06%w/w amitriptyline hydrochloride over a 30-day period (Fig. 3a) indicating the potential of the system to modulate drug liberation kinetics over an extended duration which is needful for a potentially implantable delivery system. The mathematical models applied to the drug release profiles were based on R-values, a measure of best fits, closest to one. Identified best fits as it related to the generated release kinetics were the zero order release kinetics (R2 = 0.97), Peppas and Sahlin (R2 = 0.99; k1 = 0.14; k2 = 0.59) Korsmeyer-Peppas (R2 = 0.98; n = 0.95) and Michaelis–Menten (R2 = 1.00) models. These numerical quantities indicate that drug release from the polyamide 6,10 delivery systems is related to time as well as the matrix remaining post structural collapse as a result of external wetting and dissolution which is directly linked to progressive drug liberation. Also, drug release from this unique system can be described as being more regulated by matrix relaxation than the Fickian diffusion. In addition, an n-value of 0.95 obtained from the Korsmeyer-Peppas fitting can also imply that the system is diffusion and swelling controlled often referred to as the anomalous transport [27, 51,52,53] which correlates with the predicted zero order kinetics (R2 = 0.97). Experimental in vitro release kinetics data was well substantiated by the model fitted outputs. Furthermore, the application of a zero order model fit which was based on the linearity of the drug release profile (Fig. 3a) revealed that the polyamide-based disc may continue to constantly release amitriptyline hydrochloride for approximately 240 days more. A desirable linear regression value closest to one (R2 = 0.98) was obtained showing the accuracy and reliability of the mathematical fitting approach.

Profiles depicting a drug release patterns and b changes in matrix mass as drug liberation progresses
The drug loaded polyamide disc showed the ability to undergo significant loosening, fragmentation and erosion with about 78%w/w of the matrix remaining after 30-days under in vitro biorelevant conditions. A typical profile representing matrix remaining as a measure of loosening and erosion is presented in Fig. 3b.
3.3 In vitro matrix solvation measured through electrolyte conductivity
The susceptibility of the polyamide disc to disintegration and actual dissolution leading to mass loss and generation of polar ions into aqueous media was assessed here from a micromolecular perspective. Significant changes in media conductivity trends over 24 h was observed and measured in microsiemens/centimeter (µS/cm) thus indicating the ability of the polyamide-based drug delivery system to undergo time-dependent gradual dissolution, which further substantiates the matrix loss patterns discussed earlier. Figure 4a ,b respectively illustrate a plot of conductivity changes over time and proposed polar ion disintegration mechanisms during the matrix dissolution processes. Generated conductivity patterns can be described as two-phased (Fig. 4a) characterized by an initial rapid increase in conductivity followed a relatively consistent steady phase which can be related to an early matrix breakdown as a result of rapid wetting followed a steadier influx of aqueous media molecules resulting in a slower matrix dissolution rate. This may be responsible for the initial relatively quick amitriptyline chloride release and matrix loss displayed in Fig. 3a, b. These findings suggest that the polyamide-based delivery system can possibly undergo in vivo biodegradation, which is always desired for most implantable drug carriers.

Illustrations of: a changes in conductivity over time (n = 3; standard deviation ≤ 12.89 μS/cm) and b suggested mechanisms of hydrolytic ionization resulting in conductivity readings
3.4 Water uptake efficiency and swelling analyses
The polyamide-based disc demonstrated the ability to absorb water molecules which was determined as the weight gained over a specified period. Water gain for the drug loaded polyamide disc can be described as following a 1-in-1 trend (~ 100%w/w weight gain as a result of hydration) in which case polyamide weighing about 350 mg absorbed 370.58 ± 7.88 mg of water (Fig. 5a) within 24 h. Despite this significant increase in the weight of the delivery system post hydration, the polyamide-based disc displayed minimal peripheral swelling characteristic. An initial first hour thrust (5.42 ± 1.2% w/w swelling) followed by a relatively steady increase (5.50 ± 0.72–12.10 ± 1.94% w/w swelling at 2–24 h) in matrix volumetric dimensions was noticeable (Fig. 5b). Given that minimal peripheral swelling was observed, it can be inferred that drug release from the compact disc was more controlled by polymeric disentanglement followed by dissolution and eventual outward drug diffusion. This further validates the earlier described Korsmeyer-Peppas release profile fitting that predicted drug release from the disc as being dependent on anomalous transport mechanisms. This behavior is contrary to that known for most hydrophilic polymers that are employed as rate modulators for drug delivery purposes as these carriers usually have matrix swelling as a major mechanism for controlling release [7, 52].

Graphical representations of: a water uptake by weight and b percentage peripheral swelling over time
3.5 Typical physicomechanical properties
The polyamide-based compact disc displayed measurable physicochemical strength which corroborates its robust texture in relation to its drug release sustaining potential irrespective of its minimal swelling behavior and hydrophilic nature. It displayed quantifiable resistance to deformation (Res = 64.43 ± 3.89%), performed work associated with matrix disruption (Edef = 0.05 ± 0.01 J) and physicomechanical rigidity (BH# = 20.52 ± 2.49 N/mm2 and H = 71.11 ± 3.45 N/mm) which suggests that the polyamide-based disc displays a degree of physicomechanical robustness and textural stiffness further supports the observed release retarding capabilities comparable to those reported in literature [54, 55].
3.6 Physicochemical characteristics
Typical physicochemical properties of the polyamide-based compact disc was evaluated utilizing different analytical approaches. Chemical backbone configurational elucidation showed peaks at vibrational frequencies characteristic of salient bonds and/or chemical moieties present within the structural network of the polyamide indicating the chemical and physical stability of this carrier system. The obtained FTIR spectra produced typical bands representing C–H stretch, C–O absorption, C = O vibration, aliphatic N–H and C–N stretches, CH2 wag and CH2 rock identified at 2978 cm−1, 1216 cm−1, 1711 cm−1, 3423 cm−1, 1334 cm−1, 1477 cm−1 and 769 cm−1 respectively [55] which overall validated the already established chemical structure of the polyamide (Fig. 6a).

Physicochemical characterization of the polyamide-based compact disc formulation: a chemical backbone structure, b thermal transitional changes, c semi-crystalline conformation and d characteristic surface topography
The transitional thermal events were measured with the differential scanning calorimeter. Recorded thermal events included an endothermic glass transition phase at 79.85 °C, crystallization temperature peak at 192.23 °C and melting event that began at 214.76 °C and progressed towards thermal degradation up to 329.09 °C. In addition, the polyamide-based disc exhibited multiple melting endotherms at 119.53 °C, 168.42 °C and 214.76 °C which is typical of aliphatic polyamides when in the melt-crystallized state [57, 58]. This could be associated with their known high heat of fusion which has been shown to directly impact their intrinsic entropy state. This is usually dependent on the existing dissociation energy level within their intra-molecular hydrogen bonding structure which often requires collective dissociation before visible dimensional changes, due to the presence of heat, can occur [56]. The hydrogen bonds have been reported to relatively preserve the ordered polymeric crystal lattice backbone of aliphatic polyamides even in the molten state thus limiting mobility within its continuous methylene network. It was also observed that the exothermic crystallization and multiple endothermic melting peaks exhibited slightly broadened contours which could be associated with the semi-crystalline nature of the polyamide chain configuration.
A strong fragmentation peak was identified in the neutral mode at m/z 410.5 (RI = 94.5%) corresponding to (M+). In other words, the drug free polyamide disc had an average molecular mass distribution of 410.5 g/mole which may not be necessarily related to just the chemical backbone configuration but also the ionic fragmentation patterns of the polymer itself, when bombarded by the electron beam from the mass spectrometer probably due to the stoichiometric composition of the monomers and solvent system ratio. The intensity of the intramolecular hydrogen bonding, reaction stoichiometry and synthesis processes could also impact the fragmentation drifts.
The diffractogram showed both prominently sharp, high intensity diffraction peaks at 20, 24, 33, 46, 58, 67, 76°2theta and blunt, low intensity peaks representing the presence of both crystalline and amorphous segments confirming compact disc’s semi-crystalline nature which is typical of aliphatic polyamides (Fig. 6c). In this case, the diffractogram was predominated by the crystalline segments which further supports the displayed extended release behavior. The peak position and intensity centered “search-match routine” analysis performed on the obtained diffractogram showed that the polyamide backbone was composed of carbon, hydrogen, nitrogen and oxygen atoms which relates well to its conventionally known chemical structure (Fig. 1).
The obtained surface morphology can be described as single layered, densely packed with characteristic minimally porous intersecting longitudinal ridges (Fig. 6d). The nature of the polyamide-based compact disc exterior microstructure may possibly limit water molecule penetration through the polymeric chain network which explains the observed minimal burst effect, slow drug release kinetics and retarded matrix loss observed during dissolution and erosion (Fig. 3a).
3.7 Short-term environmental stability studies
The stability of the amitriptyline hydrochloride contained in the polyamide disc was evaluated under preset incubator temperature (30 ± 2 °C) and humidity (65 ± 5%) conditions for a period 60 days representing average day-to-day environmental storage conditions. Amitriptyline hydrochloride content measured at days 0, 30 and 60 days are illustrated in Fig. 7. Summarily, this preliminary study showed that changes in drug content tested on the different days were not significant (p-values > 0.05) indicating the relative stability of the compact disc as a drug carrier. Besides, the drug loaded formulation retained their original physical appearance (colour and texture) throughout the evaluation period.

Measured amitriptyline hydrochloride content within the polyamide disc over 0, 30 and 60 days
4 Conclusion and future perspectives
This proof-of-concept study details the stoichiometry-based chemical synthesis of a typical aliphatic polyamide, poly [Imino-1,6-Hexanediylimino (1,10–Dioxo-1,10-Decanediyl)] and its eventual formulation into an erodible, extended release, amitriptyline hydrochloride loaded, compact drug delivery matrix. Further, the polyamide disc was characterized using physicochemical and physicomechanical methods with the aim of understanding its unique properties and how this complements its extended drug release capabilities especially using a hydrophilic drug molecule, amitriptyline hydrochloride, as a model. Summarily, the polyamide-based compact disc displayed consistent, well-sustained in vitro drug release kinetics and erodible tendencies under biorelevant conditions with highly robust physicochemical and physicomechanical characteristics which complemented it’s in vitro performance. In conclusion, polyamide 6,10 prepared in this study can possibly function as a useful pharmaceutical excipient for the development of stable, extended release drug carriers such as implantable systems. The formulation’s ability to load, regulate and extend the release of amitriptyline chloride (model drug) in vitro together with its erodible, physicochemical and physicomechanical characteristics support the need for further development. Further investigations will encompass extensive drug release (over 30 days), extended stability investigations under diverse storage conditions, pharmacokinetic, efficacy and bioerosion studies using biorelevant in vitro methods and experimental animal models (e.g. pigs) that closely represent the human system. Moreover, the need to perform detailed in vitro and in vivo toxicity profiling of the polyamide-based compact disc and its degradation products utilizing normal cells and animal models are also essential for understanding the biocompatibility of this polymeric drug carrier. Considering the flexible and monolithic nature of the polyamide disc, it would be beneficial to expand its use as a carrier to other drugs and bioactive agents.
References
Bruschi ML (2015) Strategies to modify the drug release from pharmaceutical systems. Woodhead Publishing, Cambridge (CA)
Paolini MS, Fenton OS, Bhattacharya C, Andresen JL, Langer R (2019) Polymers for extended-release administration. Biomed Microdevices 21(2):45
Sponchioni M, Palmiero UC, Moscatelli D (2019) Thermo-responsive polymers: applications of smart materials in drug delivery and tissue engineering. Mater Sci Eng C 102:589–605
Batool A, Arshad R, Razzaq S, Nousheen K, Kiani MH, Shahnaz G (2020) Formulation and evaluation of hyaluronic acid-based mucoadhesive self-nanoemulsifying drug delivery system (SNEDDS) of tamoxifen for targeting breast cancer. Int J Biol Macromol 152(1):503–515
Behar-Cohen F (2019) Recent advances in slow and sustained drug release for retina drug delivery. Expert Opin Drug Deliv 16(7):679–686
Tiwari G, Tiwari R, Sriwastawa B, Bhati L, Pandey S, Pandey P, Bannerjee SK (2012) Drug delivery systems: an updated review. Int J Pharm Invest 2(1):2–11
Jamzad S, Tutunji L, Fassihi R (2005) Analysis of macromolecular changes and drug release from hydrophilic matrix systems. Int J Pharm 292(1–2):75–85
Uchegbu IF, Schatzlein AG (2006) Polymers in drug delivery. CRC Press, Florida (FL)
Guo X, Zhao Z, Chen D, Qiao M, Wan F, Cun D, Sun Y, Yang M (2019) Co-delivery of resveratrol and docetaxel via polymeric micelles to improve the treatment of drug-resistant tumors. Asian J Pharm Sci 14(1):78–85
Stewart S, Domínguez-Robles J, Donnelly R, Larrañeta E (2018) Implantable polymeric drug delivery devices: classification, manufacture, materials, and clinical applications. Polym 10(12):1379
Janićijević Ž, Ninkov M, Kataranovski M, Radovanović F (2019) Poly (DL-Lactide-co-ε-Caprolactone)/Poly (Acrylic Acid) composite implant for controlled delivery of cationic drugs. Macromol Biosci 19(2):1800322
Padmakumar S, Paul-Prasanth B, Pavithran K, Vijaykumar DK, Rajanbabu A, Sivanarayanan TB, Kadakia E, Amiji MM, Nair SV, Menon D (2019) Long-term drug delivery using implantable electrospun woven polymeric nanotextiles. Nanomed Nanotechnol 15(1):274–284
Pons-Faudoa FP, Ballerini A, Sakamoto J, Grattoni A (2019) Advanced implantable drug delivery technologies: transforming the clinical landscape of therapeutics for chronic diseases. Biomed Microdevices 21(2):47
Cha GD, Kang D, Lee J, Kim DH (2019) Bioresorbable electronic implants: history, materials, fabrication, devices, and clinical applications. Adv Healthc Mater 8(11):1801660
Langer R (1990) New methods of drug delivery. Science 249(4976):1527–1533
Rajgor N, Patel M, Bhaskar VH (2011) Implantable drug delivery systems: an overview. Surg Neurol Int 2(2):91–95
Schlesinger E, Johengen D, Luecke E, Rothrock G, McGowan I, van der Straten A, Desai T (2016) A tunable, biodegradable, thin-film polymer device as a long-acting implant delivering tenofovir alafenamide fumarate for HIV pre-exposure prophylaxis. Pharm Res 33(7):1649–1656
Le Devedec F, Boucher H, Dubins D, Allen C (2018) Factors controlling drug release in cross-linked poly (valerolactone) based matrices. Mol Pharm 15(4):1565–1577
Jones NA, Atkins EDT, Hill MJ, Cooper SJ, Franco L (1997) Chain-folded lamellar crystals of aliphatic polyamides. investigation of nylons 4 8, 4 10, 4 12, 6 10, 6 12, 6 18 and 8 12. Polym 38(11):2689–2699
Gaymans RJ, Sikkema DJ (1989) Comprehensive polymer science: aliphatic polyamides, vol 5. Pergamon Press Plc, Oxford, pp 357–373
Kiely DE, Chen L, Lin TH (1994) Hydroxylated nylons based on unprotected esterified D-glucaric acid by simple condensation reactions. J Am Chem Soc 116(2):571–578
Chattaraj SC, Swarbrick J, Kanfer I (1995) A simple diffusion cell to monitor drug release from semi-solid dosage forms. Int J Pharm 120(1):119–124
Cui X, Liu Z, Yan D (2004) Synthesis and characterization of novel even–odd nylons based on undecanedioic acid. Eur Polym J 40(6):1111–1118
Chi E, An M, Yao G, Tian F, Wang Z (2017) The influence of epitaxial crystallization on the mechanical properties of polyamide 66/reduced graphene oxide nanocomposite injection bar. Crystals 7(12):384
Dolorico AM, Tayyani R, Ong HV, Gaster RN (2003) Shortterm and longterm visual and astigmatic results of an opposing 10–0 nylon double running suture technique for penetrating keratoplasty. J Am Coll Surg 197(6):991–999
Lee MJ, DePoli PA, Casas LA (2003) Aesthetic and predictable correction of the inverted nipple. Aesthet Surg J 23(5):353–356
Bougherara H, Zdero R, Dubov A, Shah S, Khurshid S, Schemitsch EH (2011) A preliminary biomechanical study of a novel carbon–fibre hip implant versus standard metallic hip implants. Med Eng Phys 33(1):121–128
Wang X, Jolliffe A, Carr B, Zhang Q, Bilger M, Cui Y, Wu J, Wang X, Mahoney M, Rojas-Pena A, Hoenerhoff MJ (2018) Nitric oxide-releasing semi-crystalline thermoplastic polymers: preparation, characterization and application to devise anti-inflammatory and bactericidal implants. Biomater Sci 6(12):3189–3201
Xu Y, Li H, Wu J, Yang Q, Jiang D, Qiao B (2018) Polydopamine-induced hydroxyapatite coating facilitates hydroxyapatite/polyamide 66 implant osteogenesis: an in vitro and in vivo evaluation. Int J Nanomed 13:8179–8193
Torres D, Seijo B, García-Encina G, Alonso M, Vila-Jato JL (1990) Microencapsulation of ion-exchange resins by interfacial nylon polymerization. Int J Pharm 59(1):9–17
Ostad SN, Gard PR (2000) Cytotoxicity and teratogenicity of chlorhexidine diacetate released from hollow nylon fibres. J Pharm Pharmacol 52(7):779–784
Vyas SP, Venugopalan ASP, Venkatesan N (2000) Preparation and characterization of microencapsulated gelospheres for controlled oral theophylline delivery. J Microencapsul 17(6):767–775
Chu LY, Liang YJ, Chen WM, Ju XJ, Wang HD (2004) Preparation of glucose-sensitive microcapsules with a porous membrane and functional gates. Coll Surf B Biointerfaces 37(1–2):9–14
Kolawole OA, Pillay V, Choonara YE, du Toit LC, Ndesendo VM (2010) The influence of polyamide 6, 10 synthesis variables on the physicochemical characteristics and drug release kinetics from a monolithic tablet matrix. Pharm Dev Technol 15(6):595–612
Kolawole OA, Pillay V, Choonara YE (2007) Novel polyamide 6, 10 variants synthesized by modified interfacial polymerization for application as a rate-modulated monolithic drug delivery system. J Bioact Compat Polym 22(3):281–313
Adeleke OA, Choonara YE, Kumar P, du Toit LC, Tomar LK, Tyagi C, Pillay V (2013) Evaluation of the impacts of formulation variables and excipients on the drug release dynamics of a polyamide 6, 10-based monolithic matrix using mathematical tools. AAPS PharmSciTech 14(4):1349–1359
Abdullah AM, Rahim TNAT, Hamad WNFW, Mohamad D, Akil HM, Rajion ZA (2018) Mechanical and cytotoxicity properties of hybrid ceramics filled polyamide 12 filament feedstock for craniofacial bone reconstruction via fused deposition modelling. Dent Mater 34(11):e309–e316
Choi JW, Yun BH, Jeong CM, Huh JB (2018) Retentive properties of two stud attachments with polyetherketoneketone or nylon insert in mandibular implant overdentures. Int J Oral and Maxillofac Implants 33(5):1079–1088
del Burgo LS, Ciriza J, Espona-Noguera A, Illa X, Cabruja E, Orive G, Hernández RM, Villa R, Pedraz JL, Alvarez M (2018) 3D Printed porous polyamide macrocapsule combined with alginate microcapsules for safer cell-based therapies. Sci Rep 8(1):8512
Dória RGS, Freitas SHD, Hayasaka YDB, Hage MCFNS, Strefezzi RDF, Carregaro AB, Reginato GM, Ambrósio CE, Müller AF (2018) Evaluation of polyamide surgical mesh as an abdominal ventral implant in rabbits. Acta Cir Bras 33(5):454–461
Olkhov A, Gur’ev V, Akatov V, Mastalygina E, Iordanskii A, Sevastyanov VI (2018) Composite tendon implant based on nanofibrillar polyhydroxybutyrate and polyamide filaments. J Biomed Mater Res A 106(10):2708–2713
Chen G, Yin M, Liu W, Xin B, Bai G, Wang J, Wang J, Gao X, Wang Y, Liu C, Cheng L (2019) A novel height-adjustable nano-hydroxyapatite/polyamide-66 vertebral body for reconstruction of thoracolumbar structural stability after spinal tumor resection. World Neurosurg 122:e206–e214
Li Y, Yan D, Zhu X (2001) Crystal forms of nylon 10 12 crystallized from melt and after solution casting. Eur Polym J 37(9):1849–1853
Lundborg G, Dahlin L, Dohi D, Kanje M, Terada N (1997) A new type of “bioartificial” nerve graft for bridging extended defects in nerves. J Hand Surg 22(3):299–303
Manzo RH, Olivera ME, Amidon GL, Shah VP, Dressman JB, Barends DM (2006) Biowaiver monographs for immediate release solid oral dosage forms: amitriptyline hydrochloride. J Pharm Sci 95(5):966–973
Giannola LI, De Caro V, Giandalia G, Siragusa MG, Tripodo C, Florena AM, Campisi G (2007) Release of naltrexone on buccal mucosa: permeation studies, histological aspects and matrix system design. Eur J Pharm Biopharm 67(2):425–433
Briscoe BJ, Sinha SK (1999) Hardness and normal indentation of polymers: mechanical properties and testing of polymers. Springer, Dordrecht, NL
Sundararajan G, Roy M (2001) Encyclopedia of materials: science and technology. Ref Mod Mater Sci Mater Eng 2001:3728–3736
Mills D (2016) Pneumatic conveying design guide, third edition: Introduction to pneumatic conveying and guide (chapter one). Butterworths, Oxford, UK
Kopp S (2006) Stability testing of pharmaceutical products in a global environment. RAJ Pharm 5:291–293
Karatas A, Baykara T (2001) Studies on indomethacin inserts prepared by water-soluble polymers: II. the relation between dissolution rate and swelling behaviour. Farmaco 56(3):197–202
Siepmann J, Peppas NA (2001) Mathematical modeling of controlled drug delivery. Adv Drug Deliv Rev 48(2–3):137
Yang X, Trinh HM, Agrahari V, Sheng Y, Pal D, Mitra AK (2016) Nanoparticle-based topical ophthalmic gel formulation for sustained release of hydrocortisone butyrate. AAPS PharmSciTech 17(2):294–306
Chou SF, Woodrow KA (2017) Relationships between mechanical properties and drug release from electrospun fibers of PCL and PLGA blends. J Mech Behav Biomed 65:724–733
Wong RSH, Dodou K (2017) Effect of drug loading method and drug physicochemical properties on the material and drug release properties of poly (ethylene oxide) hydrogels for transdermal delivery. Polym 9(7):286
Coates J (2000) Encyclopedia of analytical chemistry: interpretation of infrared spectra, a practical approach. Wiley, Chichester, UK
Cui X, Yan D (2005) Preparation, characterization and crystalline transitions of odd–even polyamides 11, 12 and 11, 10. Eur Polym J 41(4):863–870
Murthy NS (2006) Hydrogen bonding, mobility, and structural transitions in aliphatic polyamides. J Polym Sci Poly Phys 44(13):1763–1782
Acknowledgements
The author acknowledges the South African Medical Research Council for the award of funding. OA thanks Professors Pillay and Danckwerts at the University of the Witwatersrand, Johannesburg, South Africa for their valuable dialogues and guidance on some aspects of the work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Adeleke, O.A. In vitro characterization of a synthetic polyamide-based erodible compact disc for extended drug release. SN Appl. Sci. 2, 2152 (2020). https://doi.org/10.1007/s42452-020-03954-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-03954-x