
Abstract
Novel Schiff base of N-(2,6-dibenzylidenecyclohexylidene)-N′-(2,4-dinitrophenyl)hydrazine derivatives were synthesized by the condensation of 2,4-dinitrophenylhydrazine with substituted 2,6-dibenzylidenecyclohexanone. The synthesized compounds were characterized by UV-Visible, Fourier transform infrared, nuclear magnetic resonance and elemental analysis studies. Density functional theory was performed to predict structural geometries of compounds. The structures were optimized using B3LYP/6-311G+(2d,p) basis sets. The energies of HOMO and LUMO were found −6.23109 eV and −3.21595 eV for 4a, −5.77287 eV and −2.95528 eV for 4b, −5.28554 eV and −2.76835 eV for 4c respectively. These results revealed the possibility of intramolecular charge transfer during excitation of the molecules. Other quantum chemical parameters of compound were also calculated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
A compound in which the C=O is replaced by a C=N is known as an imine or Schiff base. It was first discovered by a German chemist, Hugo Schiff [1]. Hydrazone is one kind of Schiff base and it is a class of organic compounds having the basic structure R1R2C=NNH- [2]. High physiological activity is the most important properties of hydrazones and is an important class of compounds containing an azomethine group (–NHN=C–) having two different nature of connected nitrogen atoms and a C=N double bond that is conjugated with a lone pair electron of the terminal nitrogen atom [3,4,5]. Extensive studies have revealed that the nitrogen atom of azomethine group having the lone pair of electrons in its sp2 hybridized orbital is considered responsible the chemical and biological activities [6, 7]. Schiff base ligands are the important ligands in coordination chemistry owing to their ease of preparation, structural variety and polydenticity. These ligands show miscellaneous uses comprising electro-chemical behavior and biological field [8]. The metal complexes of Schiff base ligands are also promising features since the uses of Schiff base ligands enormously become widespread upon coordination with metal ions [9, 10]. The coordination mode adopted by hydrazone depends on various factors such as tautomerism, various reactions conditions, nature and number of the substituents on hydrazone skeleton and stability of the complex formed. The structural changes of the different segment attached to the hydrazine will affect the metal binding of the ligand and exhibit fascinating coordination modes with transition metal ions and formed different types of complexes [11, 12]. Membrane sensors based on Schiff bases also act as chelating ionophores in trace analysis for organic pollutants, metal ions and biological substances [13]. Particularly the 2,4-dinitrophenylhydrazones derivatives based Schiff bases have been shown to be potentially DNA-damaging and mutagenic agents [14]. In addition, the 2,4-dinitrophenylhydrazones derivatives exhibit good nonlinear optical (NLO) and crystalline properties [15, 16]. Owing to their significant applications, these compounds have attracted much attention to the researchers. But still, there are few reports based on both experimental and computational study of 2,4-dinitrophenylhydrazone derivatives based Schiff bases. Therefore, in order to obtain further information about 2,4-dinitrophenylhydrazone derivatives such as N-(2,6-dibenzylidenecyclohexylidene)-N′-(2,4-dinitrophenyl)hydrazine derivatives (Scheme 1), the compounds were synthesized and characterized by UV-Visible, Fourier transform infrared (FTIR), nuclear magnetic resonance (NMR) and elemental analysis studies. The structures were optimized by density functional theory (DFT) method. Furthermore, quantum chemical calculation, molecular orbital energy, mulliken charge and frontier orbital constitution were performed to divulge the energy of the examined compounds.

Preparation of substituted N-(2,6-dibenzylidenecyclohexylidene)-N′-(2,4-dinitrophenyl)hydrazine
2 Results and discussion
Substituted bis-chalcone based hydrazones were synthesized by condensation of substituted bis-chalcones with hydrazine as previously shown in Scheme 1. The products were identified on the basis of UV-Visible, FTIR, NMR spectra and elemental analysis. The FTIR spectra of all compounds showed the characteristics stretching vibration of the NH group at 3263–3270 cm−1. This band along with the C=N absorption band between 1612 and 1620 cm−1, is a strong evidence for the presence of an azomethine group [17]. The C=C in conjugation with C=N and C=C stretching of phenyl showed at 1585–1590 cm−1. Another important band at 1385–1392 cm−1 was attributed to stretching of C–NH. The substituted hydrazones were further examined by their 1H NMR and 13C NMR spectra in DMSO-d6. The characteristics signals for the protons of NH- were observed δ = 11.5 ppm as well as aromatic protons in the region of δ from 7.22 to 8.79 ppm. The values are in good agreement with the literature values [17]. In 13C NMR spectra, the significant signals are for azomethine carbon between δ from 143 and 150 ppm. This values are also similar as literature values [18].
3 Computational study
3.1 Geometry optimization
Optimized structures is the key materials to define its structural parameters. The examined compounds were optimized at DFT/6-311G+(2d,p) level of theory. Single point energies of the measured compounds were shown in Table 1.
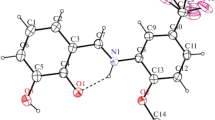
Among the optimized structure, compound N-(2,6-(4-dimethylaminobenzylidene)-N′-(2,4-dinitrophenyl)hydrazine by means of B3LYP/6-311G+(2d,p) basis set was shown in Fig. 1.

Optimized structure of N-(2,6-(4-dimethylaminobenzylidene)-N′-(2,4-dinitrophenyl)hydrazine at DFT B3LYP/6-311G+(2d,p) level of theory
Rotational isomer of the examined compounds based on N–H group relative to NO2 were also checked by the same method. It was found that the isomer where N–H and NO2 on the same side are stable and forming intramolecular hydrogen bond with the bond distance 1.86 Å.
The structural parameters such as bond lengths, bond angles and dihedral angles of the examined compounds using DFT/6-311G+(2d,p) basis set given in Table 2. The C=N bond distance for all the substituted hydrazones are similar and the value is 1.2956 ± 0.002. The values are good agreement with the experimental reported values [19]. Both benzene rings are regular hexagons with C–C bond lengths (1.40 Å) somewhat in between the normal values for a single (1.54 Å) and a double (1.33 Å) bond [20]. For simplify the calculation, the compound has three phenyl rings as designated C8–C13 (PhI), C15–C20 (PhII), C15–C20 (PhIII) and cyclohexayl rings. Dihedral angles of a molecule provided information about planarity. Generally the values of dihedral angle is 0°, 180° or 360° showed planarity of a molecule. The calculated dihedral angles between these ring planes are as PhI/PhII = 52.12, PhII/PhIII = 8.92 and PhI/PhIII = 50.88. These results revealed that the rings are in out-of-plane. The bond angles of the optimized structures are also calculated. The C3 atom in cyclohexayl ring makes bond angles, C3–C2–C13 = 118.86 Å and C3–C4–C14 = 119.71 Å. The C26 atom in phenyl ring makes bond angles, C13–C26–C28 = 117.88 Å and C13–C26–C27 = 125.97 Å as well as C14–C16–C17 = 118.09 Å and C14–C16–C18 = 125.90 Å. All the bond angles are significantly deviated from trigonal planarity. The other Schiff base derivatives found similar results which shown in Table 2.
3.2 Vibrational and NMR studies
The IR spectra of the examined compounds calculated at the DFT/6-311G+(2d,p) level of theory and scaled by 0.9688 [21]. The calculated and the observed FTIR spectra showed good agreement that shown in Table 3.
Therefore, more trustworthy consignment of IR active modes of vibration can be performed by comparing experimental FTIR intensities with the computed wavenumbers. The sharp band for N–H stretching frequencies are found in the region of 3342 cm−1.The C–H stretching vibrations of the aromatic are found in the frequency region of 3141, 3105 and 3028 cm−1. Generally, in the aromatic system, stretching vibrations of C–H occur in variable intensity at 3093, 3075, 3030, and 3017 cm−1 [22]. The C–H stretching of aliphatic system is found in the region 2981–2891 cm−1. The aromatic stretching of C–C occur at 1596–1592 cm−1. The sharp band in the frequency of 1339–1314 cm−1 and 1157–1069 cm−1 are assigned as stretching vibration of C–N bond.
The nuclear shieldings (absolute values) are computed by gauge independent atomic orbital method (GIAO) with B3LYP/6-311G+(2d,p) level of theory. The calculated 1H NMR for the examined compounds showed that the characteristics signals for the protons of NH are found between δ from 12.55 to 12.66 ppm. On the other hand for 13C NMR signals for azomethine carbon between δ from 149.51 to 149.76 ppm. These values are in good agreement with the experimental results.
3.3 Electronic properties
Frontier molecular orbital (FMO) and molecular electrostatic potential (MEP) calculations of compounds are performed by DFT/B3LYP/6-311G+(2d,p) method. Since MEP maps show three-dimensional charge distribution of molecules, they provide information about electron acceptor as well as electron donor regions [23]. These regions are represented by different colors. On MEP map, red regions constitute negative potential areas, where blue color constitute positive potential areas and finally green color refers to zero potential areas. To estimate possible intermolecular and intramolecular hydrogen bonds as well as intermolecular charge transfer (ICT), these regions have to be focused. As the MEP map for the compounds 4a–4c in Fig. 2 are examined, it could be seen that the highest negative and lowest negative regions line up according to the positioning of O and N atoms, respectively.

Molecular Electrostatic Potential (MEP) calculated at DFT B3LYP/6-311G+(2d,p) level of theory
3.4 Frontier molecular orbitals
HOMO and LUMO molecular orbitals are the frontier orbitals and they show electron donor and electron acceptor ability of the molecule, respectively as presented by Parr and Nitti [24, 25]. The EHOMO and ELUMO energies correspond to ionization energy (I) and electron affinity (A), respectively.
As Fig. 3, it can be seen that HOMO is located in the whole region except for PhII ring and LUMO is located only in PhIII. Therefore it exposed that intramolecular charge transfer is predominantly happened during excitation of the molecules. The computed energies of HOMO and LUMO is −6.23109 eV and −3.21595 eV for 4a, −5.77287 eV and −2.95528 eV for 4b, −5.28554 eV and −2.76835 eV for 4c respectively. Ionization potential is the energy absorbed as one electron is released from HOMO and its trend follows the decreasing order: 4a > 4b > 4c, while electron affinity, which is the energy released when one electron is added to LUMO decreases in the order: 4a > 4b > 4c. From these results it is concluded that 4a absorbs high energy to becoming a cation and also releases high energy to becoming an anion in relation to the others in the group. According to quantum theory, the mixing of appropriate excited state wave functions with the ground state wave function changes the electron density of the chemical entity and the mixing coefficient is inversely proportional to the excitation energy between the ground and the excited state. So, the rigid molecule has a large HOMO–LUMO gap on the other hand a soft molecule has a small HOMO–LUMO gap. For considering chemical reactivity, soft molecules with a small gap in their electron density alter more easily and hence will be more reactive than hard molecule. For the studied schiff base derivatives, compound 4c having small value of HOMO-LUMO gap indicates a soft molecule with high reactivity compared to the others.

HOMO and LUMO of substituted N-(2,6-dibenzylidenecyclohexylidene)-N′-(2,4-dinitrophenyl)hydrazine at B3LYP/6-311G+(2d,p) level of theory
Quantum chemical parameters such as chemical hardness (η) and softness (σ); chemical potential (μ) and electronegativity (χ); nucleophilicity (ε) and electrophilicity (ω) can be calculated using the following equations [26].
The values of 4a–4c compounds are; χ = −μ = 4.72 eV, 4.36 eV, 4.03 eV; η = 1/σ = 1.51 eV, 1.41 eV, 1.26 eV and ω = 1/ε = 7.39 eV, 6.76 eV, 6.44 eV respectively. According to these parameters, the chemical reactivity varies with the substituents of schiff bases. Nonlinear properties of N-(2,6-(4-dimethylaminobenzylidene)-N′-(2,4-dinitrophenyl)hydrazine is also confirmed by contour optimized structure shown in Fig. 4.

Contour structure of N-(2,6-(4-dimethylaminobenzylidene)-N′-(2,4-dinitrophenyl)hydrazine at DFT B3LYP/6-311G+(2d,p) level of theory
3.5 Mulliken charges distribution
The Mulliken charge distribution of all atoms of the optimized structure of the substituted Schiff base derivatives are calculated at the same level of theory. Mulliken charges for compound 4c shown in Fig. 5.

Mulliken charges of N-(2,6-(4-dimethylaminobenzylidene)-N′-(2,4-dinitrophenyl)hydrazine at DFT B3LYP/6-311G+(2d,p) level of theory
The calculated results demonstrate that the atomic electronegativity plays vital roles for Mulliken charge distribution. The atom having bigger electronegativity will carry large negative charges, whereas the atom having smaller electronegativity will carry less negative or positive charges in the bonding between two atoms. So, for the studied compound N (from nitro group) are less negative, H and C (non-bonding with H atoms) are all positive. N (from amine group), O and C (bonding with H atoms) are all negative. Similar results are observed for other derivatives.
4 Experimental section
All materials otherwise noted were used as purchased without further purification. UV-visible spectra of the sample were recorded on UV-180 SHIMADZU Spectrophotometer with a scanning range of 800–220 nm. FTIR spectrum was recorded on IRTracer-100, SHIMADZU, Japan over the frequency range from 4000 to 400 cm−1 using KBr pellets. Melting point was measured by Stuart’s electro thermal melting point apparatus (Model no. SMP 30). 1H NMR and 13C NMR spectra of the sample were measured with an AVANCE Bruker NMR spectrometer at 400 MHz using suitable solvent and TMS as an internal standard. The elemental analysis was performed using CE-440 elemental analyzer, Exeter Analytical, Inc.
4.1 General procedure for synthesis of substituted bis-chalcones [27]
A mixture of cyclohexanone (1.0 equivalent), substituted benzaldehyde (2.0 equivalent), and sodium hydroxide pellet (2.2 equivalent) grinded in a mortar and pestle for 20 min at room temperature and then 10% hydrochloric acid was poured in the reaction mixture. The solid product was filtered and dried. The crude product was purified by recrystallization from ethanol and ethyl acetate.
4.1.1 2,6-Bis-(4-chlorobenzylidene)cyclohexanone (3a)
Yellowish green powder, m.p.: 127 °C, yield 90%. UV (EtOH): λmax = 336. IR (KBr): 2929, 1666, 1575, 1576, 1489, 1263, 1159, 819, 798 cm−1. 1H NMR (400 MHz, CDCl3) δ = 7.73 (s, 2H, 2CH) 7.38–7.25 (m, 8H, Ar), 2.91–2.87 (m, 4H, 2CH2), 1.84–1.77 (m, 2H, CH2).
4.1.2 2,6-Bis-(4-methoxybenzylidene)cycloexanone (3b)
Pale yellow powder, m.p.: 152 °C, yield 88%. UV (EtOH): λmax = 360. IR (KBr): 2937, 1656, 1593, 1161, 1184, 833 cm−1. 1H NMR (400 MHz, CDCl3) δ = 7.76 (s, 2H, 2CH), 7.45 (d, J = 8.7 Hz, 4H, Ar), 6.93 (d, J = 8.7 Hz, 4H, Ar), 3.85 (s, 6H, 2CH3O), 2.92 (t, J = 14.2, 4H, 2CH2), 1.84–1.78 (m, 2H, CH2).
4.1.3 2,6-Bis-(4-dimethylaminobenzylidene)cyclohexanone (3c)
Yellow powder, m.p.: 136 °C, yield 85%. UV (EtOH): λmax = 450. IR (KBr): 2915, 2853, 1658, 1551, 1367, 1302, 1159, 823 cm−1. 1H NMR (400 MHz, CDCl3) δ = 7.76 (s, 2H, 2CH), 7.45 (d, J = 8.8 Hz, 4H, Ar), 6.71 (d, J = 8.8 Hz, 4H, Ar), 3.01 (s, 12H, 2N(CH3)2), 2.94 (t, J = 14.1, 4H, 2CH2), 1.83–1.79 (m, 2H, CH2).
4.2 General procedure for synthesis of N-[2,6-bis-(benzylidene)cyclohexylidene]-N′-(2,4-dinitrophenyl)hydrazine derivatives [28]
2,4-dinitrophenyl hydrazine (1.0 mmol) and 20 mL of methanol were taken in a round bottom flask. Then 1.0 mL of 10% hydrochloric acid was added to this solution. After that substituted 2,6-bis-benzylidinecyclohexanone (1.0 mmol) was added to this clear solution. The reaction mixture was reflux for 2 h and allowed to stand for overnight. The residue was separated by filtration and washed with ethanol then dried in air. The crude product was separated by column chromatography.
4.2.1 N-[2,6-bis-(4-chlorobenzylidene)cyclohexanone]-N′-(2,4-dinitrophenyl)hydrazine (4a)
Flash column chromatography on silica gel using eluent (HX/EtOAc = 9/1; v/v). Red powder, m.p.: 185 °C, yield 83%. UV (EtOH): λmax = 342. IR (KBr): 3263, 3095, 2941, 2916, 1612, 1585, 1508, 1490, 1332, 1139, 1082, 1055, 1010, 885, 827, 740, 653, 626 cm−1. 1H NMR (400 MHz, DMSO-d6) δ: = 11.5 (s, 1H, N–H), 8.9 (d, J = 8.7 Hz, 1H, Ar), 8.5 (d, J = 8.7 Hz, 1H, Ar), 8.2 (d, J = 8.7 Hz, 1H, Ar), 7.5–7.4 (m, 8H, Ar), 7.2 (s, 2H, 2CH), 2.6–2.8 (m, 4H, cyclic), 1.7 (m, 2H, cyclic). 13C NMR (125 MHz, DMSO-d6): δ = 151, 143, 137, 134, 134, 133.5, 132, 129, 28, and 22. Anal. calcd for C26H20Cl2N4O4 (523.37): C, 59.57; H, 3.81; N, 10.64; O, 12.18. Found C, 59.67; H, 3.85; N, 10.71; O, 12.23.
4.2.2 N-[2,6-bis-(4-methoxybenzylidene)cyclohexanone]-N′-(2,4-dinitrophenyl)hydrazine (4b)
Flash column chromatography on silica gel using eluent (HX/EtOAc = 9/1; v/v). Coffee color powder, m.p.: 225 °C, yield 65%. UV (EtOH): λmax = 384. IR (KBr): 3269, 3107, 2931, 2835, 1612, 1585, 1506, 1475, 1249, 1170, 1130, 1076, 1024, 831, 740, 628 cm−1. 1H NMR (400 MHz, DMSO-d6) δ: = 11.5 (s, 1H, N–H), 8.4 (d, J = 8.7 Hz, 1H, Ar), 8.0–8.1 (m, 4H, Ar), 7.7 (s, 1H, Ar), 7.2 (s, 1H, Ar) 7.0–7.1 (m, 4H, Ar), 6.9 (s, 2H, 2CH), 3.8 (s, 6H, 2CH3O), 1.6–1.8 (m, 4H, cyclic), 1.2 (m, 2H, cyclic). 13C NMR (125 MHz, DMSO-d6): δ = 150, 145, 137, 131.5, 130, 129, 127, 123, 117, 114, and 57. Anal. calcd for C28H26N4O6 (514.53): C, 65.31; H, 5.04; N, 10.81; O, 18.60. Found C, 65.36; H, 5.09; N, 10.89; O, 18.66.
4.2.3 N-[2,6-bis-(4-dimethylaminobenzylidene)cyclohexanone]-N′-(2,4-dinitrophenyl)hydrazine (4c)
Flash column chromatography on silica gel using eluent (HX/EtOAc = 9/1; v/v). Black powder, m.p.: 245 °C, yield 75%. UV (EtOH): λmax = 439. IR (KBr): 3271, 3091, 2931, 2835, 1612, 1602, 1502, 1413, 1327, 1305, 1249, 1076, 1024, 829, 740, 628 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 11.5 (s, 1H, N–H), 8.8 (s, 1H, Ar), 8.6 (s, 1H, Ar), 8.3–8.4 (m, 1H, Ar), 8.1 (d, J = 8.7 Hz, 4H, Ar), 7.8 (d, J = 8.7 Hz, 4H, Ar), 6.8 (s, 2H, 2CH), 3 (s, 12H, 4CH3), 2 (s, 4H, cyclic), 1.27 (s, 2H, cyclic). 13C NMR (125 MHz, DMSO-d6): δ = 151, 144, 136.5, 130, 129, 128.5, 123.5, 121, 116.5, and 112. Anal. calcd for C30H32N6O4 (540.61): C, 66.59; H, 5.92; N, 15.51; O, 11.79. Found C, 66.65; H, 5.97; N, 15.55; O, 11.84.
5 Computational method
Theoretical calculations were carried out using the Gaussain 16 program suite [29]. The geometries was entirely optimized by the DFT-B3LYP/6-311G+(2d,p) level of theory. Vibrational normal-mode analyses were performed at the same level to ensure that each optimized structure was a true minimum on the potential energy surface. Unscaled B3LYP/6-311G+(2d,p) frequencies were used to obtain thermochemical quantities, the thermal enthalpy and free energy corrections. After optimizations, the Mulliken charge and properties of frontier molecular orbitals of the studied compounds were investigated using the results calculated at B3LYP/6-311G+(2d,p) level of theory.
6 Conclusions
Hydrazone schiff base derivatives were synthesized in a general method for the addition of 2,4-dinitrophenylhydrazine and substituted 2,6-dibenzylidenecyclohexanone based bis-chalcones in the presence of acid. The characteristics signal for the proton of NH, the δ value 11.5 ppm in 1H NMR and signals are for azomethine carbon between δ from 143 and 150 ppm in 13C NMR spectra were observed. Theoretical studies were performed by DFT method using B3LYP/6-311G+(2d,p) level of theory. Nonlinear structure of the compounds were found through optimizing the structure. The energy of HOMO and LUMO molecular orbital showed the possibility of intramolecular charge transfer as well as hydrogen bond. Quantum chemical parameters of compounds were estimated using EHOMO and ELUMO. It was found that chemical potential (μ) and electronegativity (χ); chemical hardness (η) and electrophilicity (ω) follow the order 4a > 4b > 4c whereas chemical softness (σ) and nucleophilicity (ε) follow the reverse order 4a < 4b < 4c. The results revealed that substituent effect plays a vital role on stability, reactivity and excitation of the compounds.
References
Schiff H (1869) Untersuchungen über Salicinderivate. Justus Liebigs Ann Chem 150:193–200. https://doi.org/10.1002/jlac.18691500206
Lygaitis R, Getautis V, Grazulevicius JV (2008) Hole-transporting hydrazones. Chem Soc Rev 37:770–788. https://doi.org/10.1039/b702406c
Kucukoglu K, Gul HI, Taslimi P, Gulcin I, Supuran CT (2019) Investigation of inhibitory properties of some hydrazone compounds on hCA I, hCA II and AChE enzymes. Bioorg Chem 86:316–321. https://doi.org/10.1016/j.bioorg.2019.02.008
Patra D, Paul S, Sepay N, Kundu R, Ghosh T (2018) Structure-activity relationship on DNA binding and anticancer activities of a family of mixed-ligand oxidovanadium (V) hydrazone complexes. J Biomol Struct Dyn 36:4143–4155. https://doi.org/10.1080/07391102.2017.1409652
Al Zoubi W, Al-Hamdani AAS, Ahmed SD, Ko YG (2018) A new azo-Schiff base: synthesis, characterization, biological activity and theoretical studies of its complexes. Appl Organomet Chem 32:3895. https://doi.org/10.1002/aoc.3895
Wang X, Yin J, Shi L, Zhang G, Song B (2014) Design, synthesis, and antibacterial activity of novel Schiff base derivatives of quinazolin-4(3H)-one. Eur J Med Chem 77:65–74. https://doi.org/10.1016/j.ejmech.2014.02.053
Mukherjee AJ, Zade SS, Singh HB, Sunoj RB (2010) Organoselenium chemistry: role of intramolecular interactions. Chem Rev 110:4357–4416. https://doi.org/10.1021/cr900352j
Al Zoubi W, Ko YG (2017) Schiff base complexes and their versatile applications as catalysts in oxidation of organic compounds: Part I. Appl Organomet Chem 31:3574. https://doi.org/10.1002/aoc.3574
Radunsky C, Kösters J, Müller J (2015) Chromogenic behaviour of a family of hydrazine and hydrazone metal complexes. Inorg Chim Acta 428:14–20. https://doi.org/10.1016/j.ica.2015.01.012
Fekri R, Salehi M, Asadi A, Kubicki M (2019) Synthesis, characterization, anticancer and antibacterial evaluation of Schiff base ligands derived from hydrazone and their transition metal complexes. Inorg Chim Acta 484:245–254. https://doi.org/10.1016/j.ica.2018.09.022
Malik MA, Dar OA, Gull P, Wani MY, Hashmi AA (2018) Heterocyclic Schiff base transition metal complexes in antimicrobial and anticancer chemotherapy. MedChemComm 9:409–436. https://doi.org/10.1039/c7md00526a
Zhang JP, Zhang YB, Lin JB, Chen XM (2012) Metal azolate frameworks: from crystal engineering to functional materials. Chem Rev 112:1001–1033. https://doi.org/10.1021/cr200139g
Al Zoubi W, Al Mohanna N (2014) Membrane sensors based on Schiff bases as chelating ionophores–a review. Spectrochim Acta A 132:854–870
Jasinski JP, Braley AN, Chidan Kumar CS et al (2011) (E)-1-(2,4-Dinitrophenyl)-2-(2-fluorobenzylidene) hydrazine. Acta Crystallogr E 67:1200–1201. https://doi.org/10.1107/S1600536811014383
Kaya Y, Yilmaz VT, Buyukgungor O (2016) Synthesis, spectroscopic, structural and quantum chemical studies of a new imine oxime and its palladium(II) complex: hydrolysis mechanism. Molecules 21:52. https://doi.org/10.3390/molecules21010052
Basavarajappa KV, Arthoba Nayaka Y, Purushothama HT et al (2020) Optical, electrochemical and current–voltage characteristics of novel coumarin based 2,4-dinitrophenylhydrazone derivatives. J Mol Struct 1199:126946. https://doi.org/10.1016/j.molstruc.2019.126946
Suydam FH (1963) The C=N stretching frequency in azomethines. Anal Chem 35:193–195. https://doi.org/10.1021/ac60195a024
Hussain MM, Rahman MM, Arshad MN, Asiri AM (2017) Hg2+ sensor development based on (E)-N′-nitrobenzylidene-benzenesulfonohydrazide (NBBSH) derivatives fabricated on a glassy carbon electrode with a Nafion matrix. ACS Omega 2:420–431. https://doi.org/10.1021/acsomega.6b00359
Feilchenfeld H (1959) A relation between the lengths of single, double and triple bonds. J Phys Chem 63:1346. https://doi.org/10.1021/j150578a603
Mary YS, Raju K, Yildiz I et al (2012) FT-IR, FT-Raman, SERS and computational study of 5-ethylsulphonyl-2-(o-chlorobenzyl)benzoxazole. Spectrochim Acta A 96:617–625. https://doi.org/10.1016/j.saa.2012.07.006
Merrick JP, Moran D, Radom L (2007) An evaluation of harmonic vibrational frequency scale factors. J Phys Chem A 111:11683–11700. https://doi.org/10.1021/jp073974n
Kumar A, Srivastava AK, Gangwar S, Misra N, Mondal A, Brahmachari G (2015) Combined experimental (FT-IR, UV-visible spectra, NMR) and theoretical studies on the molecular structure, vibrational spectra, HOMO, LUMO, MESP surfaces, reactivity descriptor and molecular docking of Phomarin. J Mol Struct 1096:94–101. https://doi.org/10.1016/j.molstruc.2015.04.031
Kafer D, El Helou M, Gemel C, Witte G (2008) Packing of planar organic molecules: interplay of van der Waals and electrostatic interaction. Cryst Growth Des 8:3053–3057. https://doi.org/10.1021/cg800195u
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516. https://doi.org/10.1021/ja00364a005
Nitti A, Signorile M, Boiocchi M et al (2016) Conjugated thiophene-fused isatin dyes through intramolecular direct arylation. J Org Chem 81:11035–11042. https://doi.org/10.1021/acs.joc.6b01922
Pearson RG (1989) Absolute electronegativity and hardness: applications to organic chemistry. J Org Chem 54:1423–1430. https://doi.org/10.1021/jo00267a034
Badal MMR, Ashekul Islam HM, Maniruzzaman M, Abu Yousuf M (2020) Acidochromic behavior of dibenzylidene cyclohexanone-based bischalcone: experimental and theoretical study. ACS Omega 5:22978–22983. https://doi.org/10.1021/acsomega.0c02556
Behforouz M, Flynt MS, Bolan JL (1985) 2,4-Dinitrophenylhydrazones: a modified method for the preparation of these derivatives and an explanation of previous conflicting results. J Org Chem 50:1186–1189. https://doi.org/10.1021/jo00208a009
Frisch MJ, Trucks GW, Schlegel HB et al (2016) Gaussian 16 Revision C01. Gaussian Inc, Wallingford CT
Acknowledgements
The authors are acknowledging Khulna University of Engineering & Technology for financial support and necessary facilities. The authors are also grateful to Prof. Dr. Manabu Abe, Graduate School of Advanced Science and Engineering, Hiroshima University, Japan, for supporting the NMR analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary material
Tables S1–S3
(DOCX 22 kb)
Rights and permissions
About this article

Cite this article
Badal, M.M.R., Hossain, M.Z., Maniruzzaman, M. et al. Synthesis, identification and computational studies of novel Schiff bases N-(2,6-dibenzylidenecyclohexylidene)-N′-(2,4-dinitrophenyl)hydrazine derivatives. SN Appl. Sci. 2, 1914 (2020). https://doi.org/10.1007/s42452-020-03745-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-03745-4