
Abstract
Gossypol is a polyphenolic component found in pigment glands of whole cottonseed, it is toxic in its free-form (FG) to livestock farm animals. It can be degraded through the preparation and extraction of the tested sample, leading to inaccurate quantification of FG content in animal diets. The primary size of the laboratory samples, the particle size of the analytical samples, and interval of time between grinding and solvent extraction were tested as uncertainties of the sample preparations. The efficiency of extraction, accuracy, linearity, matrix effects, limits of detection and quantification were assessed in the chemical analysis. The chemical analysis contributes as a minor source of uncertainty in analysis, despite the occurrence of matrix effects. Solvent soaking time can be considered as an effective uncertainty source; at least 16 h of soaking in acetone should be applied to extract FG from glands. Trueness and precision are the accuracy parameters most affected in the steps prior to the chemical analysis. The reduction of particle size as the increase of the interval of time between grinding and chemical analysis affected the trueness of results. On the other hand, precision was affected by the sample size of analytical and laboratory sample preparation. The best practices to reduce the uncertainty in FG analysis increase the confidence of cottonseed use as an animal feed source regarding its toxicity, improving animal health and the safety of animal products intended to human consumption.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Gossypol is a polyphenolic binaphthyl dialdehyde pigment found in whole cottonseed and cottonseed cake [1], it exists in two forms within plants, a toxic free form (FG) and a non-toxic bound plant protein form [2]. The toxic effects of FG are cumulative, causing many reproductive and systemic disease syndromes for monogastric livestock and pre-ruminant animals, which are frequently negatively influenced by even minimal quantities of FG [3]. In general, ruminant animals have more resistance to toxic effects than monogastric, however, they are susceptible to the FG adverse effects [4]. Although cottonseed is not part of the human diet, exposure can occur through the consumption of animal products contaminated with gossypol residues. The transfer of gossypol residues occurs through animal metabolism and has already been described in the literature [5, 6], although there are no studies of gossypol screening in foods or exposure estimates.
Cottonseed cake is considered a good protein-rich source; however, the presence of FG limits its use in livestock feeds. The maximum allowed concentration of FG in cottonseed cake for animal diets are 100 ppm for poultry and calves, and 500 ppm for goats and sheep (except kids and lambs), and cattle (except calves) according to the regulation 2002/32 EU [7]. This strict maximum permitted levels for FG required reliable analytical methods that enable accurate quantification of FG at very low levels.
There are several tools for quality control to select accurate analytical methods that can be applied for FG quantification [8], wherein the most recommended methods are based on High-Performance Liquid Chromatography (HPLC) due to the sensitivity and accuracy of results. According to the Association Oil Chemists Society (AOCS), FG is defined as the portion of gossypol extractable by organic solvents as acetone [9]. However, even after full validation of a methodology, the FG results may have a degree of uncertainty associated with evaluated values. This is because the FG can be simply degraded under certain conditions through selection, preparation and extraction of the tested sample, e.g. exposure to temperature, oxygen as well as extraction with organic solvents [10]. Moreover, grinding of the samples also can rupture the FG glands, promoting the protein-complex formation of gossypol, consequently converting the toxic FG form to the non-toxic bounded form [1]. Time advancing between the grinding of the tested sample and FG extraction may also increase the risk of FG loss. Although these factors are among the uncertainties that can interfere with accurate quantification of FG content in various cottonseed samples and consequently the safety for livestock consumption, they have not been evaluated in previously published methodologies for FG analysis [1, 3, 11, 12]. The uncertainty of a result is defined as a range that characterizes the dispersion of the values that reasonably could be attributed to measurement; it is different from the error which is the difference between an individual result and the true value [13]. Therefore, the objective of the current study is to identify the uncertainty sources of FG measurement by HPLC quantification method through preparation and quantification of the experimental samples. These are including primary sample weight, the drift of the system characters between calibration and measurement, linearity, precision, trueness, etc. to determine which of them have a significant effect on result uncertainty.
2 Material and methods
2.1 Chemicals
Acetic acid and analytical standard of (±)-gossypol (≥ 95% purity HPLC, product no. G8761) from cotton seeds were obtained from Sigma-Aldrich, Inc. (St. Louis, USA). Methanol and chloroform were HPLC grade, and acetone was of analytical grade. High purity water was obtained by using a Milli-Q water purification system.
2.2 Uncertainties sources from cottonseed sampling and sample preparation
A homogeneous representative cottonseed sample (5 kg) of cultivar IAC 25-RMD was collected in the same harvesting period from Agronomic Research Institute (IAC, São Paulo, Brazil).
2.2.1 Laboratory and analytical sample size
To distinguish the effects of the primary laboratory sample size, different subsample weights of 10, 25, 50 and 100 g were taken from the primary homogenized whole cottonseed samples in triplicates to prepare the laboratory ground samples. Each subsample was manually de-hulled, weighed and ground through a 1 mm screen. From each cottonseed subsample, analytical samples of 0.3 g were extracted by acetone soaking for 16 h at room temperature (23 °C) [14]. Extraction procedure was started soon after the grinding of subsamples. The solution was filtered by vacuum filtration, and the solid residue was washed three times with 2 mL of acetone. The liquid phase was collected and dried under nitrogen stream in a water-bath at 40 °C and re-suspended in chloroform: acetic acid (10 mL, 95:5, v/v).
To determine the analytical sample size in the seed-by-seed analysis as a source of uncertainty, a different number of whole cottonseeds pools 10, 25, and 75 were randomly collected in triplicates from 5 kg sample, manually de-hulled and weighted and ground using a pestle while they were immersed in acetone, and started acetone extraction immediately as described above. Each pool of whole cottonseeds resulted in three analytical sample sizes of 1.0, 2.5, and 5.8 g, respectively.
2.2.2 The particle size of the sample after grinding
To verify the analytical variability associated with the particle size of cottonseed after grinding, two subsamples were ground through different screens to obtain two ranges of particle sizes: particle < 0.25 mm (group I) and 0.25 < particle < 1 mm (group II). From cottonseed ground, the particles that passed through the 1 mm mesh was collected and passed through a sieve of 0.25 mm. Replicate samples were collected from the material retained on the 0.25 mm mesh (particle size range 0.25–1 mm), and from the material that passed through the 0.25 mm mesh (particle size < 0.25 mm). Six replicates of each particle size group were allowed for acetone extraction immediately as earlier described.
2.2.3 Rate of FG loss after grinding
The rate of FG loss promoted by grinding was measured in the first 72 h after sample preparation. A hundred grams of de-hulled cottonseed sample was ground at 0.25 mm. Then triplicates of 0.3 g were extracted by acetone soaking after 0, 1, 3, 6, 9, 12, 24, 48, and 72 h of the grinding. All subsamples remained at ambient temperature (23 °C) while protected from light until the soaking extraction. Replicates of time 0 were immersed in acetone immediately after finishing grinding, thus samples of time 0 were considered as 100% FG. The results of FG from other extraction times were expressed as a percentage of the FG content at time zero.
2.3 Uncertainty source from the analytical procedure
The analytical method evaluation was carried out according to the following parameters: linearity, matrix effects, accuracy, and limits of detection and quantification.
2.3.1 The linearity of calibration and matrix effects
The linearity was evaluated by the analytical curve and regression analysis by the least-squares method. The analytical curve was prepared by dilutions of an analytical standard of FG in chloroform: acetic acid (99:1, v/v). Seven solutions of 0.01, 0.03, 0.06, 0.125, 0.25, 0.50, and 1.00 mg FG ml−1 were injected in triplicate in HPLC.
Matrix effects were investigated by comparing the slopes of the matrix-matched calibration curve and the solvent calibration curve [15]. In a cottonseed meal extract without detectable levels of FG, a suitable concentration of FG standard was added to provide seven solutions with concentration ranging from 0.01 up 1.00 mg FG ml−1 used for matrix-matched. The criterion for the acceptance of the points for each calibration solution was a peak area relative standard deviation of no more than 10% between the triplicates.
2.3.2 Precision, trueness, and limits of detection and quantification
The precision of the extraction was measured by relative standard deviations (RSD) from the repeatability test. Six replicates of a cottonseed sample (containing natural FG) were ground, extracted, and used to obtain the RSD. The trueness was measured by the recovery test (10 replicates) adding 0.28 mg g−1 of FG standard in a sample without detectable levels of FG.
The limit of detection (LOD) was set up as a relation between 3.3 the standard deviation of the response of the first point and the slope of the analytical standard curve. The limit of quantitation (LOQ) was set up like 10 times the LOD.
2.3.3 The efficiency of extraction regarding solvent soaking time
The effect of solvent soaking time on the efficiency of FG extraction was verified in three experimental treatments, the first one was used to verify the efficiency of the extraction along soaking time in recovery tests, while the second and third ones were conducted to evaluate the effect of solvent soaking time on the extraction of natural FG in whole cottonseed sample.
For the first treatment, a cottonseed meal sample (without FG detectable concentration) was fortified with 0.28 mg g−1 of FG standard. In the second treatment, a whole cottonseed sample was dry ground (1 mm mesh) and extracted in triplicates. To evaluate the effect of the soaking time on the FG glands that remained intact within the cottonseed particles after grinding, in the third treatment, cottonseed was ground in acetone by 15 s to remove the gossypol that was released from broken glands in the grinding process. Soon after, the acetone was filtered by vacuum pressure, and the solid residue of cottonseed was sieved at 1 mm mesh and collected in replicates; then the FG extraction was immediately determined. All experimental treatments were triplicates and extracted by soaking in acetone for 1, 2, 4, 8, and 16 h.
2.4 High-performance liquid chromatography
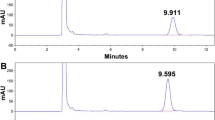
The FG was measured by liquid chromatography in an Agilent 1100 HPLC Series liquid chromatography (USA) equipped with a quaternary pump, automatic injector, photodiode array detector, ODS-C18 Zorbax column (250 × 4.6 mm, 5 µm particle). Five microliters of sample were injected in the column at a controlled temperature of 23º C. The UV–Vis wavelength for quantification was 254 nm. Figure 1 illustrates the FG chromatograms in the chromatographic system used in this study. The elution was achieved from modification of a [15] method. Prior experiment, the analytical quality of method was checked (repeatability, recovery test, and limit of detection (LOD) and (LOQ)). Aliquots of all the experimental solutions were injected into the chromatograph using the conditions of the preparation of the calibration curve. Peaks of FG were identified by comparing the retention times and spectrum analysis using the Chem Station software (Agilent, 3.1.1 version, revision 3).

Chromatogram of a recovery test showing FG peak eluted at 6.9 min (a cottonseed meal sample was added of FG standard prior to the extraction). The table illustrates the elution system programming and the control of analytical quality (rsd correspond to the relative standard deviation of 5 injections of FG standard solution)
2.5 Statistical analysis
The data were subjected to analysis of variance using the general linear model’s procedure using Statistical Analysis System software (SAS Institute Inc., Care, NC, USA). The rate of FG loss due to the grinding process and the efficiency of extraction results were tested by the regression model, where the FG recovery rate was analyzed as a function of time of acetone soaking. The verification of outliers, the suitability of sample size, and the assumption of homogeneity of variances and model fit were carried out. The Box-Cox method was used for the transformation of the original data [16] for the efficiency of the extraction test, where FG content = (x)3.4. The particle size test was evaluated as y = f (x), where y is the FG concentration and x is the particle size. The FG concentration of the laboratory sample size and the analytical sample (seed-by-seed analysis) were evaluated as a function of the mass of sample and number of seeds, respectively. Pearson correlation was used to verify the association between FG concentrations and the mass of samples for laboratory sampling and the association between FG concentration and the mass of groups of seeds in analytical sampling. Differences between experimental treatments were declared significant at P ≤ 0.05 using Tukey’s procedure for multiple comparisons.
3 Results and discussion
3.1 Uncertainties sources from cottonseed sampling and sample preparation
3.1.1 Laboratory and analytical sample size
Table 1 shows the effects of the primary laboratory sample size on FG concentrations of cottonseeds. The primary laboratory sample size did not affect (P > 0.05) the FG concentrations since no significant (P = 0.895) correlation was detected between the FG content and the size of the laboratory sample. This result shows that the trueness of FG quantification cannot be impaired by reducing the size of the laboratory samples. However, the variability of analysis seems to decrease with the increase in the size of the sample. This suggested that the precision among replicates may be impaired as sample size decrease.
Sampling is the main contributor to overall uncertainty sources of any measurement. Although many recommendations describe the optimal sampling practice in several applications, the sampling uncertainties are hardly quantified or documented [17, 18]. The official method of AOCS recommended a ground laboratory primary sample of 50 g of de-hulled cottonseed for FG quantification [19]. Glands of FG may have a highly heterogeneous distribution in size and density within the seeds [20, 21], thus differences in FG concentrations among the replicates of the experimental laboratory samples would be expected. Despite this, the current results revealed that until a 10 g de-hulled ground homogenized sample can be used as a primary laboratory sample for FG quantification.
The official method of AOCS [9, 19] recommended different sizes of analytical samples based on the expected FG content in the experimental sample, e.g. if FG concentration is predicted between 0.2 up 2 mg g−1, one g of the sample should be taken as an analytical sample. To increase the capacity of detection of the method, an increase in the size of the analytical sample is suggested until 5 g of cottonseed to detect 0.1 mg g−1 or less. Thus the size of the analytical sample can be considered as an uncertainty source for FG quantification. In the current study, three analytical sample sizes of 1.0, 2.5, and 5.8 g corresponded to 10, 25, and 75 seeds, respectively, were evaluated as a source of uncertainty in a seed-by-seed analysis. Results in Table 2 showed that the FG content of seed has not been affected (P > 0.05) by the size of the analytical sample. The coefficient of variation was similar to the results of using 10 g of laboratory sample, which was grounded before analysis (Table 1). The variability found in our study was similar to levels reported by literature [21]. The correlation analysis between FG content and the number of seeds found a highly significant (P = 0.02) correlation (R = 0.817) to the group of 75 seeds, which total weight corresponds to 5.9 g. On the other hand, this correlation disappeared (P > 0.05) in the small analytical size samples (Table 2).
Therefore it shows that a group of 75 seeds (corresponding to 5 g) seems to be more representative of the entire seeds sample either due to the high variability observed among seed weight also as to the positive correlation between FG and seed weigh observed. The FG concentrations were ranged between 3.2 and 3.5 mg g−1, thus according to AOCS [19], a one g analytical sample is sufficient to detect accurately the FG concentrations. However, our results suggested that a higher size sample (5 g) with high seed numbers are recommended to obtain accurate FG data, even for samples predicted to have more than 2 mg g−1 FG concentrations.
3.1.2 The particle size of the sample after grinding
The FG loss in the grinding step due to the rupture of FG glands was checked in two range of particle size: particle < 0.25 mm (group I) and 0.25 < particle < 1 mm (group II). The results from the group I showed a significant (P < 0.05) reduction of 61% in the FG content (1.98 mg g−1 ± 0.21) compared to group II (5.15 mg g−1 ± 0.54). This result reflects the different amount of FG extracted from glands that remain unbroken after grinding in both groups (Fig. 2). The largest particles support the maintenance of more number of intact glands where FG remains protected inside even after grinding. Thus, in the extraction by acetone soaking the solvent can subsequently open the glands and extract the FG. As gossypol glands have a range of sizes between 50–400 µm [21, 22], seed particles with sizes below 250 µm are much less likely to contain intact FG glands than particles with sizes between 250 and 1000 µm.

Effect of particle size on free gossypol loss. The range line on bars represents the standard deviation of replicates. CV: coefficient of variation. Dots lines (mean) represents the average of replicates in each group of particle size
The most important characteristic of gossypol is its capacity to exist in dual form. The storage of FG inside the glands of gossypol in cottonseeds is responsible for its biological and biochemical properties [1, 3]. Once released from glands, FG may change its biological activity by complex-formation, and become the inactive form of gossypol (bound gossypol), which is non-extractable by the organic solvents [9]. From an animal nutritional point of view, FG losing is desirable in the cottonseed meal to reduce its toxicity. Although along with this detoxification by the gossypol protein-complex formation [1], the protein available for digestion of cottonseed meal can be reduced, consequently adversely affect the whole animal performance [4]. Through oil extraction, the cottonseeds can be exposed to heating, moisture, and pressure that lead to promoting the detoxification of FG by its conversion to BG [1]. Similarly, reducing the particle size of the cottonseed meal can be investigated as a potential tool to improve FG reduction. Ultrafine grinding provides a reduction of 30% of FG when compared to the traditional process of grinding, in which the particle size is around 0.25–0.42 mm. The high shear strength, the collision, friction, and impulse within particles during the process could be the reasons for the loss of FG, due to the destruction of its gland structure [23].
Conversely, the reduction of FG during the sample preparation for chemical analysis is always undesirable. The underestimation of FG analytical results may pose a risk to animal health by the mismatch of gossypol intake in the diet. In this context, the particle size is a parameter that may have a profound impact on the determined FG content of whole cottonseeds. Thus, the current results suggested that the particle size is a very pronounced uncertainty source to detect the FG concentrations, and 0.25–1 mm particle size produced more realistic results than the smaller size.
3.1.3 Rate of FG loss after grinding
Figure 3 shows the effects of the interval of time between grinding and extraction of cottonseed samples. There was a significant decrease (P < 0.05) of FG concentrations by the increase of the time between grinding and extraction. The regression curve fitting a non-linear cubic adjustment was suitable to relationship FG loss and the delay time before acetone soaking. (y = 6.38 − 0.1345 x + 0.0033 x2 − 0.0000246 x3, where y = FG loss over time, x = delay time before acetone soaking and R2 = 0.86). At the first hour after grinding, the FG reduction was 5%, while the cumulative reduction was 19, 27 and 34% at 6, 24, and 72 h after grinding. Thus it seems that the FG loss rate was higher (P < 0.05) through the first hour than thereafter, and this FG loss may be continued after 72 h.

Effect of increasing the interval of time between grinding and extraction of cottonseed samples on free gossypol loss. The bars deviations correspond to the standard deviations of replicates
The reduction of the losing of the FG by the advancing time probably occurs due to the exhausting of the epsilon-amino groups of lysine from cottonseed [24]. The aldehyde groups from FG molecules react with amino groups present in proteins leading to the protein-complex formation of gossypol [1]. Since the grinding promotes the rupture of the glands and releases FG to be reacted, a delay between grinding and extraction can increase the lack of trueness of results. The precision of results may also be negatively impacted when the delay between grinding and extraction increases among replicates.
Thus, to reduce the lack of trueness and precision of FG concentrations, the primary delay between sample preparation and chemical analysis should be avoided. These resulted elucidate that the time after grinding and before chemical extraction was a potential source of uncertainty in FG measurement from cottonseeds.
3.2 Chemical analysis and quantification
3.2.1 Analytical method performance
FG retention time was 6.09 min for solutions of calibration and matrix-matched calibration curve injected (see supplementary material). Figure 4 shows the calibration curve and matrix-matched calibration curve.

Calibration curve (red line) and matrix-matched curve (blue line). The difference between slope of curves illustrate the matrix-effect
The calibration curve was linear either in the solvent curve (y = 15.707x + 86.787; R2 = 0.998) as in matrix-matched curve (y = 12.433x + 24.875; R2 = 0.999). A medium (− 50% < C % < 20%, where C is the relation between the slope of calibration curve and matrix-matched curve expressed as a percentage) matrix effects (C % = 20.7) was observed [15]. Matrix effects have been described as an important suppressor or enhancer of signal in liquid or gas chromatography (CG) coupled with MS techniques [25, 26]. These effects may occur due to the analyte or to the matrix and its intensity may be reduced when the sample is diluted [15]. Currently, is possible to observe a decline of the slope of the matrix-matched curve regarding the slope of the solvent curve, showing that the FG response in the detector is reduced due to the matrix effect. Thus the quantification of analyte should be carried out using a matrix-matched curve. The LOD was 0.00137 mg g−1 and the LOQ was set up as 0.01137 mg g−1. The repeatability (RSD) of the methodology was 0.54 and the mean of recoveries was 98% ± 9. Both recovery and repeatability results were suitable to produce good accuracy according to Horwitz criteria [26].
3.2.2 Solvent soaking time effect
The efficiency of solvent extraction in different soaking times when FG was an external addition or naturally presented in the cottonseeds is presented in Fig. 5.
The results showed that one hour of acetone soaking was not enough to extract all the FG content of the sample, even when FG is externally added (1). Only 48% of the added FG standard was recovered after 1 h of acetone soaking while no differences were observed for the FG extraction efficiency among the other tested soaking times (88%). The regression analysis showed in quadratic adjustment that the interval between 8 and 16 h of acetone soaking has a better efficiency of the FG extraction (y = 820847 + 1055724.39 x − 54715.45 x2, where y = FG recovery, x = time of acetone soaking and R2 = 0.54). Therefore, our results show that a period of 8 to 16 h of acetone soaking is the best compromise between the rate of extraction and the degradation of the FG externally added. To verify whether the natural FG would have a similar response regarding soaking time, the same procedure was carried out using samples contained natural FG. Results of Fig. 5 showed that FG concentration among replicates in different soaking times did not show the difference after dry grinding of cottonseed. Also, there was not a significant (P > 0.05) regression adjustment with the advance of the soaking time. Thus it seems that the external FG may not show the same degree of difficulty of extraction as the natural one. The naturally existed compounds might be strongly bonded to the matrix of the sample. Thus, natural compounds have a higher strength of molecular interaction with the matrix than the external added to the sample.

Efficiency of soaking time. The range line on bars represents the standard deviation of replicates. Black bars (External FG (1)) represent replicates of cottonseed meal with external addition of free gossypol (FG) standard (recovery test). Gray bars (Natural FG (2)) represent replicates of cottonseed meal obtained by dry grinding. Blue bars (Natural FG (3)) represent replicates of cottonseed meal obtained grinding in acetone to remove the extern gossypol from broken glands in the grinding process. The polynomial line represents the regression curve for black bars (External FG (1))
Immediately after grinding, the released FG can also be readily extracted by organic solvents before forming a protein-complex. In dry grinding of cottonseed, the FG released from the glands remains outside of the mass of particles. Thus it was hypothesized that the affinity of the solvent to obsess the FG is higher by the immediate addition of the organic solvent rather than reacting with the proteins. The results obtained in this experiment comprise not only the gossypol which remained inside glands within the particles but also the FG previously released from glands by grinding process.
Treatment 3 represented the efficiency of different soaking time regarding the extraction of FG which reminded only inside glands after sample grinding (Fig. 5). Cottonseed sample was ground in acetone to remove the FG which was released from the broken glands. The results showed that the highest (P < 0.05) FG concentrations were obtained in 16 h soaking time. It seems that the time of interaction between solvent and particles of cottonseed which is needed to remove FG from the inside glands is higher than when FG is “free” in the sample (e.g. Treatment 1). The distribution of glands inside particles may have a high variability of the size and density [20, 21]. Thus, to extract FG from glands, at least 16 h of soaking in acetone should be applied. It is important to highlight this statement is valid when only solvent soaking is applied as a method of extraction.
Thus it is assumed that 1 h of soaking time was not enough time to extract deep FG from particles (Treatment 3), since it presented the lowest extraction efficiency compared to 16 h of extraction. The addition of the external FG does not allow for measuring the capacity of the solvent to open the FG glands, the efficiency of time of soaking could be better evaluated by tests with native FG. According to our results, 16 h had shown suitable for sample extraction both in recovery tests as in natural FG from cottonseed.
Figure 6 concluded the uncertainty sources for FG quantification by HPLC which still have not been described in protocols for gossypol analysis.

Cause-effect diagram of uncertainty sources identified on free gossypol analysis. Arrows represent the effect on trueness and/or precision due to the uncertainty source evaluated (single arrow indicates the occurrence of the effect while arrow with cross-sectional indicates the absence of the effect)
Through sample preparation, the particle size and time between grinding and before acetone soaking were found to be the main sources of uncertainty which affect the trueness but not the precision of analytical measurement. Conventional uncertainty sources in the chemical analysis have less importance among the sources of uncertainty identified. A medium matrix effect was observed, in sense of FG signal suppression in HPLC analysis. The accuracy of the method measured by recovery and repeatability tests has a low contribution to overall uncertainty sources, even though the efficiency of extraction showed low robustness regarding acetone soaking time.
4 Conclusions
To enhance the precision and trueness of FG quantification results, a laboratory sample of 100 g cottonseed with particle size between 0.25 and 1 mm can be recommended. Analytical sampling must be done following the homogenization of the laboratory sample, without delay time before chemical extraction. Chemical analysis has a low contribution to the variability of results compared to sample preparation. The gossypol storage inside glands is an intrinsic feature of this compound responsible by the major variability in FG results. Analytical procedures that promote the rupture of gossypol glands must be carefully controlled to reduce the uncertainty in FG analysis. Best practices to reduce the uncertainties of FG analysis increase the confidence of cottonseed use as an animal feed source to improve either animal health as well as the safety of animal products (milk or meat-free of gossypol residues) for human consumption.
Availability of data and materials
All relevant data are within the article.
References
European Food Safety Authority [EFSA] (2008) Scientific Opinion. Gossypol as an undesirable substance in animal feed. Scientific Opinion of the Panel on Contaminants in the Food Chain (Question No EFSA-Q-2005–222). EFSA J. https://doi.org/10.2903/j.efsa.2009.908
Abou-Donia MB (1989) In: Cheeke PR (ed) Toxicants of plant origin, vol 4. CRC Press, Florida
Wang X, Howell CP, Chen F, Yin J, Jiang Y (2009) Gossypol—a phenolic compound from cotton plant. Adv Food Nutr Res. https://doi.org/10.1016/S1043-4526(09)58006-0
Arieli A (1998) Whole cottonseed in dairy cattle feeding: a review. Animal Feed Sci Technol 72:97–110
Nikokyris PN, Kandylis K, Deligiannis K (1999) Effects of varying levels of dietary free gossypol in whole cotton seed on physiological responses of growing-fattening lambs. J Sci Food Agric. https://doi.org/10.1002/(SICI)1097-0010(199911)79:14<1969:AID-JSFA464>3.0.CO;2-T
Kim HL, Calhoun MC, Stipanovic RD (1996) Accumulation of gossypol enantiomers in ovine tissues. Comp Biochem Phys. https://doi.org/10.1016/0305-0491(95)02061-6
European Union EU (2002) Directive 2002/32/EC of the european parliament and of the council of 7 may 2002 on undesirable substances in animal feed. Off J Eur Commun Legis 140:10–22
Magnusson B, Ornemark U (2014) Eurachem guide: the fitness for purpose of analytical methods—a laboratory guide to method validation and related topics, 2edn. https://www.eurachem.org/images/stories/Guides/pdf/MV_guide_2nd_ed_EN.pdf. Accessed 12 Sept 2019.
American Oil Chemists Society AOCS (1990) Official methods and recommended practices of the American Oil Chemists’ Society 2 Aa 1–38
Kenar JA (2006) Reaction chemistry of gossypol and its derivatives. J Am Oil Chem Soc 83:269–302
Li C, Zhao T, Li C, Mei L, Yu E, Dong Y, Chen J, Zhu S (2017) Determination of gossypol content in cottonseeds by near infrared spectroscopy based on Monte Carlo uninformative variable elimination and nonlinear calibration methods. Food Chem 221:990–996
Hron RJ, Kuk MS, Wan PJJ (1996) Quick method for estimating free gossypol in cottonseed, meats, collets, and extracted meals. J Am Oil Chem Soc 73:199–202
Ramsey MH, Ellison SRL (2019) Eurachem/CITAC guide: Measurement uncertainty arising from sampling—a guide to methods and approaches. https://www.eurachem.org/index.php/publications/guides/musamp. Accessed 30 Sept 2019.
Wang MZ (1987) Analysis of gossypol by high performance liquid chromatography. J Ethnopharmacol 20:1–11
Econommou A, Botitsi H, Spyros A, Tsipi DJ (2009) Determination of multi-class pesticides in wines by solid-phase extraction and liquid chromatography-tandem mass spectrometry. J Chromatogr A 1216:5856–5867
Box GEP, Cox DR, Stat JR (1964) An analysis of transformations. J Royal Stat Soc Ser B 26:211–252
Zhang J, Zhang C (2012) Sampling and sampling strategies for environmental analysis. Int J Environ Anal Chem 92:466–478
Whitaker TB, Johansson AS (2005) Sampling uncertainties for the detection of chemical agents in complex food matrices. J Food Prot 68:1306–1313
American Oil Chemists’ Society [AOCS] (1990) Official methods and recommended practices of the American Oil Chemist’s Society 2 Ba 7–58.
Bolek Y, Fidan MS, Oglakci M (2010) Distribution of gossypol glands on cotton (Gossypium hirsutum L.) genotypes. Not Bot Horti Agrobot Cluj-Napoca 38:81–87
Benbouza H, Lognay G, Palm R, Bandoin JP, Mergeal G (2002) Development of a visual method to quantify the gossypol content in cotton seeds. Crop Sci 42:1937–1942
Gardner HK Jr, Hron RJ, Vix HLE (1976) Removal of pigment glands (gossypol) from cottonseed. Am Assoc Cereal Chem 53:549–560
Dai C, Ma H, Zhang L, Zhu S, Yin X, He R (2016) Effects of ultrafine grinding and pulsed magnetic field treatment on removal of free gossypol from cottonseed meal. Food Bioprocess Technol 9:1494–1501
Damaty SM, Hudson BJF (1979) The interaction between gossypol and cottonseed protein. J Sci Food Agric 30:1050–1056
Gosetti F, Mazzucco E, Zampieri D, Gennaro MCJ (2010) Signal suppression/enhancement in high-performance liquid chromatography tandem mass spectrometry. J Chromatogr A 1217:3929–3937
Gonzales AG, Herrador MA, Asuero AG (2010) Intra-laboratory assessment of method accuracy (trueness and precision) by using validation standards. Talanta 82:1995–1998
Acknowledgements
This work was supported by the São Paulo Research Foundation, São Paulo, Brazil (Grant 51265-9, 10630-1, São Paulo Research Foundation (FAPESP)). The authors are grateful to Dr. Maria Antonia Calori-Domingues and to Prof. Dr. Pedro Esteves Duarte Augusto for the valuable comments and suggestions for improving our manuscript
Author information
Authors and Affiliations
Contributions
AdCR: Conceptualization, Methodology, Investigation, Validation, Formal Analysis, Writing—Original Draft, Writing—Review & Editing, Visualization. ALA: Resources, Supervision. Project Administration, Resources, Writing—Review & Editing. CTdSD: Formal Analysis (Statistical analysis) and Review. YAS: Formal Analysis, Writing—Review & Editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
de Cássia Romero, A., Abdalla, A.L., dos Santos Dias, C.T. et al. Assessment of uncertainty sources of free gossypol measurement in cottonseed by high-performance liquid chromatography. SN Appl. Sci. 2, 1766 (2020). https://doi.org/10.1007/s42452-020-03541-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-03541-0