
Abstract
Calcification of Joints and Arteries (CALJA) is a rare disease that leads to chronic arthritis and lower limb claudication due to hydroxyapatite crystal deposition. The disease is caused by mutations in the 5-nucleotidase (NT5E) gene, which is responsible for pyrophosphate metabolism. Only 23 cases have been described so far. In this case report, we describe a new case of CALJA and provide a literature review. A 65-year-old woman was referred to the Rheumatology Unit with the diagnosis of seronegative oligo-arthritis. She complained of lower limb claudication, which was becoming progressively worse. Doppler ultrasound revealed bilateral obliteration of the popliteal and femoral arteries, and X-rays of the knees, hands, and feet showed extensive periarticular calcific deposits. The results of the NT5E gene analysis were positive for an inactivating variant, leading to the diagnosis of CALJA. The clinical features of CALJA are caused by hydroxyapatite crystal deposition at the periarticular and vascular levels due to abnormalities of pyrophosphate metabolism. Currently, no specific treatment is available, although a trial on the use of etidronate is ongoing. Patients with CALJA are often treated with immunosuppressant agents in the suspect of inflammatory rheumatologic diseases. Our case is the first in which clinical symptoms and a steady increase of inflammatory markers improved only after colchicine therapy initiation. It is crucial for the rheumatologist to recognize the features CALJA and keep it in mind in the differential diagnosis of patients with lower limb arterial insufficiency and arthritis or early osteoarthritis with joint calcification.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hereditary ectopic mineralization disorders are a group of four diseases caused by mutations of genes involved in inorganic pyrophosphate (PPi) metabolism [1, 2]. These include Pseudoxanthoma elasticum (PXE), Generalized Arterial Calcification of Infancy (GACI), Familial Idiopathic Basal Ganglia Calcification (IBGC), and Calcification of Joints and Arteries (CALJA) [3,4,5]. The latter, also known as Arterial Calcification due to Deficiency of Cluster of Differentiation 73 (CD73) (ACDC), is an autosomal recessive disease that typically manifests in adulthood. Loss-of-function mutations of the ecto-5′-nucleotidase (NT5E), encoding CD73, lead to increased activity of non-tissue specific alkaline phosphatase (TNAP and decreased PPi, a potent mineralization inhibitor [6]. The feature of the disease is periarticular deposition of hydroxyapatite crystals almost limited to the arteries of the lower limbs and the small joints of hands and feet, resulting in arthritis, early-onset osteoarthritis, and intermittent claudication of the lower limbs [2, 7,8,9]. Patients are often misdiagnosed as having rheumatoid arthritis or seronegative spondyloarthritis due to the rarity of CALJA. Therefore, rheumatologists should consider this disease in the differential diagnosis of seronegative arthritis to avoid incorrect treatment [8]. To date there are no therapies for this condition [10, 11].
We described a case of a CALJA patient with a persistent high level of inflammatory markers before administration of colchicine. We also carried out a comprehensive review of the available literature on the disease. Medline databases (PubMed) were searched using the following keywords: “Calcification of Joints and Arteries,” “CALJA,” “Arterial Calcification due to the Deficiency of CD73,” “ACDC,” “NT5E mutations,” and “Heritable Ectopic Mineralization Disorders.” Informed consent was obtained from all individual participants included in the study. We included all articles published in English language since the first description of CALJA in 2011. Thirty-nine articles were retrieved as pertinent to the object of this paper.
Case Presentation
In May 2022, a 65-year-old Caucasian woman previously diagnosed with seronegative oligo-arthritis was referred to the Rare Bone Diseases Clinic of the University Hospital of Pisa. She had been experiencing lower limb pain while walking for several months, which had rapidly worsened, extending to both calves and right thigh, limiting her ability to walk to a maximum of two hundred meters. These symptoms had not been present before. At physical examination, peripheral pulse was not detectable at lower limbs, and ankle-brachial index was reduced. Since adolescence, she had suffered of recurrent migrant asymmetrical non-erosive arthritis of hands, wrists, feet, ankles, and knees, in the absence of radiological signs of microcrystalline arthritis. The arthritis episodes did not involve more than two joints at the same time, and they usually lasted from weeks to months, with mild swelling and pain, which was detected during several physical examinations. At the time, articular X-rays were negative, whereas articular ultrasound was never performed. Laboratory findings over time revealed slightly increased erythrocyte sedimentation rate and C-reactive protein, even in the absence of joints inflammation (Fig. 1). Rheumatoid factor, anti-citrullinated peptide antibodies, anti-nuclear antibodies, HLA-B27, and Lyme disease serological test were normal or negative, as well as lipid profile, glycemia, complete blood count, creatinine, liver enzymes, and calcium-phosphorus metabolism. Her parents were second cousins, and her medical history was unremarkable. At the age of 20 years, the diagnosis of seronegative oligo-arthritis was made; the symptoms did not resolve with chronic glucocorticoid and immunosuppressant therapy. The pattern of articular involvement remained unchanged during the next 20 years.

Trends in the values of inflammatory markers before and after colchicine treatment, administered after May 2022
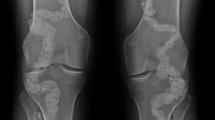
In May 2022, an arterial color Doppler ultrasound showed chronic obliteration of superficial femoral, posterior, and anterior tibial and lateral plantar arteries bilaterally. X-rays of hands and feet revealed the presence of articular and periarticular new calcific deposits suggestive for hydroxyapatite crystals (Fig. 2); X-rays of the knees revealed extensive calcifications of the femoral and popliteal arteries, which were never documented before (Fig. 2). Arterial color Doppler ultrasound of upper limbs and abdominal vessels were negative. Therefore, CALJA was suspected. Genetic analysis revealed a mutation of the ecto-5’-nucleotidase gene (NT5E), specifically a novel homozygous variant NM_002526.3:c.751+1G>A, thereby confirming the clinical suspicion of CALJA (Fig. 3). This variant is not reported in Genome Aggregation Database (gnomAD exome) nor in the mutation databases (HGMD, ClinVar, LOVD3). The variant is predicted to be likely pathogenic, based on ACMG guidelines for genomic variant classification [12]. Moreover, a likely pathogenic variant has been described in the adjacent splice site (c.751+2T>C, LOVD Variant #000721128). We applied clinical exome sequencing NextSeq 550 Dx (Illumina) platform, using the TruSight One Expanded Sequencing Panel (Illumina). The sequencing achieved good coverage of the target regions (98.8% average total coverage at 20×) with average depth 291.6×. Variant annotation was performed with QIAGEN CLC Genomics Workbench; variant prioritization was performed with QIAGEN Clinical Insights. Sanger sequencing validated the findings. Genetic investigation was not performed in the patient’s relatives due to the absence of similar symptoms.

Multiple periarticular calcifications of hands and feet and calcification of the popliteal arteries

Novel heterozygous NT5E mutation c.751+1G>A. Partial DNA sequences of NT5E exon 3 showing the presence of the variant in a homozygous state. Sequence analysis (forward and reverse strand are shown)
The patient was then treated with colchicine 1 mg per day, which ameliorated articular symptoms and normalized inflammatory markers (Fig. 1), and aspirin 100 mg; however, lower limb claudication was unaffected, but the patient is not currently eligible for surgical revascularization. To our knowledge, this represents the fifth case of CALJA to be described in an Italian patient, after those published by St. Hilaire et al. and Avruscio et al. [7, 13]. This is the first case in which a steady increase of inflammatory markers was documented, and in which a significant improvement of articular symptoms and a normalization of inflammatory markers after initiation of colchicine treatment were described.
Discussion
The genetic and molecular basis of CALJA was first reported by St Hilaire et al. in 2011 [7, 14]. However, seven case reports published between 1912 and 1992 [15,16,17,18,19] described patients with symptoms compatible with CALJA, but genetic testing was not performed. A patient with intermediate clinical features between pseudoxanthoma elasticum (PXE) and CALJA was described in the absence of NT5E mutations [20].
To the best of our knowledge, 23 patients (6 males and 17 females) with a confirmed NT5E mutation have been reported since 2011 (Table 1). The prevalence has been estimated to be approximately 1/2.500.000 in the East Asian population for the homozygous variant of NT5E NM_002526: c.3G>C (p.Met1?) [14]. However, according to Ichikawa et al., CALJA may be more frequent in Asian populations [8].
Alterations in the mechanisms regulating the calcification processes, e.g., mutations in the enzymes involved in the metabolism of PPi [1, 3, 21], can result in heterotopic calcifications and lead to the “ectopic mineralization diseases” [3, 11, 22, 23]. CALJA is an autosomal recessive disease caused by mutations in the NT5E gene, which encodes for the glycosylphosphatidylinositol-linked plasma membrane ectoenzyme CD73 [3, 24]. To date, 8 different genetic variants have been described [14, 25]. The transmembrane protein CD73 is expressed by stromal cells, endothelial cells, and lymphocytes and catalyzes the hydrolysis of adenosine monophosphate to adenosine and inorganic phosphate (Pi) [6,7,8]. Adenosine normally inhibits tissue non-specific alkaline phosphatase (TNAP), which catalyzes the hydrolysis of PPi to Pi. Mutations in the NT5E gene increase TNAP activity, reducing PPi [26] and causing mineralization at ectopic sites [7]. However, the pathogenesis of CALJA is still unclear, as mouse models do not fully replicate human pathophysiology [27,28,29,30].
As in our patient, articular manifestations often appear during adolescence [2, 7, 8] and include intermittent monoarthritis and early of small and large joints due to the formation of periarticular calcifications [1, 2, 8, 14] mainly composed of hydroxyapatite or calcium phosphate crystals and, to a minimal extent, of calcium pyrophosphate crystals, as demonstrated by Cudrici et al. [2].
Among the 23 patients, 16 (70%) had joint pain related to periarticular calcifications which may regress over time [1;2]. Axial involvement has been described in the form of multiple osteophytes, syndesmophytes, intervertebral space reduction, disc calcifications, and anterior and posterior longitudinal ligament calcification [2]. Although 2 cases (9%) of involvement of upper limb arteries are reported [18, 28, 31], the disease usually affects the arteries of the lower limbs, probably due to the distribution of the adenosine receptors [7] or due to the protective role of CD73 against the mechanical stress [30]. The most affected districts in CALJA are the iliac, femoral, tibial, and popliteal arteries [32].
Natural history is characterized by insidious progression of vascular symptoms such as intermittent claudication, reduction of the ankle-brachial index, and appearance of ischemic ulcers [14, 28] or aneurysmal dilatations [2, 33]. The most important complication of the disease is the occlusion of calcified arteries [9]. Twenty (87%) out of the 23 patients with CALJA reported intermittent claudication. Typically, the trunk and coronary arteries are spared [2, 7, 13]; however, 4 cases (17%) with late-onset calcific aortic stenosis have been recently described [14, 33]. The latter finding may be related to a possible association between CD73 deletion with calcific valve stenosis [34]. Furthermore, some NT5E polymorphisms seemed to contribute to the pathogenesis of calcific valves [25]. In one case, the presence of Raynaud’s phenomenon was reported [13].
Given the rarity of the disease, there are no defined diagnostic criteria. The clinical suspicion is suggested by the appearance of microcrystalline arthritis or early osteoarthritis in association with evidence of lower limb ischemia [2]. The rheumatologist is often the first specialist to evaluate these patients [2], but there is usually a delay in the diagnosis. In fact, among the 23 patients described, the mean age at diagnosis was 59 years.
In CALJA, laboratory findings are typically normal [2, 7]. An increase in inflammatory markers could be observed during episodes of arthritis, with normal values during the intercritical phases [2]. In our patient, however, they remained persistently high over the years. To our knowledge, this is the first case of CALJA in which a constant increase in ESR and CRP has been recorded.
Plain radiographs of hands and feet show periarticular calcifications with features of hydroxyapatite crystals [1, 9]. At the knee level, it is also possible to appreciate widespread vascular calcifications which, especially in young subjects, should lead to suspicion of the disease. Axial involvement with ligamentous calcification and bone bridging has been described in some patients; therefore, evaluation by X-rays of the spine is recommended. The radiological features may resemble DISH, however without fulfilling its criteria [2].
Peripheral arterial insufficiency can be evaluated by the ankle-brachial index and by eco-color Doppler of lower limb arteries. CT angiography can show the extent of vascular calcifications [7, 14, 35]. The diagnosis is confirmed by genetic analysis [20].
Arthritis is usually responsive to treatment with nonsteroidal anti-inflammatory drugs or low-dose glucocorticoids [2, 8]. On the other hand, there is no therapy that can reduce arterial and joint calcifications in CALJA [10;30]; vascular involvement is irreversible, and often these patients undergo bypass surgery [1, 7, 14]. In our patient, treatment with colchicine was effective in reducing inflammatory markers and articular symptoms. This finding might suggest a role for colchicine in reducing the number of episodes of hydroxyapatite arthritis and, in line with what has been reported in some recent studies, could reduce vascular inflammation and thus also be used in cardiovascular prevention in patients with CALJA [36,37,38].
The main studies that have investigated strategies to reduce the size of calcifications focus on the use of intravenous etidronate in the context of GACI and PXE [10, 11, 39], which has been shown to be effective in the treatment of GACI [7, 11, 26, 40, 41]. Currently, a trial on the efficacy and safety of using etidronate in CALJA is ongoing (NCT01585402; last update 04 October 2022).
Dietary calcium and magnesium supplementation have been proven to be poorly effective on ectopic mineralization in mouse models of PXE. [10, 42].
Methotrexate, which can increase the levels of extracellular adenosine [43], has not proven to be effective in the treatment of CALJA [2], including the present case.
Other therapies, e.g., sodium thiosulfate and 4-phenylbutyrate, have been proposed, but currently are being investigated only in the context of PXE, GACI, and IBGC [5, 7, 10, 11].
We described the case of a 65-year-old female patient with CALJA who had been previously diagnosed with seronegative oligo-arthritis. CALJA is a rare autosomal recessive mineralization disorder characterized by the deposition of calcium phosphate crystals in periarticular sites and arterial walls of the lower limbs. In our patient, the administration of colchicine permitted a complete relief of articular symptoms and the normalization of inflammatory markers. Although it is extremely rare, knowledge of this disease helps rheumatologists in the differential diagnosis of arthritis and can contribute to a better understanding of the pathogenetic mechanisms underlying mineralization.
Data Availability
Data used and/or analyzed during the current study are available from the corresponding author on reasonable question.
Code Availability
Not applicable.
References
Gutierrez LB, Link T, Chaganti K, Motamedi D. Arterial calcification due to CD73 deficiency (ACDC): imaging manifestations of ectopic mineralization. Skeletal Radiol. 2016 45():1583-1587. https://doi.org/10.1007/s00256-016-2465-9. Epub 2016 Aug 26.
Cudrici CD, Newman KA, Ferrante EA, Huffstutler R, Carney K, Betancourt B, Miettinen M, Siegel R, Katz JD, Nesti LJ, St Hilaire C, Lakshmipathy D, Wen H, Bagheri MH, Boehm M, Brofferio A. Multifocal calcific periarthritis with distinctive clinical and radiological features in patients with CD73 deficiency. Rheumatology (Oxford). 2021 ;61(1):163-173. https://doi.org/10.1093/rheumatology/keab270.. PMID: 33744914; PMCID: PMC8742829
Nitschke Y, Rutsch F. Genetics in arterial calcification: lessons learned from rare diseases. Trends Cardiovasc Med. 2012;22(6):145–9. https://doi.org/10.1016/j.tcm.2012.07.011.
Saeidian AH, Youssefian L, Huang J, Touati A, Vahidnezhad H, Kowal L, Caffet M, Wurst T, Singh J, Snook AE, Ryu E, Fortina P, Terry SF, Schoenecker JG, Uitto J, Li Q. Genetic heterogeneity of heritable ectopic mineralization disorders in a large international cohort. Genet Med. 2022 24(1):75-86. https://doi.org/10.1016/j.gim.2021.08.011. Epub 2021 Nov 30. PMID: 34906475; PMCID: PMC8943706.
Li Q, Uitto J. Heritable ectopic mineralization disorders: pathomechanisms and potential treatment. J Investig Dermatol Symp Proc. 2018;19(2):S106–7. https://doi.org/10.1016/j.jisp.2018.10.007.
Minor M, Alcedo KP, Battaglia RA, Snider NT. Cell type- and tissue-specific functions of ecto-5'-nucleotidase (CD73). Am J Physiol Cell Physiol. 2019 ;317(6):C1079-C1092. https://doi.org/10.1152/ajpcell.00285.2019. Epub 2019 Aug 28. PMID: 31461341; PMCID: PMC6957383.
St Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, Gill F, Carlson-Donohoe H, Lederman RJ, Chen MY, Yang D, Siegenthaler MP, Arduino C, Mancini C, Freudenthal B, Stanescu HC, Zdebik AA, Chaganti RK, Nussbaum RL, Kleta R, Gahl WA, Boehm M. NT5E mutations and arterial calcifications. N Engl J Med. 2011 ;364(5):432-442. https://doi.org/10.1056/NEJMoa0912923.. PMID: 21288095; PMCID: PMC3049958
Ichikawa N, Taniguchi A, Kaneko H, Kawamoto M, Sekita C, Nakajima A, Yamanaka H. Arterial Calcification Due to Deficiency of CD73 (ACDC) As one of rheumatic diseases associated with periarticular calcification. J Clin Rheumatol. 2015;21(4):216–20. https://doi.org/10.1097/RHU.0000000000000245.
Lakshmipathy DR, Cudrici CD, Dyda F, Xu W, Ferrante EA, Nguyen DT, Carney KM, Rollison S, Chen MY, Nesti LJ, Boehm M, Brofferio A, Wen H. Morphology and chemical identity of periarticular and vascular calcification in a patient with the rare genetic disease of arterial calcification due to deficiency of CD73 (ACDC). Radiol Case Rep. 2020 ;15(10):1883-1886. https://doi.org/10.1016/j.radcr.2020.07.056.. PMID: 32874378; PMCID: PMC7452020
Luo H, Li Q, Cao Y, Uitto J. Therapeutics development for pseudoxanthoma elasticum and related ectopic mineralization disorders: update 2020. J Clin Med. 2020 ;10(1):114. https://doi.org/10.3390/jcm10010114.. PMID: 33396306; PMCID: PMC7795895
Shimada BK, Pomozi V, Zoll J, Kuo S, Martin L, Le Saux O. ABCC6, pyrophosphate and ectopic calcification: therapeutic solutions. Int J Mol Sci. 2021 ;22(9):4555. https://doi.org/10.3390/ijms22094555.. PMID: 33925341; PMCID: PMC8123679
Standards and Guidelines for the Interpretation of Sequence Variants. A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Avruscio G, Massussi M, Adamo A, Brusco A. Challenging arterial calcification disease associated with rare NT5E gene mutation. BMJ Case Rep. 2020 ;13(6):e235365. https://doi.org/10.1136/bcr-2020-235365.. PMID: 32532917; PMCID: PMC7295373
Azuma N, Uchida T, Kikuchi S, Sadahiro M, Shintani T, Yanagi K, Higashita R, Yamashita A, Makita Y, Kaname T. NT5E genetic mutation is a rare but important cause of intermittent claudication and chronic limb-threatening ischemia. Circ J. 2020 ;84(7):1183-1188. https://doi.org/10.1253/circj.CJ-20-0153. Epub 2020 Jun 11.
Magnus-Levy A. Ueber ungewöhnliche Verkalkung der Arterien (Aterienverkalkung ohne primäre Arteriosklerose?). Dtsch Med Wochenschr 1914; 40: 1305-1309 (in German).
Levitin J. A case of arterial and periarticular calcinosis of unknown etiology. Radiology. 1945;44:489–94.
Nosaka H, Yamasaki G, Yamamoto K, Hayashi Y. A case of extensive calcification of the peripheral arteries. Yonago Acta Med. 1974;18:191–7.
Mori H, Yamaguchi K, Fukushima H, Oribe Y, Kato N, Wakamatsu T, Uzawa H. Extensive arterial calcification of unknown etiology in a 29-year-old male. Heart Vessels. 1992;7(4):211–4. https://doi.org/10.1007/BF01744607.
Sharp J. Heredo-familial vascular and articular calcification. Ann Rheum Dis. 1954 13(1):15-27. https://doi.org/10.1136/ard.13.1.15. . PMID: 13149051; PMCID: PMC1030367
Devriese M, Legrand A, Courtois MC, Jeunemaitre X, Albuisson J. Pseudoxanthoma elasticum with prominent arterial calcifications evoking CD73 deficiency. Vasc Med. 2019 ;24(5):461-464. https://doi.org/10.1177/1358863X19853360. Epub 2019 Jun 4.
Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, Terkeltaub R, Millan JL. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci U S A. 2002 ;99(14):9445-9449. https://doi.org/10.1073/pnas.142063399. Epub 2002 Jun 24. PMID: 12082181; PMCID: PMC123160.
McCarthy EF, Sundaram M. Heterotopic ossification: a review. Skeletal Radiol. 2005 34(10):609-619. https://doi.org/10.1007/s00256-005-0958-z. Epub 2005 Aug 25.
Qian S, Regan JN, Shelton MT, Hoggatt A, Mohammad KS, Herring PB, Seye CI. The P2Y2 nucleotide receptor is an inhibitor of vascular calcification. Atherosclerosis. 2017 257:38-46. https://doi.org/10.1016/j.atherosclerosis.2016.12.014. Epub 2016 Dec 15. PMID: 28038380; PMCID: PMC5348113.
Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5′-nucleotidase (CD73). Purinergic Signal. 2006 2(2):351-360. https://doi.org/10.1007/s11302-005-5302-5. Epub 2006 Jun 1. PMID: 18404475; PMCID: PMC2254482.
Kochan Z, Karbowska J, Gogga P, Kutryb-Zajac B, Slominska EM, Smolenski RT. Polymorphism in exon 6 of the human NT5E gene is associated with aortic valve calcification. Nucl NucleotNucleic Acids. 2016;35(10-12):726–31. https://doi.org/10.1080/15257770.2016.1180393.
Jin H, St Hilaire C, Huang Y, Yang D, Dmitrieva NI, Negro A, Schwartzbeck R, Liu Y, Yu Z, Walts A, Davaine JM, Lee DY, Donahue D, Hsu KS, Chen J, Cheng T, Gahl W, Chen G, Boehm M. Increased activity of TNAP compensates for reduced adenosine production and promotes ectopic calcification in the genetic disease ACDC. Sci Signal. 2016;9(458):ra121. https://doi.org/10.1126/scisignal.aaf9109 PMID: 27965423; PMCID: PMC5489239.
Li Q, Price TP, Sundberg JP, Uitto J. Juxta-articular joint-capsule mineralization in CD73 deficient mice: similarities to patients with NT5E mutations. Cell Cycle. 2014;13(16):2609-2615. https://doi.org/10.4161/15384101.2014.943567.. PMID: 25486201; PMCID: PMC4614381
Zhang Z, He JW, Fu WZ, Zhang CQ, Zhang ZL. Calcification of joints and arteries: second report with novel NT5E mutations and expansion of the phenotype. J Hum Genet. 2015 ;60():561-564. https://doi.org/10.1038/jhg.2015.85. Epub 2015 Jul 16.
Fausther M, Lavoie EG, Goree JR, Baldini G, Dranoff JA. NT5E mutations that cause human disease are associated with intracellular mistrafficking of NT5E protein. PLoS One. 2014 Jun;9(6):e98568. https://doi.org/10.1371/journal.pone.0098568.
Moorhead WJ 3rd, Chu CC, Cuevas RA, Callahan J 4th, Wong R, Regan C, Boufford CK, Sur S, Liu M, Gomez D, MacTaggart JN, Kamenskiy A, Boehm M, St Hilaire C. Dysregulation of FOXO1 (Forkhead box O1 protein) drives calcification in arterial calcification due to deficiency of CD73 and is present in peripheral artery disease. Arterioscler Thromb Vasc Biol. 2020 ;40(7):1680-1694. https://doi.org/10.1161/ATVBAHA.119.313765. Epub 2020 May 7. PMID: 32375544; PMCID: PMC7310306.
Yoshioka K, Kuroda S, Takahashi K, Sasano T, Furukawa T, Matsumura A. Calcification of joints and arteries with novel NT5E mutations with involvement of upper extremity arteries. Vasc Med. 2017 ;22(6):541-543. https://doi.org/10.1177/1358863X17724263. Epub 2017 Aug 19.
Markello TC, Pak LK, St Hilaire C, Dorward H, Ziegler SG, Chen MY, Chaganti K, Nussbaum RL, Boehm M, Gahl WA. Vascular pathology of medial arterial calcifications in NT5E deficiency: implications for the role of adenosine in pseudoxanthoma elasticum. Mol Genet Metab. 2011 103(1):44-50. https://doi.org/10.1016/j.ymgme.2011.01.018. Epub 2011 Feb 3. PMID: 21371928; PMCID: PMC3081917.
Uchida T, Yamashita A, Ishizawa A, Sadahiro M, Azuma N, Kaname T. NT5E mutation in sisters who underwent aortic valve replacements for aortic stenosis. Interact Cardiovasc Thorac Surg. 2022 34(1):45-48. https://doi.org/10.1093/icvts/ivab229. Epub 2021 Aug 25. PMID: 34999808; PMCID: PMC8932508.
Zukowska P, Kutryb-Zajac B, Jasztal A, Toczek M, Zabielska M, Borkowski T, Khalpey Z, Smolenski RT, Slominska EM. Deletion of CD73 in mice leads to aortic valve dysfunction. Biochim Biophys Acta Mol Basis Dis. 2017 1863(6):1464-1472. https://doi.org/10.1016/j.bbadis.2017.02.008. Epub 2017 Feb 10.
de Nijs T, Albuisson J, Ockeloen CW, Legrand A, Jeunemaitre X, Schultze Kool LJ, Riksen NP. Isolated arterial calcifications of the lower extremities: a clue for NT5E mutation. Int J Cardiol. 2016 J1;212:248-250. https://doi.org/10.1016/j.ijcard.2016.03.068. Epub 2016 Mar 19.
Bouabdallaoui N, Tardif JC, Waters DD, Pinto FJ, Maggioni AP, Diaz R, Berry C, Koenig W, Lopez-Sendon J, Gamra H, Kiwan GS, Blondeau L, Orfanos A, Ibrahim R, Grégoire JC, Dubé MP, Samuel M, Morel O, Lim P, Bertrand OF, Kouz S, Guertin MC, L'Allier PL, Roubille F. Time-to-treatment initiation of colchicine and cardiovascular outcomes after myocardial infarction in the colchicine cardiovascular outcomes trial (COLCOT). Eur Heart J. 2020 ;41(42):4092-4099. https://doi.org/10.1093/eurheartj/ehaa659.. PMID: 32860034; PMCID: PMC7700755
Hennessy T, Soh L, Bowman M, Kurup R, Schultz C, Patel S, Hillis GS. The low dose colchicine after myocardial infarction (LoDoCo-MI) study: a pilot randomized placebo controlled trial of colchicine following acute myocardial infarction. Am Heart J. 2019 215:62-69. https://doi.org/10.1016/j.ahj.2019.06.003. Epub 2019 Jun 14.
Nidorf SM, Fiolet ATL, Eikelboom JW, Schut A, TSJ O, Bax WA, Budgeon CA, JGP T, Mosterd A, Cornel JH, Thompson PL. LoDoCo2 investigators. The effect of low-dose colchicine in patients with stable coronary artery disease: the LoDoCo2 trial rationale, design, and baseline characteristics. Am Heart J. 2019 Dec;218:46–56. https://doi.org/10.1016/j.ahj.2019.09.011 Epub 2019 Oct 20.
Uitto J, Li Q, van de Wetering K, Váradi A, Terry SF. Insights into pathomechanisms and treatment development in heritable ectopic mineralization disorders: summary of the PXE international biennial research symposium-2016. J Invest Dermatol. 2017 ;137(4):790-795. https://doi.org/10.1016/j.jid.2016.12.014. PMID: 28340679; PMCID: PMC5831331
Rutsch F, Böyer P, Nitschke Y, Ruf N, Lorenz-Depierieux B, Wittkampf T, Weissen-Plenz G, Fischer RJ, Mughal Z, Gregory JW, Davies JH, Loirat C, Strom TM, Schnabel D, Nürnberg P, Terkeltaub R; GACI Study Group. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet. 2008 1(2):133-140. https://doi.org/10.1161/CIRCGENETICS.108.797704. PMID: 20016754; PMCID: PMC2794045.
Ramjan KA, Roscioli T, Rutsch F, Sillence D, Munns CF. Generalized arterial calcification of infancy: treatment with bisphosphonates. Nat Clin Pract Endocrinol Metab. 2009;5(3):167–72. https://doi.org/10.1038/ncpendmet1067.
Li Q, van de Wetering K, Uitto J. Pseudoxanthoma elasticum as a paradigm of heritable ectopic mineralization disorders: pathomechanisms and treatment development. Am J Pathol. 2019 189(2):216-225. https://doi.org/10.1016/j.ajpath.2018.09.014. Epub 2018 Nov 7. PMID: 30414410; PMCID: PMC6412714.
Chan ES, Cronstein BN. Methotrexate--how does it really work? Nat Rev Rheumatol. 2010;6(3):175–8. https://doi.org/10.1038/nrrheum.2010.5.
Funding
Open access funding provided by Università di Pisa within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
MM contributed to conception and design of the study and contributed to write the manuscript. He is the guarantor.
M. Maffi prepared the first draft of the paper, performed the literature search, contributed to the acquisition of data, and was in charge of writing the manuscript.
GDM contributed to the writing of the manuscript and to performing language editing and proofreading.
AM, BT, and CMA contributed to the acquisition and the interpretation of genetic analysis and contributed to write the manuscript.
MRM and M. Mosca actively contributed to drafting the manuscript.
All the authors revised the work critically for intellectual content.
All the authors agree to be accountable for the work and to ensure that any questions relating to the accuracy and integrity of the paper are investigate and properly resolved.
Corresponding author
Ethics declarations
Ethics Approval
The procedures followed were in accordance with the Helsinki Declaration of 1975/83 and Ethics Committee “Area Vasta Nord Ovest” (CEAVNO) with the number 14478.
Consent for Publication
Informed consent was obtained from all individual participants included in the study.
Conflict of Interest
M.Mosca. Advisor for Astra Zeneca, Glaxo Smith Kline, Lilly, UCB, Biogen. Speaker for Janssen, UCB, GSK, Astra Zeneca, Abbvie. Other authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Medicine
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maffi, M., De Mattia, G., Mazzoni, M.R. et al. Calcification of Joints and Arteries (CALJA) Is a Rare Cause of Arthritis and Lower Limb Ischemia: Case Report and Literature Review. SN Compr. Clin. Med. 5, 145 (2023). https://doi.org/10.1007/s42399-023-01485-1
Accepted:
Published:
DOI: https://doi.org/10.1007/s42399-023-01485-1