
Abstract
IgG4-related disease (IgG4-RD) is an immune-mediated fibroinflammatory condition with heterogeneous organ-specific manifestations. IgG4-related hypophysitis results from pituitary involvement and represents a rare neuro-ophthalmic complication of IgG4-RD, but the presentation as pituitary abscess is exceptional. We report the case of a 38-year-old otherwise healthy woman with a 3-year history of relapsing pituitary sterile abscess repeatedly treated with neurosurgery and antimicrobials. Histological re-examination of pituitary biopsy specimens revealed a dense inflammatory infiltrate rich in IgG4-positive plasma cells. Serum IgG4 levels were normal and circulating plasmablasts were increased. A diagnosis of IgG4-related hypophysitis was made and the patient treated with high-dose steroid pulse therapy with subsequent reduction of the pituitary lesion and resolution of symptoms. This case highlights how the diagnostic intuition on the basis of histopathology has changed management with prompt dramatic response to steroid therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
IgG4-related disease (IgG4-RD) is an immune-mediated fibroinflammatory condition with heterogeneous organ-specific manifestations. Pituitary involvement represents a known neuro-ophthalmic complication of IgG4-RD [1, 2]. IgG4-related hypophysitis more frequently occurs in men than women with a median age of 62 years in males and 38 years in females [3]. Patients usually present with central diabetes insipidus, signs and symptoms of anterior hypopituitarism, headache, and visual impairment (mainly bitemporal hemianopsia) as manifestations of pituitary mass effects in the sellar area or a thickened pituitary stalk [3,4,5]. Constitutional symptoms (fever, general malaise, fatigue, appetite loss, weight loss) can also occur [3, 6]. We herein report the case of a 38-year-old otherwise healthy woman with a recent history of pituitary sterile abscess and current mild headache and mild-moderate fatigue.
Case Report
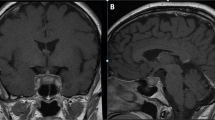
Three years before the current presentation, while she was on a summer vacation in Mexico, the patient complained of sudden severe frontal headache, profound asthenia, diffuse myalgia, dizziness and nausea, without fever, vomiting, or altered vision. She reported no recent trauma. Her past medical history was unremarkable. After 2 weeks, she went back to her home region in Italy and because of constant headache she presented to the emergency department of this hospital for further evaluation. On arrival, a lumbar puncture was performed and cerebrospinal fluid analysis resulted inconsistent for meningitis, revealing only mild increase of white blood cells (11 per μL, reference range 0–5 per μL). Complete blood count with differential was within normal limits and inflammatory markers were negative; other laboratory tests are reported in Supplementary Table 1. Computed tomography (CT) of the head revealed a pituitary mass occupying the sella turcica. Subsequent magnetic resonance imaging (MRI) of the head confirmed a pituitary mass measuring 15 × 19 × 19 mm with peripheral enhancement and mild compression and dislocation of the optic chiasm (Fig. 1). She underwent transsphenoidal neurosurgery with drainage of purulent material, consistent with a pituitary abscess, and the headache abated. Microbiologic cultures of abscess drainage fluid were negative for bacteria, mycobacteria, or fungi. Antineutrophil cytoplasmic antibodies (ANCA), anti-MPO, and anti-PR3 antibodies were negative; other laboratory test results are shown in Supplementary Table 1. Biopsy specimen of the pituitary mass showed a mild inflammatory process. The patient underwent broad-spectrum antimicrobial therapy that was continued orally for at least 4 months. After surgery, she began treatment with cortone acetate and desmopressin for 4 months with complete endocrine function recovery. Nine months after surgery (2 years before the current presentation), severe headache recurred. MRI of the head showed an intrasellar cystic formation measuring about 17 mm in greatest dimension with mixed component (liquid and coarse internal septa), which was suspected for a possible recurrence of the previous pituitary lesion. She underwent a second transsphenoidal drainage of yellowish purulent material whose microbiologic cultures and molecular analyses were negative for bacteria, mycobacteria, protozoa, helminths, or fungi. This time, histologic examination of a biopsy specimen of the pituitary lesion revealed predominantly dense mononuclear inflammatory infiltrate with very limited necrotic/haemorrhagic areas, a few neutrophils, foamy macrophages, and fibrosis. Testing for ANCA was repeatedly negative and IgG4 level was 62 mg/dL (reference range 8–82); other laboratory test results are shown in Supplementary Table 1. Eight months later, she complained of a severe frontal headache and she was again admitted to the Infectious Diseases Unit of this hospital. Imaging findings were similar to those found in previous MRIs. Broad-spectrum antimicrobial therapy was started and continued intravenously in the following months. Nonetheless, a couple of months later, the headache relapsed. MRI of the head showed signs of relapse-progression of the abscess lesion measuring up to 12 × 14 mm. A third neurosurgery was performed 10 months after the previous operation. The patient maintained discrete clinical conditions, but complained of episodes of headache and asthenia. MRI performed 9 months later revealed a mild increase of the pituitary lesion. At this point, histological re-examination of pituitary biopsy specimens was performed and revealed dense inflammatory infiltrate rich in polyclonal IgG4-positive plasma cells with fibrosis (Fig. 2). Immunohistochemically, there was both kappa and lambda light chains positivity, the ratio of IgG4-positive plasma cells to IgG-positive plasma cells was greater than 40%, and more than 10 IgG4-positive plasma cells per high-power field (HPF) in more than 3 different HPF were observed. Storiform fibrosis and obliterative phlebitis were absent. Serum IgG4 levels were within normal limits (28 mg/dL, reference range 8–82). Circulating plasmablasts (CD20-CD27+ CD38+ +) were increased (4700 per mL, reference range 0–600 per mL, 1.5% of CD19+ cells), as evaluated by flow cytometry. Other laboratory test results are shown in Supplementary Table 1. Whole-body 18F-FDG-PET/CT was negative for abnormal uptake. A diagnosis of probable IgG4-RD was made and it was concluded for IgG4-related hypophysitis (Supplementary Table 2) [7, 8].

Initial MRI of the head. Coronal (A) and transverse (B) gadolinium-enhanced T1-weighted images show a pituitary cystic lesion with fluid content and intense peripheral enhancement

Biopsy specimen of the hypophysis. Hematoxylin and eosin staining shows dense mononuclear inflammatory infiltrate with plasma cells, foam cells and fibrosis (40× magnification) (A). Immunohistochemical staining for IgG4 shows numerous IgG4-positive plasma cells (ratio of IgG4+ to IgG+ plasma cells > 40%, > 10 IgG4 + plasma cells per HPF assessed in ×3 HPF) (40× magnification) (B)
Discussion
Hypophysitis is a chronic inflammation of the pituitary gland that comprises a heterogeneous spectrum of conditions. It can be classified on the basis of anatomical location of the pituitary involvement (adenohypophysitis, infundibuloneurohypophysitis, panhypophysitis), of the histologic features (lymphocytic, granulomatous, xanthomatous, IgG4-related, necrotizing, and mixed forms), and of the cause: primary forms are limited to the gland (isolated or as part of a multiorgan systemic disease), whereas secondary forms develop secondary to systemic diseases (granulomatosis with polyangiitis, tuberculosis, sarcoidosis, syphilis), immunomodulatory treatments (immune checkpoints inhibitors, interferon-α), or sellar diseases (germinoma, Rathke cleft cyst, pituitary adenoma, craniopharyngioma) [8,9,10,11]. IgG4-related hypophysitis represents 30% of all primary forms [12], is characterized by a mononuclear infiltration enriched in lymphocytes and plasma cells, with more than 10 IgG4-positive cells per HPF and typical morphological features (storiform fibrosis or obliterative phlebitis) [7, 8, 13]. Non-infectious pituitary abscess or sterile abscess are a controverse entity, indeed in some Authors’ opinion, sterile abscess results from liquefaction of necrosis of an infarcted pituitary gland or the contents of an atypical pituitary cyst [14, 15]. However—as in the current case—patients typically present with no fever, no leukocytosis, and culture results are negative. Notably, abscess is an extremely rare manifestation of IgG4-related hypophysitis, as only one case has been reported to our knowledge to date [16]. In the current case, both clinically and radiologically the pituitary lesion was compatible with an abscess: during the surgeries, yellowish purulent material was drained from the pituitary lesion (in fact, the initial clinical orientation was a pyogenic abscess or an abscess formation in pituitary adenoma) and on imaging studies the lesion showed well-defined irregular borders with peripheral enhancement after the administration of intravenous contrast material, exerting mass effect of adjacent structures.
In a case with atypical single organ manifestation and normal serum IgG4 levels, the diagnosis of IgG4-RD heavily relies on histopathological evaluation of the resected tissue (Supplementary Table 2). Nonetheless, IgG4-RD with exclusive pituitary involvement with normal serum IgG4 levels has been described [17].
The patient was treated with high-dose pulse methylprednisolone (10 mg/kg/day for three consecutive days/month) for 3 months and symptoms abated. One month later, an MRI of the head revealed reduction of the pituitary lesion, measuring 9 × 10 mm and appearing as a cystic lesion with fluid content and hyperintense walls. Moreover, 1 month after the first pulse steroid therapy, circulating plasmablasts (CD20-CD27+ CD38+ +) declined to 1400 per mL (0.4% of CD19 + cells). It has been found that circulating plasmablasts are increased in patients with IgG4-RD even when IgG4 levels are normal and correlate with disease activity and treatment response [18,19,20]. Although increased circulating plasmablasts are not considered a diagnostic criterion of IgG4-RD, their presence in this case could be a marker of such diagnosis, particularly considering their reduction after steroid treatment [21]. In the following months, two more high-dose boluses of methylprednisolone were administered. At a follow-up MRI performed 3 months later, the lesion was further reduced, measuring 5 × 6 mm (Fig. 3) and the patient was still asymptomatic. Two months later, high-dose pulse methylprednisolone (10 mg/kg/day for three consecutive days) was administered as maintenance. At 6-month follow-up, the patient was in good clinical conditions (Supplementary Table 3).

MRI of the head before and after steroid treatment. Sagittal gadolinium-enhanced T1-weighted images before (A) and after high-dose steroids pulse therapy (B) display significant decrease of the pituitary lesion
In conclusion, pituitary abscess is a rare presentation of IgG4-RD; however, IgG4-related hypophysitis should be considered in the differential particularly in young women [22]. Since IgG4 serum levels can be normal, histopathological examination is crucial for diagnosis. Early recognition and prompt treatment are essential due to the risk of progression from an inflammatory to a fibrotic stage poorly responsive to steroid therapy [23]. Patients with severe signs and symptoms of sella compression could be treated with surgery, although relapse is common. Induction treatment with high-dose glucocorticoids usually determines an excellent response. In case of resistance or as a steroid-sparing agent in remission maintenance, rituximab (anti-CD20 monoclonal antibody) is suggested [24,25,26].
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Chwalisz BK, Stone JH. Neuro-ophthalmic complications of IgG4-related disease. Curr Opin Ophthalmol. 2018;29(6):485–94.
Umehara H, Okazaki K, Nakamura T, et al. Current approach to the diagnosis of IgG4-related disease - Combination of comprehensive diagnostic and organ-specific criteria. Mod Rheumatol. 2017;27(3):381–91.
Amirbaigloo A, Esfahanian F, Mouodi M, et al. IgG4-related hypophysitis. Endocrine. 2021;73(2):270–91.
Li Y, Gao H, Li Z, et al. Clinical Characteristics of 76 Patients with IgG4-Related Hypophysitis: A Systematic Literature Review. Int J Endocrinol. 2019;2019:5382640.
Uccella S, Amaglio C, Brouland JP, et al. Disease heterogeneity in IgG4-related hypophysitis: report of two histopathologically proven cases and review of the literature. Virchows Arch. 2019;475(3):373–81.
Shikuma J, Kan K, Ito R, et al. Critical review of IgG4-related hypophysitis. Pituitary. 2017;20(2):282–91.
Umehara H, Okazaki K, Kawa S, et al. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD. Mod Rheumatol. 2021;31(3):529–33.
Leporati P, Landek-Salgado MA, Lupi I, et al. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab. 2011;96(7):1971–80.
Angelousi A, Alexandraki K, Tsoli M, et al. Hypophysitis (Including IgG4 and Immunotherapy). Neuroendocrinology. 2020;110(9–10):822–35.
Lojou M, Bonneville JF, Ebbo M, et al. IgG4 hypophysitis: Diagnosis and management. Presse Med. 2020;49(1):104016.
Jessel S, Weiss SA, Austin M, et al. Immune Checkpoint Inhibitor-Induced Hypophysitis and Patterns of Loss of Pituitary Function. Front Oncol. 2022;8(12):836859.
Bando H, Iguchi G, Fukuoka H, et al. The prevalence of IgG4-related hypophysitis in 170 consecutive patients with hypopituitarism and/or central diabetes insipidus and review of the literature. Eur J Endocrinol. 2013;170(2):161–72.
Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181–92.
Liu Y, Liu F, Liang Q, et al. Pituitary abscess: report of two cases and review of the literature. Neuropsychiatr Dis Treat. 2017;13:1521–6.
Nordjoe YE, Aubin Igombe SR, Laamrani FZ, Jroundi L. Pituitary abscess: two case reports. J Med Case Rep. 2019;13(1):342.
Hadjigeorgiou GF, Lund EL, Poulsgaard L, et al. Intrachiasmatic abscess caused by IgG4-related hypophysitis. Acta Neurochir (Wien). 2017;159(11):2229–33.
Yuen KCJ, Moloney KJ, Mercado JU, Rostad S, et al. A case series of atypical features of patients with biopsy-proven isolated IgG4-related hypophysitis and normal serum IgG4 levels. Pituitary. 2018;21(3):238–46.
Wallace ZS, Mattoo H, Carruthers M, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74(1):190–5.
Lanzillotta M, Della-Torre E, Milani R, et al. Effects of glucocorticoids on B-cell subpopulations in patients with IgG4-related disease. Clin Exp Rheumatol. 2019;37 Suppl 118(3):159–66.
Lanzillotta M, Della-Torre E, Milani R, et al. Increase of circulating memory B cells after glucocorticoid-induced remission identifies patients at risk of IgG4-related disease relapse. Arthritis Res Ther. 2018;20(1):222.
Fox RI, Fox CM. IgG4 levels and plasmablasts as a marker for IgG4-related disease (IgG4-RD). Ann Rheum Dis. 2015;74(1):1–3.
Sosa GA, Bell S, Christiansen SB, et al. Histologically confirmed isolated IgG4-related hypophysitis: two case reports in young women. Endocrinol Diabetes Metab Case Rep. 2014;2014:140062.
Khosroshahi A, Wallace ZS, Crowe JL, et al. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Rheumatol. 2015;67(7):1688.
Ebbo M, Grados A, Samson M, et al. Long-term efficacy and safety of rituximab in IgG4-related disease: Data from a French nationwide study of thirty-three patients. PLoS ONE. 2017;12(9):e0183844.
Campochiaro C, Della-Torre E, Lanzillotta M, et al. Long-term efficacy of maintenance therapy with Rituximab for IgG4-related disease. Eur J Intern Med. 2020;74:92–8.
Katz G, Stone JH. Clinical Perspectives on IgG4-Related Disease and Its Classification. Annu Rev Med. 2022;73:545–62.
Acknowledgements
Thanks is due to all persons who participated in the care of the patient.
Funding
Open access funding provided by Università degli Studi di Firenze within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
LS and DC wrote the manuscript. All Authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1975 Helsinki declaration and its later amendments or comparable ethical standards.
Consent to Participate
Informed consent was obtained from the patient to participate.
Written Consent for Publication
Written consent was obtained from the patient for publication of this case report and any accompanying images.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Medicine
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salvati, L., Tinghi, F., Ammannati, F. et al. Pituitary Abscess as Manifestation of IgG4-Related Hypophysitis: A Case Report. SN Compr. Clin. Med. 4, 170 (2022). https://doi.org/10.1007/s42399-022-01250-w
Accepted:
Published:
DOI: https://doi.org/10.1007/s42399-022-01250-w