
Abstract
Kawasaki disease (KD) is an acute, self-limiting febrile illness of childhood associated with vasculitis, mainly of the medium-sized arteries. The clinical significance and impact of this condition arise from its predilection for the coronary arteries. The criteria for classic Kawasaki disease are clearly defined, but many children present with atypical forms, and clinicians need to consider this possibility. Although most diagnosed cases respond to intravenous immunoglobulin (IVIG) and aspirin, some have proven resistant to the standard treatment. This article aims to provide a brief overview of Kawasaki disease, focusing on the resistant/refractory cases, and the treatment options available for such cases.
Similar content being viewed by others

Introduction
Kawasaki disease (KD) is an acute, self-limiting febrile illness of childhood associated with vasculitis, mainly of the medium-sized arteries, the clinical significance and impact arising from its predilection for the coronary arteries. It is the leading cause of acquired heart disease in children in developed countries [1] and the second most common vasculitis in children after Henoch-Schönlein purpura [2].
In most children (75–85%), the symptoms subside without any long-term sequelae. In the remaining 15–25% of children, however, coronary artery involvement can lead to complications. The coronary involvement could be an asymptomatic mild dilatation or aneurysm formation, but at the other end of the spectrum, it can be very severe with giant aneurysms in the coronary arteries associated with thrombosis, myocardial infarction, and sudden death [3]. Although mortality during the acute phase is extremely low, fatalities can occur later in life in late childhood or adulthood due to myocardial infarction. Intravenous immunoglobulin (IVIG) treatment is a time-tested first-line treatment for KD with a proven role in the prevention of coronary artery aneurysms (CAA). Nonetheless, 10–20% of KD patients fail to respond to IVIG, and this refractory group is more prone to coronary artery complications, making the management of this sub-group challenging.
Types of Kawasaki Disease
Classic Kawasaki Disease
KD is defined as classic, typical, or complete when the child’s symptoms and signs satisfy the diagnostic criteria. The diagnostic criteria [4] put forward by American Heart Association (AHA) include five or more days of fever, four or more of the five principal clinical features, and exclusion of other diseases with similar findings. The five principal clinical features are:
-
1.
Bilateral non-exudative bulbar conjunctivitis
-
2.
Erythema and cracking of lips, strawberry tongue, and/or erythema of oral and pharyngeal mucosa
-
3.
Cervical lymphadenopathy (≥1.5 cm diameter), usually unilateral
-
4.
Polymorphous rash
-
5.
Erythema and edema of the hands and feet in the acute phase and/or periungual desquamation in the subacute phase
The clinical features of KD tend to appear sequentially, and they are listed above order. One or more may have resolved by the time the child presents to the clinician. Hence, a detailed history is crucial to avoid missing diagnostic criteria at the time of presentation.
Atypical Kawasaki Disease
KD is classified as incomplete (atypical) when the symptoms and signs of the child satisfy some but not all diagnostic criteria for complete KD. Incomplete cases are not uncommon (15–20% of all KD patients) especially in children younger than 6 months or older than 5 years. This group presents with a higher incidence of coronary artery abnormalities, long-term sequelae, and is resistant to standard treatment. This might be due to the age group characteristics and delay in diagnosis. Maintaining a high index of suspicion is crucial in identifying such cases.
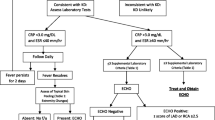
The AHA scientific statement on Kawasaki disease has proposed an algorithm to aid the diagnosis of incomplete KD cases [1]. Children with prolonged fever and elevated inflammatory markers (CRP and/or ESR) are further evaluated. Three or more listed laboratory findings or positive echocardiogram findings in children with 5 or more days of fever with at least two principal clinical features can be diagnosed as incomplete KD and started on standard treatment. Children with more than 7 days of fever with no other explanation can also be diagnosed as incomplete KD using the same criteria.
The six listed laboratory findings for diagnosis of incomplete KD are anemia for age, platelet count > 450,000, serum albumin < 3 g/dL, elevated ALT levels, total WBC count > 15,000/mm3, and urine WBC > 10/hpf.
Febrile children with features of atypical Kawasaki disease, Kawasaki disease shock syndrome, or toxic shock syndrome have been reported during the current COVID-19 pandemic and labeled as multisystem inflammatory syndrome (MIS-C) [5].
Etiology and Pathophysiology of KD
Despite years of research, the exact etiology of KD is not yet known. The most widely accepted view is that it is the result of an exaggerated immune response in genetically predisposed children in whom environmental factors including infections could be a potential trigger. The result of this exaggerated response is an imbalance between pro- and anti-inflammatory pathways causing vascular damage.
The first phase of vascular involvement occurs within 2 weeks of the onset of fever. This is characterized by infiltration of neutrophils progressively into the layers of arteries from intima to media reaching until the outermost layer of adventitia resulting in artery dilatation and aneurysm formation [1, 6, 7]. Increased production of nitric oxide secondary to upregulation of NO synthase enzyme (iNOS) present in vascular smooth muscle cells and leukocytes during the inflammatory response also contributes to the arterial dilatation. The second phase is the subacute chronic arteritis characterized by infiltration of CD8+ T cells, IgA+ plasma cells, monocytes, and macrophages. This phase can be variable and can last up to months or even years in a small subset. The third phase is the luminal myofibroblast proliferation (LMP), derived from the smooth muscle cells of the media layer of blood vessels (Fig. 1). This transformation is triggered by pro-inflammatory cytokines like IL-1β and TNF-α released by the infiltrates in the second phase. The occurrence of myocardial infarction later in life is either the result of sequential thrombosis in the damaged coronaries or the progressive stenosis caused by LMP.

Noval Rivas M, Arditi M. Kawasaki disease: pathophysiology and insights from mouse models. Nat Rev Rheumatol.2020;16(7):391-405.doi:10.1038/s41584-020-0426-0. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7250272 on 23/12/2020
The theory of genetic predisposition is supported by epidemiological observations of increased incidence of KD in certain races (Japan, Korea, and Taiwan and Asian races in the USA ) [8] and within the same family (ten-times more risk of KD in siblings of patients with KD than the general population and two-times higher risk in children with parental history of KD) [9]. Further studies have substantiated this theory by detecting HLA associations (DRB1, B5, Bw51) and gene polymorphism (ITPKC—inositol 1,4,5-triphosphate 3-kinase) associated with susceptibility to KD [10].
The theory of environmental factors and infections playing a role in triggering KD is supported by the epidemiological data of seasonal (winter and spring seasons) [11] increase in the incidence of KD and clustering of cases. Several studies have investigated the role of different infectious agents, but no specific infectious agent is consistently associated with KD.
Standard Treatment of KD
The currently followed standard first-line treatment for KD is IVIG (2gm/kg) [8] and high-dose aspirin (80 to 100 mg/kg/day or 30 to 50 mg/kg/day in 4 divided doses) until the child is afebrile for 48 h or day 14 of illness followed by low-dose aspirin (3 to 5 mg/kg/day) for 6 to 8 weeks when repeat echocardiography confirms the absence of CAA.
IVIG treatment has conclusively been shown to decrease the incidence of CAA in multiple trials and meta-analysis [12]. The possible mechanisms of action of IVIG include modulation of cytokine production, neutralization of toxins or other pathogenic agents, augmentation of regulatory T-cell activity, and suppression of antibody synthesis [13]. Aspirin has a significant anti-inflammatory effect at high doses and an antiplatelet effect at low doses. But it has not shown any significant reduction in coronary artery involvement [14].
Refractory/Resistant KD
Refractory Kawasaki disease is diagnosed when the child has persistent fever or relapse of fever, 36 h after completion of IVIG treatment [1]. It is vital to evaluate other causes of fever including infections and systemic onset juvenile idiopathic arthritis (sJIA) before diagnosing refractory KD. Differentiating between sJIA and KD can be a diagnostic challenge. Measuring IL-18 levels can be helpful since it is significantly raised in sJIA [15] although the availability of this investigation in clinical practice can be a constraint. Serum ferritin levels are also markedly elevated in sJIA compared with KD [16]. It is particularly important to look out for features of macrophage activation syndrome (MAS) as it may occur during the acute phase of Kawasaki disease (KD) [17] and can present as a complication of sJIA as well [18]. The incidence of resistance to IVIG treatment varies and ranges between 10 and 20% of KD cases [11]. Persistence or increase in inflammatory markers after initial IVIG treatment is also an indicator of refractory disease.
Predictors of Refractory KD
There is a significantly higher incidence of coronary artery abnormalities including aneurysm formation in children with refractory KD. It would be immensely helpful if the children at higher risk of developing resistance can be identified from the onset. This will enable the clinician to consider adjunctive treatment along with IVIG for the first course of treatment and to plan further aggressive treatment after the initial standard treatment regime. Multiple scoring systems have been devised to serve this purpose, Kobayashi [19] and Egami [20] from Japan being the prominent ones. Unfortunately, the sensitivity and specificity of these scoring systems outside the geographical area of development are low [21]. Variation of basic genetic profiles in different populations could be a reason for the discrepancy.
Multiple meta-analyses [22, 23] and systemic reviews have been conducted to identify risk factors for developing refractory KD. The common predictors in many of these studies include raised CRP, ESR, total bilirubin, AST, ALT, pro-BNP, and polymorphonuclear neutrophil (PMN). Also, decreased serum sodium and albumin levels were consistently reported.
Treatment of Refractory KD
There are no well-defined guidelines in managing refractory KD to date, and management remains controversial five decades since the condition was first reported. The following interventions have been effective in decreasing inflammatory markers and abatement of fever, but none of them, so far, have proved unequivocally successful in decreasing the incidence of coronary artery abnormalities.
Intravenous Immunoglobulin
Many centers advocate a second dose of IVIG at a dose of 2 g/kg [24], adding to a cumulative dose of 4g/kg of IVIG. The risk of hemolysis and the possible requirement for blood transfusion need to be considered with this approach.
Corticosteroids
The treatment options of pulse dose of steroids and low dose adjuvant therapy [25] have been evaluated over the years and ruled out as the first-line treatment for KD due to a lack of effectiveness in preventing coronary artery complications.
A meta-analysis [26] including the multicentric RAISE trial (Randomized Controlled Trial to Assess Immunoglobulin Plus Steroid Efficacy for Kawasaki Disease) reported that a combination of corticosteroid with standard-dose IVIG as an initial treatment in high-risk patients reduces the incidence of CAA and IVIG resistance [25]. The low accuracy of scoring systems to identify high-risk patients outside Japan, nevertheless, precludes universal acceptance for this strategy.
Multiple case series and observational studies [27,28,29] have demonstrated symptomatic improvement and decreased incidence of CAA abnormalities in IVIG-resistant children treated with pulse dose of steroids. Methylprednisolone in the dose of 30–50mg/kg/day for three consecutive days is the recommended schedule. This can be administered to children who are at high risk for complications from the second dose of IVIG or those who failed to respond to the second dose of IVIG. This pulse dose can be administered as a stand-alone option or can be followed up by an oral prednisolone tapering dose over 2 weeks.
Infliximab
Given the characteristic elevation of TNF in KD, TNF antagonists like infliximab have been tried as a treatment option. Infliximab binds to and inhibits TNF α, preventing the release of pro-inflammatory cytokines and interleukins. It has been shown to bring in quicker resolution of fever symptoms in children with IVIG resistance compared with the second dose of IVIG. No differences in CAA incidence were noted in a study by Mori et al. [30] but a later study by Gyu Hur et al. [31] suggests that the early administration of infliximab may reduce the incidence of significant CAA in patients with IVIG-resistant KD. Infliximab (5mg/kg single infusion) can be considered as a treatment option in children with IVIG resistance instead of the second dose of IVIG or pulse methylprednisolone.
Cyclosporin
Cyclosporin suppresses immune activation by inhibiting the assembly and release of IL-2 and inhibiting the activation of T cells. Cyclosporin (administered 12 hourly with a total daily dose of 3mg/kg iv or 4–8mg/kg oral, levels can be monitored) can be considered in those resistant cases not responding to the second dose of IVIG or pulse methylprednisolone or infliximab [32]. Cyclosporin dose should be tapered and stopped when the child is afebrile or after 2 weeks.
Anakinra
Since IL-1-induced inflammation was observed in coronary artery vasculitis in KD, Anakinra, a short-acting competitive inhibitor of IL-1 receptor, was used in trials to treat resistant KD [32]. Further trials are still in progress to evaluate the efficacy of this agent.
Cyclophosphamide
Cytotoxic agents like cyclophosphamide are relatively toxic and should be reserved for very refractory cases under exceptional circumstances [33].
Plasma exchange
Plasma exchange or plasmapheresis has shown effectiveness in managing resistant KD [34]. The blood is centrifuged, and the filtered plasma containing the inflammatory cytokines is replaced with albumin. Owing to the complexity of the procedure and potential complications, it is considered as a last resort where all other modalities have failed.
The treatment options for refractory Kawasaki disease are summarized in the following table (Table 1) from the 2017 AHA scientific statement on KD [1].
Conclusion
Prompt institution of the first-line treatment and moving on to the next treatment option when no clinical response is noted are crucial in preventing long-term morbidity and mortality in children with refractory KD. Since such cases are rare, it is imperative to involve the experts in the field (pediatric rheumatologist) whenever possible to guide the management approach.
Change history
15 February 2021
A Correction to this paper has been published: https://doi.org/10.1007/s42399-021-00812-8
References
McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation. 2017;135:e927–99.
Gardner-Medwin JMM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. 2002;360(9341):1197–202.
Holve TJ, Patel A, Chau Q, Marks AR, Meadows A, Zaroff JG. Long-term Cardiovascular Outcomes in Survivors of Kawasaki Disease. Pediatrics. 2014;133(2):e305–11.
JCS Joint Working Group. Guidelines for diagnosis and management of cardiovascular sequelae in Kawasaki disease (JCS 2013). Circ J. 2014;78:2521–62.
Feldstein LR, Rose EB, Horwitz SM, Collins JP, Newhams MM, Son MBF, et al. Multisystem inflammatory syndrome in U.S. children and adolescents. N Engl J Med. 2020;383:334.
Noval Rivas M, Arditi M. Kawasaki disease: pathophysiology and insights from mouse models. Nat Rev Rheumatol. 2020;16(7):391–405. https://doi.org/10.1038/s41584-020-0426-0.
Singh S, Vignesh P, Burgner. The epidemiology of Kawasaki disease: a global update. D Arch Dis Child. 2015;100(11):1084–8.
Takahashi K, Oharaseki T, Yokouchi Y. Update on etio and immunopathogenesis of Kawasaki disease. Curr Opin Rheumatol. 2014;26(1):31–6.
Yoon KL. Update of genetic susceptibility in patients with Kawasaki disease. Korean J Pediatr. 2015;58(3):84–8.
Makino N, Nakamura Y, Yashiro M, Ae R, Tsuboi S, Aoyama Y, et al. Descriptive epidemiology of Kawasaki disease in Japan, 2011-2012: from the results of the 22nd nationwide survey. J Epidemiol. 2015;25(3):239–45.
Durongpisitkul K, Gururaj VJ, Park JM, Martin CF. The prevention of coronary artery aneurysm in Kawasaki disease: a meta-analysis on the efficacy of aspirin and immunoglobulin treatment. Pediatrics. 1995;96:1057–61.
Wang Y, Wang W, Gong F, Fu S, Zhang Q, Hu J, et al. Evaluation of intravenous immunoglobulin resistance and coronary artery lesions in relation to Th1/Th2 cytokine profiles in patients with Kawasaki disease. Arthritis Rheum. 2013;65(3):805–14.
Downie ML, Manlhiot C, Collins TH, Chahal N, Yeung RSM, McCrindle BW. Factors associated with development of coronary artery aneurysms after Kawasaki disease are similar for those treated promptly and those with delayed or no treatment. Int J Cardiol. 2017;236:157–61.
Takahara T, Shimizu M, Nakagishi Y, Kinjo N, Yachie A. Serum IL-18 as a potential specific marker for differentiating systemic juvenile idiopathic arthritis from incomplete Kawasaki disease. Rheumatol Int. 2015;35(1):81–4.
Mizuta M, Shimizu M, Inoue N, Kasai K, Nakagishi Y, Takahara T, et al. Serum ferritin levels as a useful diagnostic marker for the distinction of systemic juvenile idiopathic arthritis and Kawasaki disease. Mod Rheumatol. 2016;26(6):929–32.
Latino GA, Manlhiot C, Yeung RS, Chahal N, McCrindle BW. Macrophage activation syndrome in the acute phase of Kawasaki disease. J Pediatr Hematol Oncol. 2010;32(7):527–31.
Shimizu M, Nakagishi Y, Inoue N, Mizuta M, Ko G, Saikawa Y, et al. Interleukin-18 for predicting the development of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Clin Immunol. 2015;160(2):277–81.
Kobayashi T, Inoue Y, Takeuchi K, Okada Y, Tamura K, Tomomasa T, et al. Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease. Circulation. 2006;113(22):2606–12.
Egami K, Muta H, Ishii M, Suda K, Sugahara Y, Iemura M, et al. Prediction of resistance to intravenous immunoglobulin treatment in patients with Kawasaki disease. J Pediatr. 2006;149(2):237–40.
Davies S, Sutton N, Blackstock S, Gormley S, Hoggart CJ, Levin M, et al. Predicting IVIG resistance in UK Kawasaki disease. Arch Dis Child. 2015;100(4):366–8.
Baek JY, Song MS. Meta-analysis of factors predicting resistance to intravenous immunoglobulin treatment in patients with Kawasaki disease. Korean J Pediatr. 2016;59:80–90.
Li X, Chen Y, Tang Y, Ding Y, Xu Q, Sun L, et al. Predictors of intravenous immunoglobulin-resistant Kawasaki disease in children: a meta-analysis of 4442 cases. Eur J Pediatr. 2018;177:1279–92.
Burns JC, Capparelli EV, Brown JA, Newburger JW, Glode MP. Intravenous gamma-globulin treatment and retreatment in Kawasaki disease: US/Canadian Kawasaki Syndrome Study Group. Pediatr Infect Dis J. 1998;17:1144–8.
Campbell AJ, Burns JC. Adjunctive therapies for Kawasaki disease. J Infect. 2016;72:S1–5.
Chen S, Dong Y, Yin Y, Krucoff MW. Intravenous immunoglobulin plus corticosteroid to prevent coronary artery abnormalities in Kawasaki disease: a meta-analysis. Heart. 2013;99:76–82. https://doi.org/10.1136/heartjnl-2012-302126.
Kobayashi T, Saji T, Otani T, Takeuchi K, Nakamura T, Arakawa H, et al. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, open-label, blinded-endpoints trial. Lancet. 2012;379:1613–20.
Furukawa T, Kishiro M, Akimoto K, Nagata S, Shimizu T, Yamashiro Y. Effects of steroid pulse therapy on immunoglobulin-resistant Kawasaki disease. Arch Dis Child. 2008;93:142–6. https://doi.org/10.1136/adc.2007.126144.
Miura M, Tamame T, Naganuma T, Chinen S, Matsuoka M, Ohki H. Steroid pulse therapy for Kawasaki disease unresponsive to additional immunoglobulin therapy. Paediatr Child Health. 2011;16:479–84.
Mori M, Imagawa T, Hara R, Kikuchi M, Hara T, Nozawa T, et al. Efficacy and limitation of infliximab treatment for children with Kawasaki disease intractable to intravenous immunoglobulin therapy: report of an open-label case series. J Rheumatol. 2012;39:864–7.
Hur G, Song MS, Sohn S, Lee HD, Kim GB, Cho HJ, et al. Infliximab treatment for intravenous immunoglobulin-resistant Kawasaki disease: a multicenter study in Korea. Korean Circ J. 2019;49(2):183–91.
Suzuki H, Terai M, Hamada H, Honda T, Suenaga T, Takeuchi T, et al. Cyclosporin A treatment for Kawasaki disease refractory to initial and additional intravenous immunoglobulin. Pediatr Infect Dis J. 2011;30:871–6.
Cohen S, Tacke CE, Straver B, Meijer N, Kuipers IM, Kuijpers TW. A child with severe relapsing Kawasaki disease rescued by IL-1 receptor blockade and extracorporeal membrane oxygen- ation. Ann Rheum Dis. 2012;71:2059–61. https://doi.org/10.1136/annrheumdis-2012-201658.
Wallace CA, French JW, Kahn SJ, Sherry DD. Initial intravenous gammaglobulin treatment failure in Kawasaki disease. Pediatrics. 2000;105:E78.
Imagawa T, Mori M, Miyamae T, Ito S, Nakamura T, Yasui K, et al. Plasma exchange for refractory Kawasaki disease. Eur J Pediatr. 2004;163:263–4.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Table 1 was revised.
This article is part of the Topical Collection on Medicine
Rights and permissions
About this article

Cite this article
Abraham, D., Kalyanasundaram, S. & Krishnamurthy, K. Refractory Kawasaki Disease—a Challenge for the Pediatrician. SN Compr. Clin. Med. 3, 855–860 (2021). https://doi.org/10.1007/s42399-021-00775-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42399-021-00775-w