
Abstract
Two novel nonlinear optical isomorphous crystals of proline derivatives with alkyne functionality have been obtained (Boc-L-ProNH(CH2)2CCH and Boc-cis-4-fluoro-L-ProNH(CH2)2CCH). Both derivatives, which differ only by the substitution of a H atom to a F atom, adopt the same polar and columnar right-handed helix arrangement in the crystalline state. In addition, adjacent polar helical columns all point in the same direction, thus generating a macrodipole and a crystalline system conducive for second harmonic generation (SHG) properties. This isomorphous crystal system constitutes an interesting tool to study the effect of the fluorine atom on the dipole moment and on the first hyperpolarizability. Starting from the PBC optimized geometries of the crystals, the macroscopic second-order nonlinearity, χ(2), of the newly synthesized crystals has been estimated by quantum chemical calculations. These χ(2) responses are of the same order of magnitude as those of inorganic proline derivatives while smaller than those observed in crystals of push–pull π-conjugated molecules.
Graphic Abstract

Similar content being viewed by others


1 Introduction
A better understanding of the structure–property relationship is central in the process of designing new materials. In the search of materials exhibiting improved nonlinear optical (NLO) properties, the link between the first hyperpolarizability (β) rising at the molecular level and the emerging macroscopic property is not wholly understood. Thanks to their detailed molecular organization, crystals offer a great opportunity to address a better comprehension of such connexion and consequently to provide a rational design approach [1,2,3,4]. Comparable approach has been performed by two of us in crystal derivatives of (S)-2-(α-methylbenzylamino)-5-nitropyridine (MBANP). It has been shown that the second-order NLO response changes substantially as a function of the substitution of the 2-amino-5-nitropyridine unit by one methyl group, owing to effects on the molecular geometry, to the in-crystal polarizing field, and to the type of packing in the crystal structure [5]. Among potential candidates, isomorphous crystal derivatives provide also very interesting opportunities to better grasp the structure-properties relationship as small molecular changes have great impact on NLO properties [6]. We propose in this study to combine synthesis of isomorphous amino acid crystals derivatives with quantum chemical calculations to better understand the impact of adding a non-aromatic electronegative atom like fluorine on NLO properties.
Among NLO properties the second harmonic generation (SHG) is certainly the most studied property. Designing organic materials with highest SHG activities is thus a very active research area [7]. Among polar crystals with their intrinsic asymmetry, natural amino acids offer very interesting avenues to explore as they are homochiral. Indeed, non-centrosymmetry is a condition sine qua non to achieve second-order NLO responses [8]. Moreover, most of them are transparent, and can be easily tunable. Despite its potential, and contrary to proline salts and proline based semiorganic compounds [9,10,11], purely organic proline derivatives were not greatly studied for their ability to exhibit second-order NLO responses [12]. Proline presents a rigid core which tends to limit the number of available conformations and leads in nature to helical structure such as collagen whose symmetry of the supramolecular structure is compatible with efficient SHG response [13,14,15,16]. Nevertheless, besides molecules such as substituted helicenes and helicene-like compounds [17, 18] and other oligomeric helical strands [19, 20], obtaining polar material structure with a helical structure remains difficult to achieve [21]. In this study, we propose the synthesis and computational characterization of two novel proline derivatives which yield polar and helical isomorphous crystals. The two studied proline derivatives are Boc-L-ProNH(CH2)2CCH and Boc-cis-4-fluoro-L-ProNH(CH2)2CCH that will be designed all along the text by BocProyne-H and BocProyne-4F, respectively (Fig. 1). They differ only by the presence of a fluorine atom in the pyrrolidine ring. Both derivatives adopt the same columnar right-handed helix arrangement in the crystalline state (isomorphism). They thus both lead to strong macrodipoles and to SHG response according to our calculations. To the best of our knowledge, these crystals contain the first non-aromatic fluoro crystalline derivative that exhibit non-zero second-order NLO response.

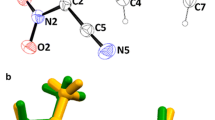
Structure of new proline derivatives BocProyne-H and BocProyne-4F
2 Results and Discussion
2.1 Synthesis
BocProyne-H was obtained in a straightforward and efficient manner from commercially available N-Boc-L-Pro-OSu and but-3-yn-1-amine (Scheme 1). BocProyne-4F was obtained from commercial N-Boc-cis-4-fluoro-L-Pro, but-3-yn-1-amine, DIPEA and HBTU has coupling agent.

Synthesis of BocProyne-H and BocProyne-4F
2.2 Crystallographic Studies
2.2.1 BocProyne-H
Crystallographic data of BocProyne-H, are reported in Table 1. Crystallographic structures reveal that BocProyne-H adopts a conformation in which the main polar functional groups, mainly the amide and carbamate carbonyls, are oriented approximately in the same direction (Fig. 2a). Computations yield a dipole of 7.4 Debye. The conformation of the pyrrolidine ring is the Cγ-endo conformation (Fig. 2b), where the Cγ atom and the carbonyl group are found in the same side of the plane possessing the other atoms of the ring [22, 23]. There are two types of hydrogen bonds (Fig. 2c). The strongest hydrogen bond is found between amide groups, NH⋯O=C. It mainly stabilizes the columnar 1D architecture into an infinite parallel amide chain where cooperativity effects occur [24]. The resulting right-handed helix is composed of 6 units per helical turn and a pitch of 29.21 Å/turn (Fig. 2d) [25]. Right-handed helix can be found in several biological molecules such as collagen, polyproline type I (PPI), α-helix, 310 helix, π helix, and in the double helical structure of the A and B forms of DNA [26]. The second hydrogen bond (CH⋯O=C) occurs between an alkyne function and a carbamate group of a molecule in the adjacent 1D column (Fig. 2c and 2d). Though weaker than the previous one, this nonconventional interaction is still considered as an hydrogen bond since the CH⋯O distance, 2.310 Å, is shorter than the sum of their VDW radii, 2.61 Å, and the three atoms are almost aligned (CH⋯O angle = 166.50º) [27, 28]. It results in 3D polar crystal assembly [29,30,31]. The whole crystal lattice is shown in Figs. 2d and 2e, revealing the anisotropic organization in which all the carbonyl groups point nearly in the same direction along the c axis. Such arrangement of adjacent polar helical columns, or screw axes, lead to a macroscopic dipole.

a Orientation of single-molecule BocProyne-H carbonyls; b pyrrolidine ring Crystal structure conformation of BocProyne-H; c main hydrogen bonds inside a 1D column (NH⋯O=C) and between column (CH…O=C); d profile view of two adjacent right-handed helix (6 units per turn) with the same macrodipole orientation (H bond are represented as dotted lines and the alkyne bonds are designed according the spacefilled model; e cell axes of BocProyne-H along the c axis (top view of 4 columns)
2.2.2 BocProyne-4F
Crystallographic data of BocProyne-4F are reported in Table 1. They are compared with those of BocProyne-H. The two structures present the same group space. Moreover, the axis units differ by 0.2% for the a and b lattice parameters, and by 0.4% for the c unit cell. Finally, considering the fact both structures also possess the same polar helical molecular organization, these two crystals are thus isomorphous. The presence of the fluorine atom in BocProyne-4F (Fig. 3a) affords a stronger dipole than in the case of BocProyne-H (Fig. 2a). Indeed, in an approximate way, the carbamate, amide, and also the C-F dipoles add up (Fig. 3b) to generate an electric dipole moment of 8.7 D, as evaluated from MP2/6–311 + + G(d,p) calculations. A slight difference can be found between the two crystal structures: the pitch in BocProyne-4F is 29.079 Å/turn (Fig. 3d), while it is 29.21 Å/turn for BocProyne-H (Fig. 2d). It originates from the shorter hydrogen bond NH⋯O=C for the former compound, 2.058 Å (Fig. 3c), comparatively to 2.070 Å for the latter structure (Fig. 2c).

a Orientation of single-molecule BocProyne-4F carbonyls and C-F bond; b pyrrolidine ring Crystal structure conformation of BocProyne-4F; c main hydrogen bonds inside a 1D column (NH⋯O=C) and between column (CH…O=C); d profile view of two adjacent right-handed helix (6 units per turn) with the same macrodipole orientation (H bond are represented as dotted lines and the alkyne bonds are designed according the spacefilled model; e cell axes of BocProyne-4F along the c axis (top view of 4 columns)
2.3 Multiscale Numerical Simulation (SHG Response)
Starting from the PBC optimized geometries of the crystals, the dipole moment (μ) and first hyperpolarizability (β) of BocProyne-H and BocProyne-4F have been evaluated in their crystal field, as described by the PBC/Mulliken charges. They have been compared to the corresponding responses in the same geometries but without the crystal polarizing field (Table 2). Without crystal field, substitution by F atoms leads to an increase of the dipole moment by 11% (5.14 D → 5.71 D) and a stronger increase (28%: 144 au → 185 au) of the norm of the vectorial invariant of the first hyperpolarizability tensor. This increase of β amplitude is accompanied by an increase of the angle between the μ and β vectors, from 104° to 113°. The in-crystal field enhances the amplitudes of both the dipole moment (by 40–50%) and first hyperpolarizability (by 6–9%) while the (μ, β) angle is reduced. This large μ increase agrees with the observations and analyses due to Spackman et al. [33]. Further analysis of the β tensor reveals that its dipolar character increases with the F substitution. This dipolar character is evidenced by resorting to the unit sphere representation (USR), initially proposed by Tuer et al. [34], as shown in Fig. 4. The USR patterns show a preferential direction of hyperpolarization, which is typical of dipolar responses. In the case of dominating dipolar response, the major nonlinear induced dipoles are oriented from the acceptor to the donor moieties of the molecule. On the other hand, the average molecular polarizability is equal to 182.4 a.u. (λ = 1064 nm) for both compounds, demonstrating the negligible impact of the F substitution on the linear response. This very small difference can certainly find its origin in the very small average polarizability difference between H2 (6.2 a.u.) and HF (6.4 a.u.) [35].

DrawMol [36] USR representation of the first hyperpolarizability tensor of BocProyne-H (left) and BocProyne-4F (right). The calculations were performed by using the crystal geometry but without the crystal field
From these molecular properties, the crystal linear and nonlinear optical responses were evaluated (Table 3) at representative wavelengths as well as in the static limit. Though the molecular second-order NLO responses are larger for the F-containing species, this substitution effect translates to only a very tiny increase of the dominant χ(2) tensor component: \(\chi_{333}^{\left( 2 \right)}\) of c.a. 1% (2.07 to 2.09 pm/V). An additional effect observed for χ(2) tensor is the improved contrast between the main (\(\chi_{333}^{\left( 2 \right)}\)) and secondary components (\(\chi_{113}^{\left( 2 \right)}\) = \(\chi_{223}^{\left( 2 \right)}\)). These SHG χ(2) values are 2–3 times larger than those observed for KDP at 1064 nm (0.78 pm, [37]). For comparison, it was observed that the SHG efficiencies of inorganic proline crystals range between 50 and 250% of those of KDP, as reported from Kurtz-Perry powder SHG measurements [38, 39]. Yet, the comparison between χ(2) amplitudes and Kurtz-Perry SHG efficiencies is not straightforward because these efficiencies do not only depend on the χ(2) amplitudes but also on the grain size distribution, on the refractive indices, and on phase-matchable interactions. On the other hand, these SHG χ(2) responses are much smaller than those observed in crystals of push–pull π-conjugated molecules like 2-methyl-4-nitroaniline (MNA) [40].
3 Experimental Section
3.1 Synthesis
3.1.1 BocProyne-H
To a solution of N-t-Boc-L-proline-N-hydroxysuccinimide ester (N-Boc-L-Pro-OSu, 2.052 g, 6.57 mmol) in dichloromethane (DCM, 15 mL) at 0 °C were added 1-amino-3-butyne (500 mg, 7.24 mmol), 4-DiMethylAminoPyridine (DMAP, 13 mg, 0.11 mmol) and potassium carbonate (K2CO3, 818 mg, 5.92 mmol). The reaction mixture was stirred under an argon atmosphere for three days at room temperature. DCM (25 mL) was added and the resulting mixture was washed with water (2 × 25 mL). The organic layer is filtered through a funnel with a small cotton plug and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel eluting with diethyl ether to yield BocProyne-H as a light yellowish oil (1.740 g, 99%).
Rf = 0,34 (ether/Hex 90:10); IR (v cm−1) 3307, 3243, 3094, 2974, 2933, 2869, 2119, 1678, 1657, 1557, 1401, 1364, 1250, 1159, 1122, 724, 662; 1H NMR (300 MHz, CDCl3, δ ppm) 7.14 (br, 0.5H), 6.40 (br, 0.5H), 4.37–4.17 (m, 1H), 3.57–3.28 (m, 4H), 2.49–2.28 (m, 2H), 2.23–2.03 (m, 1H), 2.02–1.95 (m, 1H), 1.94–1.75 (m, 3H), 1.46 (s, 9H); HRMS (m/z): calcd for C14H22N2O3Na [MNa+]: 289.1523; found 289.1525.
3.1.2 BocProyne-4F
To a solution of N-Boc-cis-4-fluoro-L-Proline (235 mg, 1.0 mmol) and N-DiIsoPropylEthylAmine (DIPEA, 600 µL, 3.4 mmol) in dichloromethane (DCM, 10 mL) were added 1-amino-3-butyne (76 mg, 1.1 mmol) and Hexafluorophosphate Benzotriazole Tetramethyl Uronium (HBTU, 418 mg, 1.1 mmol). The mixture was heated under reflux for 1 h and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel eluting with diethyl ether to yield the title product BocProyne-4F as an oil (121 mg, 42%).
Rf = 0.21 (100% ether); IR (v cm−1) 3307, 2932, 2119, 1703, 1642, 1505, 1379, 1142; 1H NMR (300 MHz, CDCl3, δ ppm): 5.25 (br, 0.5H), 5.08 (br, 0.5H), 4.42–4.31 (m, 1H), 3.95–3.60 (m, 3H), 3.50–3.30 (m, 2H), 2.55 (br, 1H), 2.40–2.30 (m, 2H), 2.24–2.20 (m, 1H), 1.99–1.90 (m, 1H), 1.45 (s, 9H); HRMS (m/z): calcd for C14H22FN2O3 (MH+): 285.1609; found 285.1614.
3.2 Crystallizations
BocProyne-H crystallized from an antisolvent recrystallization technique. Pentane antisolvent vapor diffusion induced crystallization of a solution of the BocProyne-H in CHCl3. Colorless crystals of BocProyne-H, suitable for X-ray analysis were obtained. A Needle-like specimen of C14H22N2O3, approximate dimensions 0.290 mm × 0.290 mm × 0.620 mm, was used for the X-ray crystallographic analysis.
A small crystal (seed) of the BocProyne-H is dipped into a solution of the BocProyne-4F in acetone/CHCl3/Hexane that was left to stand in a small vial at room temperature for several days. Colorless crystals of BocProyne-4F, suitable for X-ray analysis were obtained. A Needle-like specimen of C14H21FN2O3, approximate dimensions 0.080 mm × 0.090 mm × 0.660 mm, was used for the X-ray crystallographic analysis.
3.3 X-ray Crystallography
The X-ray intensity data were measured on a Bruker Apex DUO system equipped with a Cu Kα ImuS micro─focus source with MX optics (λ = 1.54178 Å for BocProyne-H and λ = 1.54186 Å for BocProyne-4F). The frames were integrated with the Bruker SAINT software package using a wide-frame algorithm. Data were corrected for absorption effects using the multi-scan method (SADABS). The structure was solved and refined using the Bruker SHELXTL Software Package, using the space group P 61, with Z = 6 for the formula unit. Crystallographic data are presented in Table 1.
3.4 Computational Details
Using the single crystal X-ray diffraction data as starting point, the geometries of the crystals have been fully optimized using periodic boundary conditions (PBC), density functional theory (DFT) with the B3LYP exchange–correlation functional, and the 6-31G(d,p) basis set, as implemented in the CRYSTAL09 package [41]. Only the fractional coordinates were optimized while the cell parameters were kept frozen to their experimental values. This step is necessary because the accuracy on the H atom coordinates is not sufficient. Following recent works [42], this is an efficient procedure to optimize the geometries of crystal. These PBC calculations were carried out by considering the standard parameters of CRYSTAL09 [a pruned integration grid with 75 radial points and 974 angular points, truncation criteria for the two-electron integrals set to 7 7 7 7 21 (TOLINTEG keyword), the Pack–Monkhorst shrinking factor was set to 6, the SCF convergence on the total energy was fixed at 10–7 a.u., the criteria for the RMS deviations of the gradients and the displacements were set to 3 × 10–4 a.u. and 12 × 10–4 a.u., respectively, while the maximum values of the gradients and displacements were fixed to 4.5 × 10–4 a.u. and 18 × 10–4 a.u., respectively.].
The tensor components of the linear [χ(1)] and second-order NLO [χ(2)] susceptibilities were evaluated using a multiscale approach that some of us have recently developed [43,44,45]. This method combines 1°) first principles calculations of the molecular properties (the polarizability (α) and the first hyperpolarizability (β)) with 2°) classical electrostatic interactions schemes to account for the crystal environment effects. In step 1°) the crystal dressing or polarization field effects are simulated by the point charges evaluated in the PBC calculations. To enact step 2°) and evaluate the local field effects, the molecular α and β tensor components are partitioned evenly on the non-hydrogen atoms. In step 1°), following the analysis of the convergence of the electric field amplitude as a function of the sphere radius [43] the charged surrounding is a sphere of R = 100 Å centered on the target molecule.
Following previous works, the dipole moment was determined at the MP2/6–311+ + G(d,p) level of approximation while the other molecular properties (α and β) were obtained by combining their static values evaluated at the same level of approximation and their frequency dispersion estimated using the time-dependent B3LYP/6–311 + G(d,p) method. These molecular property calculations were carried out using the GAUSSIAN09 package. To describe the β responses, they have been visualized by employing the unit sphere representation, initially proposed by Tuer et al. [34]. It consists first (i) in computing an effective induced dipole moment:
where the tensor nature of \(\beta\) is evidenced. \(\vec{E}\left( {\theta ,\phi } \right)\) is a unit vector of electric field, of which the polarization direction is defined in spherical coordinates (\(\theta\) and \(\phi\) angles) and (ii) in representing all the induced dipole moments on a sphere centered on the center of mass of the compound. This enables highlighting the directions where the second-order polarization is the strongest and its orientation (the acceptor–donor direction). These USR were plotted using the DrawMol package [36]. The χ(1) and χ(2) tensor components are reported in the abc* orthogonal reference frame (indices 1, 2, and 3). The refractive indices (n) satisfy the n2 = ε relation. The performance of this method has been substantiated in previous works where the calculated values are compared to experiment [46]. Only the non-zero, non-equivalent tensor components are reported.
3.5 Accession Codes
CCDC databank contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: + 44 1223 336033.
4 Conclusion
Over the non-centrosymmetric crystal classes, only five crystal classes—1, 2, 3, 4, and 6—exhibit chiral and polar symmetry, which could have piezoelectric, ferroelectric as well as pyroelectric properties [47]. We have reported here the synthesis, single crystal structures, and simulation, of two new proline derivatives that exhibit this type of symmetry (in our case, the scarce space group number: 169). Our new crystals show interesting features. As the two proline derivatives differ in nature only from an atom (H vs F), both crystals successfully crystallized in a similar molecular arrangement, yielding isomorphous crystals that could be compared with respect to their nonlinear structure–property relationship. The calculations show that the increase of the molecular response is not translated to the bulk properties and accounts only for a slight directionality improvement of the NLO response for the F-containing species. Nevertheless, our study paves the way for the development of SHG active crystals from isomorphous proline derivatives with various substituent at the γ position.
References
Radhakrishnan TP (2008) Molecular structure, symmetry, and shape as design elements in the fabrication of molecular crystals for second harmonic generation and the role of molecules-in-materials. Acc Chem Res 41(3):367–376. https://doi.org/10.1021/ar7002072
Corpinot MK, Bučar DK (2019) A practical guide to the design of molecular crystals. Cryst Growth Des 19:1426–1453. https://doi.org/10.1021/acs.cgd.8b00972
Yu P, Zhen Y, Dong H, Hu W (2019) Crystal engineering of organic optoelectronic materials. Chem 5:2814–2853. https://doi.org/10.1016/j.chempr.2019.08.019
Notake T, Takeda M, Okada S, Hosobata T, Yamagata Y, Minamide H (2019) Characterization of all second-order nonlinear-optical coefficients of organic N-benzyl-2-methyl-4-nitroaniline crystal. Sci Rep 9:14853. https://doi.org/10.1038/s41598-019-50951-1
Seidler T, Krawczuk A, Champagne B, Stadnicka K (2016) QTAIM-based scheme for describing the linear and nonlinear optical susceptibilities of molecular crystals composed of molecules with complex shapes. J Phys Chem C 120(8):4481–4494. https://doi.org/10.1021/acs.jpcc.5b10026
Claborn K, Kahr B, Kaminsky W (2002) Calculations of optical properties of the tetraphenyl-X family of isomorphous crystals (X = C, Si, Ge, Sn, Pb). CrystEngComm 4(46):252–256. https://doi.org/10.1039/B202304K
Solgi S, Tafreshi MJ, Ghamsari MS (2019) Nonlinear optical crystals for second harmonic generation. Crystallogr Reports 64:1138–1149. https://doi.org/10.1134/S1063774519070204
Verbiest T, Clays K, Rodriguez V (2009) Second-order nonlinear optical characterizations techniques: an introduction. CRC Press, New York
Thukral K, Vijayan N, Sonia HD, Maurya KK, Philip J, Jayaramakrishnan V (2019) Comprehensive study on l-Proline Lithium Chloride Monohydrate single crystal: a semiorganic material for nonlinear optical applications. Arab J Chem 12:3193–3201. https://doi.org/10.1016/j.arabjc.2015.08.022
Thukral K, Vijayan N, Krishna A, Singh B, Kant R, Jayaramakrishnan V, Jayalakshmy MS, Kaur M (2019) In-depth behavioral study of l-Prolinium Trichloroacetate single crystal: An efficient candidate for NLO applications. Arab J Chem 12:4887–4896. https://doi.org/10.1016/j.arabjc.2016.09.011
Thukral K, Vijayan N, Vij M, Nagaraja CM, Jayaramakrishnan V, Jayalakshmy MS, Kant R (2017) Analyses of significant features of L-Prolinium Picrate single crystal: an excellent material for non linear optical applications. Mater Chem Phys 194:90–96. https://doi.org/10.1016/j.matchemphys.2017.02.028
Venkateshan M, Suresh J (2019) Synthesis, physicochemical and quantum chemical studies on a new organic NLO crystal: cinnamoylproline. J Mol Struct 1180:826–838. https://doi.org/10.1016/j.molstruc.2018.12.071
Deniset-Besseau A, Duboisset J, Benichou E, Hache F, Brevet PF, Schanne-Klein MC (2009) Measurement of the second-order hyperpolarizability of the collagen triple helix and determination of its physical origin. J Phys Chem B 113:13437–13445. https://doi.org/10.1021/jp9046837
Chen X, Nadiarynkh O, Plotnikov S, Campagnola PJ (2012) Second harmonic generation microscopy for quantitative analysis of collagen fibrillar structure. Nat Protoc 7:654–669. https://doi.org/10.1038/nprot.2012.009
De Wergifosse M, De Ruyck J, Champagne B (2014) How the second-order nonlinear optical response of the collagen triple helix appears: a theoretical investigation. J Phys Chem C 118(16):8595–8602. https://doi.org/10.1021/jp501602a
Harczuk I, Vahtras O, Ågren H (2016) First hyperpolarizability of collagen using the point dipole approximation. J Phys Chem Lett 7(11):2132–2138. https://doi.org/10.1021/acs.jpclett.6b00721
Bossi A, Licandro E, Maiorana S, Rigamonti C, Righetto S, Stephenson GR, Spassova M, Botek E, Champagne B (2008) Theoretical and experimental investigation of electric field induced second harmonic generation in tetrathia[7]helicenes. J Phys Chem C 112(21):7900–7907. https://doi.org/10.1021/jp7117554
Coe BJ, Rusanova D, Joshi VD, Sanchez S, Vavra J, Khobragade D, Severa L, Cisarova I, Saman D, Pohl R, Clays K, Depotter G, Brunschwig BS, Teply F (2016) Helquat dyes: helicene-like push−pull systems with large second-order nonlinear optical responses. J Org Chem 81:1912–1920. https://doi.org/10.1021/acs.joc.5b02692
Méreau R, Castet F, Botek E, Champagne B (2009) Effect of the dynamical disorder on the second-order nonlinear optical responses of helicity-encoded polymer strands. J Phys Chem A 113(24):6552–6554. https://doi.org/10.1021/jp904418r
Chevalier F, Charlot M, Mongin F, Champagne B, Franz E, Clays K, Blanchard-Desce M (2016) Synthetic, optical and theoretical study of alternating ethylenedioxythiophene-pyridine oligomers: evolution from planar conjugated to helicoidal structure towards a chiral configuration. ChemPhysChem 17:4090–4101. https://doi.org/10.1002/cphc.201601057
Venkataramudu U, Sahoo C, Leelashree S, Venkatesh M, Ganesh D, Naraharisetty SRG, Chaudhary AK, Srinath S, Chandrasekar R (2018) Terahertz radiation and second-harmonic generation from a single-component polar organic ferroelectric crystal. J Mater Chem C 6:9330–9335. https://doi.org/10.1039/c8tc02638f
Rajalakshmi P, Srinivasan N, Krishnakumar RV, Razak IA, Rosli MM (2013) Supramolecular architectures of N-acetyl-L-proline monohydrate and N-benzyl-L-proline. Acta Cryst Sect C 69:1390–1396. https://doi.org/10.1107/S010827011302581X
Egli J, Schnitzer T, Dietschreit JCB, Ochsenfeld C, Wennemers H (2020) Why proline? Influence of ring-size on the collagen triple helix. Org Lett 22(2):348–351. https://doi.org/10.1021/acs.orglett.9b03528
Mahadevi AS, Sastry GN (2016) Cooperativity in noncovalent interactions. Chem Rev 116(5):2775–2825. https://doi.org/10.1021/cr500344e
The pitch of a helix is the width of one complete helix turn, measured parallel to the axis of the helix (Å/turn).
Moradi M, Babin V, Roland C, Darden TA, Sagui C (2009) Conformations and free energy landscapes of polyproline peptides. PNAS 106(49):20746–20751. https://doi.org/10.1073/pnas.0906500106
Steiner T, Desiraju GR (1998) Distinction between the weak hydrogen bond and the van der Waals interaction. Chem Commun 8:891–892. https://doi.org/10.1039/A708099I
Baillargeon P, Lussier T, Dory YL (2014) Hydrogen bonds between acidic protons from alkynes (C–H⋯O) and amides (N–H⋯O) and carbonyl oxygen atoms as acceptor partners. J Crystallogr. https://doi.org/10.1155/2014/371629
Baillargeon P, Rahem T, Amigo C, Fortin D, Dory YL (2018) Polar crystal of vanillylformamide through replacement of the alkene by an isosteric formamide group. IUCrData 3:x181630. https://doi.org/10.1107/s2414314618016309
Marmin T, Dory YL (2019) Self-Assembly of C3 symmetric rigid macrolactams into very polar and porous trigonal crystals. Chem Eur J 25:6707–6711. https://doi.org/10.1002/chem.201900802
Zhang C, Guo Y, He D et al (2021) A design principle for polar assemblies with C3-sym bowl-shaped π-conjugated molecules. Angew Chem Int Ed 60:3261. https://doi.org/10.1002/anie.202013333
The most recent figures released by the Cambridge Structural Database, on January 1, 2021 (www.ccdc.cam.ac.uk/SupportandResources/Resources/pages/Resources.aspx), are 868,614 (78 %) of structures adopt centrosymmetric space groups, 238,154 (22 %) adopt non-centrosymmetric space groups, and 179,033 (16 %) structures adopt Sohncke space groups. For the space group 169, only 711 structures (0.06%) were known over the 1,106,766 CSD structures fully defined. https://www.ccdc.cam.ac.uk/support-and-resources/ccdcresources/1d306a66ea0247e2adc0fdef013d690a.pdf
Spackman MA, Munshi P, Dittrich B (2007) Dipole moment enhancement in molecular crystals from X-ray diffraction data. ChemPhysChem 8:2051–2063. https://doi.org/10.1002/cphc.200700339
Tuer A, Krouglov S, Cisek R, Tokarz D, Barzda V (2011) Three-dimensional visualization of the first hyperpolarizability tensor. J Comput Chem 32(6):1128–1134. https://doi.org/10.1002/jcc.21694
Gough KM, Yacowar MM, Cleve RH, Dwyer JR (1996) Analysis of molecular polarizabilities and polarizability derivatives in H2, N2, F2, CO, and HF, with the theory of atoms in molecules. Can J Chem 74:1139–1144. https://doi.org/10.1139/v96-128
DrawMol V, Liégeois, UNamur, www.unamur.be/drawmol
Nikogosyan DN (1997) Properties of optical and laser-related materials: a handbook. John Wiley and Sons Inc, New York (ISBN: 978-0-471-97384-3)
Kalaiselvi D, Jayavel R (2011) Second harmonic generation of semiorganic dichlorobis(L-proline)zinc(II) single crystals for laser applications. Optoelectron Adv Mater Rapid Commun 5:58–62
Boopathi K, Babu SM, Jagan R, Athimoolam S, Ramasamy P (2018) Synthesis, crystal growth, physio-chemical characterization and quantum chemical calculations of NLO active metal–organic crystal: dibromo(4-hydroxy-l-proline)cadmium(ii) for non-linear optical applications. New J Chem 42:17464–17477. https://doi.org/10.1039/C8NJ02548G
Lipscomb GF, Garito AF, Narang RS (1981) An exceptionally large linear electro-optic effect in the organic solid MNA. J Chem Phys 75:1509–1516. https://doi.org/10.1063/1.442157
Dovesi R, Saunders VR, Roetti C, Orlando R, Zicovich-Wilson CM, Pascale F, Civalleri B, Doll K, Harrison NM, Bush IJ, D’Arco P, Llunell M (2009) CRYSTAL09 user’s manual. University of Torino, Torino
Quertinmont J, Carletta A, Tumanov NA, Leyssens T, Wouters J, Champagne B (2017) Assessing density functional theory approaches for predicting the structure and relative energy of salicylideneaniline molecular switches in the solid state. J Phys Chem C 121:6898–6908. https://doi.org/10.1021/acs.jpcc.7b00580
Seidler T, Stadnicka K, Champagne B (2013) Investigation of the linear and second-order nonlinear optical properties of molecular crystals within the local field theory. J Chem Phys 139:114105. https://doi.org/10.1063/1.4819769
Seidler T, Stadnicka K, Champagne B (2014) Linear and second-order nonlinear optical properties of ionic organic crystals. J Chem Phys 141:104109. https://doi.org/10.1063/1.4894483
Seidler T, Stadnicka K, Champagne B (2015) Erratum: linear and second-order nonlinear optical properties of ionic organic crystals. [J. Chem. Phys. 141, 104109 (2014)]. J Chem Phys 142:239901. https://doi.org/10.1063/1.4922617
Seidler T, Stadnicka K, Champagne B (2014) Second-Order nonlinear optical susceptibilities and refractive indices of organic crystals from a multiscale numerical simulation approach. Adv Opt Mater 2(10):1000–1006. https://doi.org/10.1002/adom.201400245
Halasyamani PS, Poeppelmeier KR (1998) Noncentrosymmetric oxides. Chem Mater 10(10):2753–2769. https://doi.org/10.1021/cm980140w
Acknowledgement
Financial support by the Fonds de recherche du Québec—Nature et technologies (FRQNT, Grant No. 2016-CO-194882 and grant No. 2019-CO-254502) are gratefully acknowledged. The authors acknowledge that parts of this work were supported by the PL-Grid Infrastructure. Part of the calculations were performed on the computers of the «Consortium des équipements de Calcul Intensif (CÉCI)» (http://www.ceci-hpc.be) for which the financial supports of the FNRS-FRFC, the Walloon Region, and the University of Namur (Conventions No.2.5020.11, GEQU.G006.15, U.G018.19, 1610468, and RW/GEQ2016) are acknowledged. We would like also to acknowledge Daniel Fortin for crystal identification, Tarik Rahem, Emma Couture and Mariève Proulx for their participation on this project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baillargeon, P., Seidler, T., Champagne, B. et al. Polar and Helical Isomorphous Crystals of Proline Derivatives: Influence of a Fluorine Atom on the Electric Susceptibility. Chemistry Africa 4, 553–562 (2021). https://doi.org/10.1007/s42250-021-00236-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42250-021-00236-w