
Abstract
Corrosion behaviour of stainless steel 347 was investigated in a molten nitrate salt (60 wt% NaNO3 + 40 wt% KNO3) immersion at 565 °C for up to 3000 h. A growth of stratified oxide layers consisting of NaFeO2, Fe2O3 and Fe3O4 was observed on the stainless steel surface with a constant gravimetric corrosion rate of ~ 0.4 µm/year. The feasibility of using Ni3Al coatings deposited by means of air plasma spray for suppression of corrosion was investigated. Ni3Al coatings were observed to undergo a fast oxidation with a corrosion rate of ~ 2.7 µm/year in the first 500 h, and subsequently stabilise between 500 and 3000 h with no observable changes in microstructure, composition and weight at a corrosion rate of ~ 0.02 µm/year. The results presented in this study strongly suggest that Ni3Al coating suppresses the formation of oxide layers on the surface of stainless steel substrates and can be used as protection against corrosion in the presence of molten nitrate salts, which is of relevance to thermal energy storage applications.
Similar content being viewed by others

1 Introduction
Thermal energy storage (TES) is arguably the key to further lower the levelised cost of energy (LCOE) of concentrated solar power (CSP) into a comparable level with other electricity generation technologies [1,2,3,4,5,6]. TES allows continuous power generation beyond sunset and over periods of overcast [7]. A feasible storage medium for TES is molten salt with high temperature stability, low melting point, low viscosity and high thermal conductivity [8,9,10]. Binary nitrate eutectic salts such as 60% NaNO3–40% KNO3 are commonly used as heat storage media because of their high heat capacity of ~ 1495 J kg−1 K−1 at 300 °C, high thermal conductivity of 0.55 W m−1 K−1 at 400 °C and ease of handling and storage [11,12,13,14]. One of the challenges associated with molten salts is corrosion at elevated temperatures [15,16,17,18,19,20,21]. The commonly used solution to accommodate corrosion is to use thicker walls, as it would extend the lifetime operation of the material. However, it does not solve the corrosion issue and may not be applicable for components with fixed dimensions or tight tolerance [22]. The use of high purity nitrate salt to avoid corrosion aggravation from chloride and water impurities has been proposed, although not widely implemented due to higher costs [7, 22, 23]. Ni-based superalloys have also been proposed to suppress corrosion, as they provide high temperature stability and resistance for molten salt corrosion compared to iron-based alloys [22, 24, 25]. However, they are significantly more expensive as compared to structural stainless steel [24, 25]. The use of corrosion resistant coatings has been considered as the most viable solution, as it provides barrier to suppress corrosion while maintaining the structural stability of substrate material [25,26,27,28].
Due to a large variation in operational temperature, it is best practice to match the thermal properties of the coating with substrate material to minimize cracking or spallation with temperature change [29]. For stainless steel substrate, commonly used in TES, the most suitable coating material would ideally be Fe-based, Cr-based or Ni-based. Cr-based material forms a slow growing protective CrOx under an oxidising environment. However, it is unsuitable for use in molten nitrate salts due to its high solubility [15, 17, 19, 25, 29,30,31,32]. Ni-based coating is preferred over Fe-based because of its ability to form a protective oxide layer, whilst Fe oxide is non-passivating [21]. Ni-based coatings have been reportedly used to provide wear, oxidation and hot corrosion resistance in ash and flue gases inside a coal-fire boiler [33, 34]. Among many potential different combinations of Ni-based coatings, NiAlx are favoured because of their strong adhesion, high-temperature mechanical strength, high melting temperature, high thermal conductivity, attractive stiffness, good oxidation resistance and metal-like electrical conductivity [35, 36]. Ni2Al3 coatings showed very low mass changes after 1000-h immersion in molten 60% NaNO3– 40% KNO3 salt at 580 °C [25]. NiVAl has been reported to have a corrosion rate of 0.11 nm/h after 2000-h immersion in 60% NaNO3–40% KNO3 salt at 500 °C, which is more than two orders of magnitude lower than uncoated high temperature Grade P91 9Cr-1Mo steel [22]. Compared to the aforementioned different phases of NiAlx, Ni3Al is more favourable because of lower concentration of Al to avoid brittleness after oxidation. Ni3Al coating was effective in decreasing the weight gain to about one-third as compared to uncoated Fe-based superalloy in molten Na2SO4–60% V2O5 salt at 900 °C [28]. The Ni3Al coating have been effectively used in a coal-fired boiler at 450 °C and 900 °C, as well as in molten NaNO3–(KNO3)–Na2O2 salt at 650 °C [15, 37, 38]. Numerous studies on Ni3Al coatings and their corrosion resistance in different high-temperature atmospheres have been reported in the literature. However, the long-term corrosion behaviour of Ni3Al coatings in molten binary nitrate salt at 565 °C, emulating a realistic CSP–TES environment, has not been systematically investigated [1, 5, 12, 13, 39].
The study presented herein investigates high temperature behaviour of Ni3Al coatings immersed in molten 60% NaNO3–40% KNO3 salt to address the challenge of hot corrosion on stainless steel substrates typically used in CSP–TES application. The results of this investigation show that Ni3Al yields a practically negligible change in microstructures, elemental composition and corrosion rate at a constant temperature of 565 °C for up to 3000 h. This suggests Ni3Al coatings may be highly suitable for use as protective coatings in applications with long term isothermal exposure to molten salt, such as the hot TES tanks and piping systems. Furthermore, this study provides a more suitable approach to approximate corrosion behaviour which should be adapted in other studies and applied to future coating systems.
2 Materials and methods
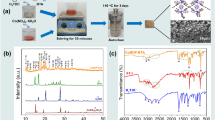
High temperature stainless steel AISI 347 circular coupons with a dimension of 10-mm diameter and 2-mm thickness (Goodfellow, temper annealed) were used as substrates. All coupons were cleaned in an ultrasonic bath of isopropyl alcohol (IPA) for 10 min at 25 °C to remove oil and dirt before coating or testing. Some of the SS347 coupons were left uncoated and denoted herein as SS347. The remaining set of coupons were coated with ~ 200-μm thick Ni3Al layer on all sides of the substrate, denoted herein as Ni3Al/SS347, using air plasma spray (Fig. 1a). Prior to the coating deposition, the substrates were grit blasted with 80 grit alumina powders to clean and roughen. The Ni3Al coatings were introduced by an air plasma spray (APS) system with a current setting of 500 A and a voltage of 35 V (Multicoat system, 9MPE-CL feeder and 9 M spray gun). N2 gas at 5600 sccm was used as carrier gas to deliver Ni3Al powder (Goodfellow NI716010, 20–45-µm particle size) at a feed rate of 67 ± 5 g/min (see Supporting Information Fig. S1). Ar and H2 gases were used as the primary and secondary plasma gases with a flow rate of 36,700 and 7000 sccm, respectively. The powders were characterised by scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) prior to APS deposition to confirm particle size distribution and elemental composition (see Supporting Information Table S1). The coatings were deposited at a spray distance of 150 mm. All corrosion behaviour assessments were carried out by immersion in eutectic solar nitrate salt mixture consisting of 60 wt% NaNO3 (Sigma-Aldrich, BioXtra, ≥ 99.0%) and 40 wt% KNO3 (Sigma-Aldrich, BioXtra, ≥ 99.0%) at a constant temperature of 565 °C for up to 3000 h unless otherwise stated. Both salts were mixed for 20 min in a ball mill (Fritsch Pulverisette 5) until a homogeneous mixture was obtained and then filled into alumina crucibles (volume of ~ 10 ml). Prior to use in the corrosion test, the mixture was fully melted by heating at 400 °C for 24 h. Each of the SS347 and Ni3Al/SS347 samples was individually immersed in a separate crucible to avoid cross corrosion and contamination. The crucibles, each containing a single piece of the test sample, were placed back in the furnace purged and filled with Ar gas to prevent gaseous phase oxidation from O2 or moisture in the air (Fig. 1a). The furnace temperature was then slowly raised to 565 °C and maintained throughout the duration of the corrosion tests. The samples were removed from the furnace after every 500-h interval to be weighed (Sartorius CP225D balance; 0.01-mg resolution) and characterised. After removal from the furnace, each sample was thoroughly cleaned in an ultrasonic bath of deionised water for 10 min at 25 °C to remove any salt residues from the surface.

(a) Schematics of air plasma spray deposition of Ni3Al coatings and molten nitrate salt tests at 565 °C. Ni3Al powder is melted in a stream of plasma at atmospheric pressure and propelled towards the substrate. Each of the SS347 and Ni3Al/SS347 coupons is then immersed in NaNO3:KNO3 salts contained in individual alumina crucibles at a constant temperature of 565 °C for up to 3000 h. (b) Time dependent area-normalised weight change (ΔM/A) of Ni3Al/SS347 and SS347 in molten nitrate salts and air at 565 °C. Here, the weight change is normalised with the surface area of the coupons. Solid and dashed lines indicate piecewise linear regressions for weight change in molten nitrate salts and air, respectively.
Scanning electron microscopy (SEM) images of sample surfaces and cross-sections were collected by secondary electron detectors (TESCAN VEGA3) under the acceleration voltage of 20 kV. Material characterisation was carried out by means of energy-dispersive X-ray spectroscopy (EDS) for elemental analysis collected by a 20-mm2 silicon drift detector (Oxford Instrument X-max with AZtec software) under acceleration voltage of 20 kV along with X-ray diffraction (XRD) for crystallography analysis (Siemens D5005 X-ray diffractometre with DIFFRACT plus software) collected under Cu Kα radiation at power setting of 40 kV and 40 mA. Cross-sectioning was carried out by mounting samples in Bakelite resin at 185 °C, grinding using sandpaper with a gradual increase from 120 to 2500 grit paper, polishing using diamond coarse polish (Met-prep, Durasilk 3-µm diamond suspension) and silica final polish (Struers, OP-S 0.04-µm colloidal silica suspension), rinsing with detergent and tap water to remove residual substances from the metallographic process, and drying with a hot air blow dryer. The arithmetic average surface roughness (Ra) was measured by a stylus profilometre (Bruker DektakXT).
2.1 Results and discussion
The overall corrosion behaviour of any metallic samples is typically assessed by gravimetric analysis in a form of mass changes per unit surface area (ΔM/A) [40]. The ΔM/A of Ni3Al/SS347 at 565 °C is observed to increase rapidly within the first 500 h to ~ 1.05 × 10−1 mg/cm2 at an apparent rate of ~ 2 × 10−4 mg/cm2/h (Fig. 1b). After 500-h immersion in molten salts, ΔM/A of Ni3Al/SS347 stabilises at a much lower rate of ~ 1.8 × 10−6 mg/cm2/h. This results in ΔM/A of just ~ 1.1 × 10−1 mg/cm2 after 3000 h. Assuming the Ni3Al coatings remain at a constant density of 6.67 × 103 mg/cm3, this is equivalent to an effective corrosion rate of ~ 2.7 µm/year within the first 500 h and a practically negligible rate of ~ 0.02 µm/year from 500 to 3000 h. In contrast, ΔM/A of SS347 increases monotonically with increasing molten salt immersion time at a rate of ~ 4.7 × 10−5 mg/cm2/h within the first 1000 h, and subsequently at a rate of ~ 3 × 10−5 mg/cm2/h to reach an ΔM/A of ~ 1.07 × 10−1 mg/cm2 after 3000 h (Fig. 1b). Assuming the SS347 substrates remain at a constant density of 8.03 × 103 mg/cm3, this is equivalent to an overall corrosion rate of ~ 0.4 µm/year in molten salts. This corrosion rate is in agreement with previous studies, where that for SS347 at 500–600 °C prior to descaling was observed between 0.1 and 2.2 µm/year (see Supporting Information Table S2) [21, 41].
We postulate that such a rapid increase in ΔM/A of Ni3Al/SS347 within the first 500 h is attributed to rapid oxidation of Al and Ni in the Ni3Al coatings without necessarily involving any uptake of Na. Note that the decomposition of nitrate salt into NO and O2 gasses in the presence of Ar atmosphere, which was used to purge and fill the test furnace, starts at a temperature of ~ 470 °C [10]. At a temperature of 565 °C, the concentration of O2 from partial decomposition of molten salt is sufficiently high to facilitate rapid uptake of O through oxidation. To verify this postulation, we carried out molten salt-free heat treatment tests in air at 565 °C for 500 h. In agreement with the test in molten salt, heat treatment in air confirms substantial increase in ΔM/A of Ni3Al/SS347 due to a rapid uptake of O [42]. As Ni3Al coatings act as an O sink, ΔM/A of 1.2 × 10−1 mg/cm2 is quickly reached within the first 500 h (Fig. 1b). A similar behaviour is also observed on SS347, where the increase rate in ΔM/A from heat treatment in air is comparable to that from immersion in molten nitrate salts within the first 500 h. This observation suggests that rapid increase in ΔM/A can be attributed mainly to the uptake of O through oxidation. The negligible increase in ΔM/A of Ni3Al/SS347 after 500 h in molten salts suggests an abrupt slowdown of O uptake once the majority of Ni and Al in the Ni3Al coating has been oxidised. In contrast, a further increase in ΔM/A of SS347 beyond 500 h suggests the oxidation process takes place continuously and has not stopped even at 3000 h.
It is important to note that the approximation of corrosion rate based on ΔM/A needs to be carried out in a piecewise manner due to the abrupt slowdown of the O uptake at 500 h in molten salts. Such an approximation should not be based solely on the final ΔM/A value measured at the maximum test time, as is typically reported in literature [31]. Both distinctive trends need to be considered to fully capture the critical information on time dependent corrosion behaviour. As an illustration, the corrosion rate of Ni3Al/SS347 would have been mistakenly reported as ~ 0.5 µm/year had it been calculated solely based on the final ΔM/A value measured at 3000 h. This is incorrect for both regimes as it does not capture the change in the O uptake that occurs at 500 h. Furthermore, there is currently no consensus in literature whether pre-descaling is required [21, 25, 30, 31, 43] or not [15, 22, 28, 34, 37, 38] in the measurement of ΔM/A. Descaling is not performed in this study to capture the full evolution of the oxide layers without damaging the substrate.
Cross-sectional SEM images of Ni3Al/SS347 suggest that the immersion in molten nitrate salts at 565 °C induces minimal changes to the microstructural morphologies of Ni3Al coatings and SS347 substrates. Ni3Al/SS347 at 0 h exhibits lamellar coating morphology with numerous un-melted AlOx particles and air cavities, which is typical for metal alloy coatings deposited by APS with high powder feed rate (Fig. 2a). APS allows direct deposition of Ni3Al coatings on SS347 substrates with good contact and without the need of bond coat. The cross-sectional SEM image of Ni3Al/SS347 at 3000 h appears similar to that of the as deposited one, albeit with less cavities within the lamellar structures due to amalgamation of some lamellae into more continuous structures at an elevated temperature (Fig. 2b). The surface SEM image of the Ni3Al/SS347 at 300 h exhibits surface features of ~ 1 μm (average size), in addition to the splats with lateral average size of ~ 5 µm in the surface SEM image of the Ni3Al/SS347, prior to molten salt immersion (0 h) (see Supporting Information Fig. S2a, b). The Ni3Al coatings exhibit no visible degradation, and no formation of corrosion product layers can be observed in the underlying SS347 substrate. This strongly suggests the potential long-term corrosion protective nature of Ni3Al coatings [25].

Cross-sectional SEM images of Ni3Al/SS347 (a, b) and SS347 (c, d) at 0 h (a, c) and 3000 h (b, d) of immersion in molten nitrate salt at 565 °C. Dashed lines indicate the interface between Ni3Al coating and SS347 substrate, while arrows indicate AlOx particles embedded within the Ni3Al coatings (a, b). No significant change in the coating morphology and no formation of corrosion layers can be observed on Ni3Al/SS347 after 3000 h. In contrast, formation of corrosion layers can be observed on the surface SS347 after 3000 h. A higher magnification SEM image (inset in d) shows the distinguishable morphologies of these corrosion layers. The dashed line in the inset indicates the interface between the corrosion layers and the original SS347 layer
In contrast, the immersion in molten salt leads to dramatic changes in microstructural morphologies near the surface of bare SS347. Corrosion product layers, which are non-existent on SS347 at 0 h (Fig. 2c), start to form on the surface immediately upon exposure to molten salt at 565 °C. Both the thickness and the number of distinguishable layers can be observed to grow with the increase in molten salt immersion time. SEM images of SS347 at 3000 h show at least four distinguishable corrosion layers with a total thickness of 40–50 μm (Fig. 2d). The observed stratification suggests each layer comprises different elemental compositions or crystalline structures. The two outermost corrosion layers exhibit distinctive porous columnar structures with numerous sub-surface microscale cavities. The morphologies of these layers closely resemble the outer and inner layers of FeOx scales formed naturally on steel alloys under strongly oxidising environment at elevated temperatures [44,45,46,47]. The corrosion layers closer to original SS347 surface appear more compact with numerous precipitates and multiphase microstructures. These layers resemble diffusion layers at which the oxide formation terminates [44,45,46,47]. The SEM image of the SS347 exhibits a typical smooth stainless steel surface with an average grain size of ~ 4 µm, prior to molten salt immersion (0 h) (see Supporting Information Fig. S2c). Changes in surface morphology of the SS347 could be observed after 3000 h, where the average size of the apparent surface features decreases to ~ 1 μm (see Supporting Information Fig. S2d). The decrease in the surface feature size is not caused by the shrinkage of the alloy grains, but rather by the formation of fine corrosion product adlayer that resembles FeOx scales on the surface of the SS347.
The rapid increase in ΔM/A of Ni3Al/SS347 along with its absence of corrosion layers motivate us to carry out elemental composition analysis, particularly around the interface between Ni3Al coatings and SS347 substrates (Fig. 3a). The spatial distribution of O obtained from qualitative cross-sectional EDS maps confirms its presence in the Ni3Al coatings and its absence in the SS347 substrates. This strongly suggests that the rapid increase in ΔM/A is indeed caused by rapid uptake of O through formation of AlOx and NiOx in the Ni3Al coatings instead of the formation of FeOx in the SS347 substrates [37, 38, 48, 49]. Note that the spatial correlation between O and Al is more pronounced than that between O and Ni throughout the coatings. This suggests AlOx is more readily formed than NiOx upon exposure to molten nitrate salts. As mentioned earlier, un-melted AlOx particles with size of tens of μm may also be inadvertently introduced during the APS deposition (indicated by arrows in Fig. 2a, b and 3a). A much lower powder feed rate is needed to minimise the quantity of these un-melted AlOx particles. Nonetheless, the oxidation of Ni3Al coatings may contribute to the suppression of inward/outward diffusion of other elements, such as Na and Fe, from/to the surrounding molten salt environment.

(a) Elemental maps and corresponding SEM image of cross-sectioned Ni3Al/SS347 at 3000 h in molten nitrate salt at 565 °C. These maps show spatial distribution of Fe, Ni, Al, O and Na at the interface between Ni3Al coating and SS347 substrate. Arrows indicate AlOx particles embedded within the Ni3Al coatings. All scale bars represent 50 μm. (b) Elemental composition in atomic percentage (at. %) of Fe, Ni, Al, O and Na of Ni3Al/SS347 taken at the area indicated by white line in the corresponding SEM image. The composition is obtained at 3000 h and presented as a function of the distance from the interface between Ni3Al coating and SS347 substrate. Positive distance corresponds to coatings (Coat) and outer mounting resin (Out), while negative distance corresponds to substrate (Sub). Solid lines are ~ 5-μm simple moving average to guide the eye.
A quantitative elemental composition analysis across the thickness of the Ni3Al coatings at 3000 h further confirms the suppression of inward and outward diffusions of Na and Fe, respectively (Fig. 3b). Na is practically non-existent in both Ni3Al coatings and SS347 substrate as its concentration is indistinguishable from the noise level. The coating-substrate interdiffusion zone, in which the concentration of Al and Ni rapidly increases to about ~ 18 at.% and ~ 60 at.%, respectively, is found to be about 7–8-μm thick. Note that the concentration of O in the Ni3Al coatings throughout its thickness is only about 7–12 at.%, which is well below the expected stoichiometric concentration for NiO and Al2O3. Such a low concentration of O suggests that Ni3Al coatings are only slightly oxidised even at 3000 h. Nonetheless, it has been suggested that the formation of NiOx and AlOx prohibits further diffusion of O and Na, even at sub-stoichiometric [38, 61]. The extremely low concentration of O of < 3 at.% in the SS347 substrate at 3000 h also suggests that the substrates remain metallic and free from oxidation. A further improvement in oxidation resistance of Ni3Al coatings can be potentially achieved by minimising the quantity of un-melted AlOx particles introduced during deposition. It is also important to mention that a small amount of FeAlOx is found on the surface of Ni3Al coatings, and further investigation in future studies is required to elucidate the origin of these oxide compounds.
In contrast to Ni3Al/SS347, the bare SS347 exhibits a formation of stratified corrosion layers with various O and Na concentrations that reach a total thickness of 30–45 μm at 3000 h in molten salts. These corrosion layers are observed to continuously grow in thickness with the increase in molten nitrate salt immersion time at a growth rate of ~ 87.6 µm/year (see Supporting Information Fig. S3a–e and S4) [22]. The qualitative cross-sectional EDS maps suggests a rapid uptake of O and Na from the molten salt along with an outward migration of Fe, Ni and Cr from the SS347 (Fig. 4a). The uptake of O and Na by SS347 at an elevated temperature leads to the formation of FeOx and NaFeOx on the outer layers, both are known to form on the surface of stainless steel substrates after molten salt immersion [31, 32, 50]. The fact that O is found much deeper in the substrate than Na, which is found only on the surface, suggests that the absorption kinetic of O is much higher than that of Na. This finding is indeed expected as O2− anions readily react with cationic Fe to form a relatively stable FeOx despite the ionic radius of O2− at ~ 0.14 nm is larger than that of Na+ at ~ 0.102 nm [51, 52]. The intercalation of Na+ cations is known to be sluggish and highly dependent on the crystalline structure of the formed FeOx [53]. The outward migration of Fe, Ni and Cr can be observed in the inner part of these corrosion layers. Such a migration is also expected as Cr is known to be highly soluble in molten nitrate salts [54].

(a) Elemental maps and corresponding SEM image of cross-sectioned SS347 at 3000 h in molten nitrate salt at 565 °C. These maps show spatial distribution of Fe, Ni, Cr, O and Na at the surface of SS347 substrate. All scale bars represent 50 μm. (b) Elemental composition in atomic percentage (at.%) of Fe, Ni, Cr, O and Na of SS347 taken at the area indicated by white line in the corresponding SEM image. The composition is obtained at 3000 h and presented as a function of the distance from the interface between corrosion layers and SS347 substrate. Positive distance corresponds to corrosion layers (Cor) and outer mounting resin (Out), while negative distance corresponds to substrate (Sub). Solid lines are ~ 5-μm simple moving average to guide the eye.
A quantitative elemental composition analysis of cross-sectioned SS347 at 3000 h suggests that the outermost layer formed immediately under the salt deposit is a combination of NaFeOx and FeOx with O:Fe at.% ratio of 4:5 and a relatively high Na content of 3–9 at.% (Fig. 4b). The subsequent FeOx layer is also found to be Fe-rich with O:Fe at.% ratio of 3:5, but with a negligible amount of Na. The inner FeOx layer is found to have an equal amount of O and Fe and contains a significant amount of Ni and Cr with a concentration range of 8–18 at.% and 23–38 at.%, respectively. It is important to note that the concentration of Ni and Cr in this layer, each at ~ 5 at.% and ~ 22 at.%, respectively, is higher than that in the substrate. Furthermore, the Fe content in the inner layer is found to be considerably low at ~ 21 at.%, which is less than half of that in the outer layers. This observation is in agreement with previous studies where the inward diffusion of O strongly correlates with the growth of Cr and Ni enrichment zone through formation of precipitates and the depletion of Fe through outward migration toward the molten salts [42, 43, 55]. These inward diffusion of O and outward migration of Fe are the main reason for the discrepancy between the growth rate of corrosion layers observed by SEM (Fig. 2d) and the calculated corrosion rate based on gravimetric analysis (Fig. 1b; Supporting Information Table S2). Note that, as discussed above, the corrosion layers’ growth rate is about two orders of magnitude higher than the gravimetrically calculated corrosion rate. The formation of NaFeOx and FeOx renders the use of constant density for corrosion rate calculation incorrect, as both have much lower densities than that of the SS347 substrate. In addition, the depletion of Fe from the substrate may counterbalance the uptake of O from the molten salts in terms of mass and thus supresses the apparent ΔM/A. Despite this discrepancy, the gravimetric analysis captures the general growth behaviour of the corrosion layers. Further investigation in future studies is required to elucidate the exact correlation between them so one can estimate the growth rate of the corrosion layers simply by gravimetrically calculated corrosion rate.
The presence of NaFeOx and FeOx on SS347 and their absence on Ni3Al/SS347 can be further observed from their respective XRD patterns (Fig. 5a). For SS347 in molten salts, the prominent diffraction peaks observed at 1000 h are attributed to Fe3O4 and (Cr, Fe)2O3. The fact that these peaks are much more prominent than those for CrFeNi, which are typical for austenitic stainless steel, suggests that the formation of Cr and Ni enrichment zone takes place within the first 1000 h. The disappearance of diffraction peaks for CrFeNi at 2000 h supports our previous observation that the growth of Cr and Ni enrichment zone occurs concurrently with depletion of Fe and leads to the formation of NaFe2O3 and Fe2NiO4 on the surface. At 3000 h, the XRD pattern of SS347 is fully dominated by peaks attributed to various oxides of Fe including Fe3O4 and (Cr, Fe)2O3, Fe2NiO4 and NaFe2O3.

(a) XRD patterns of Ni3Al/SS347 and SS347 at different molten salt exposure times: 0, 1000, 2000 and 3000 h. SS347 exhibits peaks that correspond to austenitic stainless steel (filled circle) and various oxides of Fe, including Fe3O4 and (Cr, Fe)2O3 (filled triangle), Fe2NiO4 (filled star) and NaFe2O3 (filled x mark). Ni3Al/SS347 exhibits peaks that corresponds to Ni3Al (filled diamond) and NiO (filled inverted triangle). Peaks are identified according to the International Centre for Diffraction Data (ICDD) database [31, 33, 56,57,58,59,60,61] (b) Schematic of corrosion suppression of Ni3Al coatings to the SS347 substrate in molten salts. Ni3Al coatings are rapidly oxidised and stabilised in the first 500 h. The formation of oxides within the Ni3Al coatings supresses the diffusion of Na from the molten salt or the release of Fe, Ni and Cr from the substrate for at least 3000 h. In contrast, continuous uptake of O and Na along with the release of Fe, Ni and Cr resulting in the formation of corrosion layers comprised various oxides of iron on the surface of SS347
In contrast, the XRD patterns of Ni3Al/SS347 are relatively constant for up to 3000 h in molten salts where the main distinguishable diffraction peaks are attributed to Ni3Al [33, 62,63,64]. Rapid oxidation of the Ni3Al coatings can be observed from the appearance of diffraction peaks attributed to NiO at 1000 h. Note that no peaks can be attributed to any Al2O3 phases. This suggests that the formation of AlOx observed in qualitative and quantitative elemental analysis is most likely in the amorphous form. It has been suggested that the formation of AlOx and NiOx inhibits diffusion of Na and further diffusion of O, especially if a stoichiometric oxide has been formed [38, 48]. Contrary to CrOx that is soluble in molten nitrate salts, AlOx and NiOx are stable and dense enough to create a diffusion barrier [54]. The fact that no peaks can be attributed to NaFeOx or FeOx implies that the AlOx and NiOx prevent any uptake of Na from the salts and acts as an effective diffusion barrier for Fe, Ni and Cr. The absence of any observable changes in XRD patterns from 1000 h onward agrees with our gravimetric analysis and strongly suggests that the Ni3Al coatings are stable for at least 3000 h.
2.2 Conclusion
In conclusion, Ni3Al coatings provide corrosion resistance to SS347 in the presence of molten NaNO3:KNO3 salt at 565 °C for at least 3000 h. Ni3Al coatings undergo a fast oxidation within the first 500 h of molten salt immersion with a gravimetric rate of ~ 2 × 10−4 mg/cm2/h and then stabilise at a much lower rate of ~ 1.8 × 10−6 mg/cm2/h. While this gravimetric rate is only an order of magnitude lower than that for bare SS347 of ~ 3 × 10−5 mg/cm2/h, the effect of Ni3Al coatings on microstructure and elemental composition of SS347 substrates are considerably large. Ni3Al coatings inhibit diffusion of Na and further uptake of O from the molten salts and supress outward diffusion of Fe, Cr or Ni from the SS347 substrate (Fig. 5b). On the other hand, multi-layered corrosion products consisting of various oxides of Fe including FeOx, NaFeOx, FeNiOx and CrFeOx are readily formed on the surface of SS347. These oxide layers grow continuously in the presence of molten salts and reach a total thickness of 40–45 μm within 3000 h. The results presented herein strongly suggest that the Ni3Al coatings are protective, by either delaying or stopping altogether the formation of corrosion layers, for use on stainless steel substrates. This enables the potential use of Ni3Al coatings as protective coatings to prolong the lifetime of components with long-term isothermal exposure to molten salt, such as the hot TES tanks and piping systems. Further investigation should be undertaken beyond 3000 h to elucidate the long-term corrosion behaviour of Ni3Al coatings and to correlate any morphological and compositional changes to the gravimetric analysis. These are keys to confirm the potential of Ni3Al coatings for use in next generation CSP–TES applications. Nonetheless, the approach in this study can be extended for investigation of other corrosion suppression solutions against molten salts to further advance the CSP–TES technology.
Data Availability
Data underlying this study can be accessed through the Cranfield University repository at https://doi.org/10.17862/cranfield.rd.17269424.
Abbreviations
- TES:
-
Thermal energy storage
- LCOE:
-
Levelised cost of energy
- CSP:
-
Concentrated solar power
- SS:
-
Stainless steel
- IPA:
-
Isopropyl alcohol
- APS:
-
Air plasma spray
- SEM:
-
Scanning electron microscope
- EDS:
-
Energy-dispersive X-ray spectroscopy
- XRD:
-
X-ray diffraction
- Ra :
-
Arithmetic average surface roughness
References
A.G. Fernández, B. Muñoz-Sánchez, J. Nieto-Maestre, A. García-Romero, High temperature corrosion behavior on molten nitrate salt-based nanofluids for CSP plants. Renew Energy 130, 902–909 (2019). https://doi.org/10.1016/j.renene.2018.07.018
Grogan, D. Development of molten-salt heat transfer fluid technology for parabolic trough solar power plants. Abengoa. Sol. Sunshot. Conf .Proj. Rev.
Siegel, N. P., Bradshaw, R. W., Cordaro AM Kruizenga, J. B. Thermophysical property measurement of nitrate salt heat transfer fluids. Proceedings of the ASME 2011 5th International Conference on Energy Sustainability, 1-8 (2011).
G. McConohy, A. Kruizenga, Molten nitrate salts at 600 and 680°C: thermophysical property changes and corrosion of high-temperature nickel alloys. Sol. Energy. 103, 242–252 (2014). https://doi.org/10.1016/j.solener.2014.01.028
M. Liu, N.H. Steven Tay, S. Bell, M. Belusko, R. Jacob, G. Will, W. Saman, F. Bruno, Review on concentrating solar power plants and new developments in high temperature thermal energy storage technologies. Renew. Sustain. Energy. Rev. 53, 1411–1432 (2016)https://doi.org/10.1016/j.rser.2015.09.026
Kuravi, S. Goswami, D.Y., Stefanakos, E.K., Ram, M., Jotshi, C., Trahan, J., Sridharan, P., Rahman, M., Krakow, B. Thermal energy storage for concentrating solar power plants, Clean Energy Research Center, University South Florida. (n.d.)
D. Kearney, B. Kelly, U. Herrmann, R. Cable, J. Pacheco, R. Mahoney, H. Price, D. Blake, P. Nava, N. Potrovitza, Engineering aspects of a molten salt heat transfer fluid in a trough solar field. Energy 29, 861–870 (2004). https://doi.org/10.1016/S0360-5442(03)00191-9
S. Kuravi, Y. Goswami, E.K. Stefanakos, M. Ram, C. Jotshi, S. Pendyala, J. Trahan, P. Sridharan, M. Rahman, B. Krakow, Thermal energy storage technologies and systems for concentrating solar power plants. Technol. Innov. 14, 81–91 (2013). https://doi.org/10.3727/194982412X13462021397570
J. Gasia, L. Miró, L.F, Cabeza, Review on system and materials requirements for high temperature thermal energy storage. Part 1: general requirements. Renew. Sustain. Energy. Rev. 75, 1320–1338 (2017)
R.I. Olivares, The thermal stability of molten nitrite/nitrates salt for solar thermal energy storage in different atmospheres. Sol. Energy. 86, 2576–2583 (2012). https://doi.org/10.1016/j.solener.2012.05.025
H. Benoit, L. Spreafico, D. Gauthier, G. Flamant, Review of heat transfer fluids in tube-receivers used in concentrating solar thermal systems: properties and heat transfer coefficients. Renew. Sustain. Energy. Rev. 55, 298–315 (2016). https://doi.org/10.1016/j.rser.2015.10.059
R.I. Dunn, P.J. Hearps, M.N. Wright, Molten-salt power towers: newly commercial concentrating solar storage. Proc. IEEE. 100, 504–515 (2012). https://doi.org/10.1109/JPROC.2011.2163739
A.B. Zavoico, Solar power tower - design basis document. Tech. Rep. SAND2001-2100, 148 (2001). https://doi.org/10.2172/786629
V. Encinas-Sánchez, E. Batuecas, A. Macías-García, C. Mayo, R. Díaz, F.J. Pérez, Corrosion resistance of protective coatings against molten nitrate salts for thermal energy storage and their environmental impact in CSP technology. Solar. Energy. 176, 688–697 (2018). https://doi.org/10.1016/j.solener.2018.10.083
Tortorelli, P.F., Bishop, P.S., DiStefano, J.R. Selection of corrosion-resistant materials for use in molten nitrate salts.(1989)
A. SoleimaniDorcheh, M.C. Galetz, Slurry aluminizing: a solution for molten nitrate salt corrosion in concentrated solar power plants. Sol. Energy. Mater. Solar. Cells. 146, 8–15 (2016). https://doi.org/10.1016/j.solmat.2015.11.024
J.W. Slusser, J.B. Titcomb, M.T. Heffelfinger, B.R. Dunbobbin, Corrosion in molten nitrate-nitrite salts. Jom. 37, 24–27 (1985). https://doi.org/10.1007/BF03259692
S.H. Goods, R.W. Bradshaw, Corrosion of stainless steels and carbon steel by molten mixtures of commercial nitrate salts. J. Mater. Eng. Perform. 13, 78–87 (2004). https://doi.org/10.1361/10599490417542
a. G. Fernández, M.I. Lasanta, F.J. Pérez, Molten salt corrosion of stainless steels and low-Cr steel in CSP plants. Oxid. Met. 78, 329–348 (2012). https://doi.org/10.1007/s11085-012-9310-x
Bradshaw, R.W., Goods, S.H. Corrosion resistance of stainless steels during thermal cycling in alkali nitrate. Sandia. Rep. 1–39 (2001)
Kruizenga, A.M., Gill, D.D., Laford, M., Mcconohy, G. Corrosion of high temperature alloys in solar salt at 400 , 500 , and 680 ° C. Sandia. Rep. 1–45 (2013)
M. Gurr, S. Bau, F. Burmeister, M. Wirth, E. Piedra-Gonzalez, K. Krebser, J. Preußner, W. Pfeiffer, Investigation of the corrosion behavior of NiVAl multilayer coatings in hot salt melts. Surf. Coatings. Technol. 279, 101–111 (2015). https://doi.org/10.1016/j.surfcoat.2015.07.045
R. Moore, M. Vernon, C.K. Ho, N.P. Siegel, G.J. Kolb, S.N. Laboratories, P.O.B. Albuquerque, Design considerations for concentrating solar power tower systems employing molten salt. Security 2010, 1–51 (2010). https://doi.org/10.2172/1008140
M. Spiegel, J. Mentz, High temperature corrosion beneath nitrate melts. Mater. Corros. 65, 276–281 (2014). https://doi.org/10.1002/maco.201307076
P. Audigié, V. Encinas-Sánchez, M. Juez-Lorenzo, S. Rodríguez, M. Gutiérrez, F.J. Pérez, A. Agüero, High temperature molten salt corrosion behavior of aluminide and nickel-aluminide coatings for heat storage in concentrated solar power plants. Surf. Coatings. Technol. 349, 1148–1157 (2018). https://doi.org/10.1016/j.surfcoat.2018.05.081
H. Singh, B.S. Sidhu, D. Puri, S. Prakash, Use of plasma spray technology for deposition of high temperature oxidation/corrosion resistant coatings - a review. Mater. Corros. 58, 92–102 (2007). https://doi.org/10.1002/maco.200603985
N. Cinca, J.M. Guilemany, Thermal spraying of transition metal aluminides: an overview. Intermetallics 24, 60–72 (2012). https://doi.org/10.1016/j.intermet.2012.01.020
H. Singh, D. Puri, S. Prakash, Some studies on hot corrosion performance of plasma sprayed coatings on a Fe-based superalloy. Surf. Coatings. Technol. 192, 27–38 (2005). https://doi.org/10.1016/j.surfcoat.2004.03.030
S. Kumar, V. Selvarajan, P.V.A.V.A. Padmanabhan, K.P.P. Sreekumar, Characterization and comparison between APS coatings prepared from ball milled and plasma processed nickel-aluminium powders. Mater. Sci. Eng. A. 486, 287–294 (2008). https://doi.org/10.1016/j.msea.2007.09.003
M.C. Trent, S.H. Goods, R.W. Bradshaw, Comparison of corrosion performance of grade 316 and grade 347H stainless steels in molten nitrate salt. Conf. Proc. (2016). https://doi.org/10.1063/1.4949258
A. Kruizenga, D. Gill, Corrosion of iron stainless steels in molten nitrate salt. Energy. Proc. 49, 878–887 (2013). https://doi.org/10.1016/j.egypro.2014.03.095
Bradshaw, R.W., Goods, S.H. Corrosion of alloys and metals by molten nitrates. (2001)
S.B.B. Mishra, K. Chandra, S. Prakash, B. Venkataraman, Characterisation and erosion behaviour of a plasma sprayed Ni3Al coating on a Fe-based superalloy. Mater. Lett. 59, 3694–3698 (2005). https://doi.org/10.1016/j.matlet.2005.06.050
B.S. Sidhu, S. Prakash, Degradation behavior of Ni3Al plasma-sprayed boiler tube steels in an energy generation system. J. Mater. Eng. Perform. 14, 356–362 (2005). https://doi.org/10.1361/10599490523382
J.T. Chang, a. Davison, J.L. He, a. Matthews, Deposition of Ni–Al–Y alloy films using a hybrid arc ion plating and magnetron sputtering system. Surf. Coatings. Technol. 200, 5877–5883 (2006). https://doi.org/10.1016/j.surfcoat.2005.08.138
S.C. Mishra, a. Satapathy, M. Chaithanya, P.V. Ananthapadmanabhan, K.P. Sreekumar, Wear Characteristics of plasma sprayed nickel–aluminum composite coatings. J. Reinf. Plast. Compos. 28, 2931–2940 (2009). https://doi.org/10.1177/0731684408094067
B.S. Sidhu, S. Prakash, Evaluation of the corrosion behaviour of plasma-sprayed Ni 3 Al coatings on steel in oxidation and molten salt environments at 900 8C. Surf. Coatings Technol. 166, 89–100 (2003). https://doi.org/10.1016/S0257-8972(02)00772-7
S.B. Mishra, K. Chandra, S. Prakash, Studies on erosion-corrosion behaviour of plasma sprayed Ni3Al coating in a coal-fired thermal power plant environment at 540°C. Anti. Corros. Methods. Mater. 64, 540–549 (2017). https://doi.org/10.1108/ACMM-11-2015-1592
Jonemann, M. Advanced Thermal Storage System with Novel Molten Salt. (2013)
M. Walczak, F. Pineda, Á.G. Fernández, C. Mata-Torres, R.A. Escobar, Materials corrosion for thermal energy storage systems in concentrated solar power plants. Renew. Sustain. Energy. Rev. 86, 22–44 (2018). https://doi.org/10.1016/j.rser.2018.01.010
A. Soleimani, R.N. Durham, M.C. Galetz, Corrosion behavior of stainless and low-chromium steels and IN625 in molten nitrate salts at 600 ° C. Sol. Energy. Mater. Sol. Cells. 144, 109–116 (2016)
A.G. Fernández, H. Galleguillos, E. Fuentealba, F.J. Pérez, Corrosion of stainless steels and low-Cr steel in molten Ca(NO3)2-NaNO3-KNO3 eutectic salt for direct energy storage in CSP plants. Sol. Energy. Mater. Sol. Cells. 141, 7–13 (2015). https://doi.org/10.1016/j.solmat.2015.05.004
Kruizenga, A.M., Gill, D.D., Laford, M. Materials Corrosion of high temperature alloys immersed in 600 ° C binary nitrate salt. Sandia Rep. (2013)
B. Jm.At.La. T, Influence of alloy microstructure on oxide growth in HCM12A in supercritical water. Mater. Res. Soc. Symp. Proc. Proc. 2009, 19–24 (2009)
Y. Li, D.D. Macdonald, J. Yang, J. Qiu, S. Wang, Point defect model for the corrosion of steels in supercritical water: part I, film growth kinetics. Corros. Sci. 163, 108280 (2020). https://doi.org/10.1016/j.corsci.2019.108280
T.D. Nguyen, J. Zhang, D.J. Young, Water vapour effects on corrosion of Fe-Cr and Fe-Cr-Ni alloys containing cerium and manganese in CO2 gas at 818°C. Corros. Sci. 89, 220–235 (2014). https://doi.org/10.1016/j.corsci.2014.08.029
B. Picqué, P.O. Bouchard, P. Montmitonnet, M. Picard, Mechanical behaviour of iron oxide scale: experimental and numerical study. Wear 260, 231–242 (2006). https://doi.org/10.1016/j.wear.2005.03.037
S. Saladi, J.V. Menghani, S. Prakash, Characterization and evaluation of cyclic hot corrosion resistance of detonation-gun sprayed Ni-5Al coatings on Inconel-718. J Therm. Spray. Technol. 24, 778–788 (2015). https://doi.org/10.1007/s11666-015-0235-1
K. Mehmood, M.A. Rafiq, A. Nusair Khan, M.M. Rauf, Microstructural study of as sprayed and heat treated Ni 3 Al coatings deposited by air plasma spraying technique. IOP Conf. Ser. Mat. Sci. Eng. 2016, 146 (2016). https://doi.org/10.1088/1757-899X/146/1/012050
M. Kumar, H. Singh, N. Singh, Study of Ni-20Cr coatings for high temperature applications - a review. Arch. Metallur. Mater. 58, 523–528 (2013). https://doi.org/10.2478/amm-2013-0030
R.D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta. Cryst. 1976, 751–767 (1976). https://doi.org/10.1107/S0567739476001551
L.H. Ahrens, The use of ionization potentials part 1 Ionic radii of the elements. Geochim. Cosmochim. Acta. 2, 155–169 (1952). https://doi.org/10.1016/0016-7037(52)90004-5
Y. Takeda, J. Akagi, A. Edagawa, M. Inagaki, S. Naka, A preparation and polymorphic relations of sodium iron oxide (NaFeO2). Mater. Res. Bull. 15, 1167–1172 (1980). https://doi.org/10.1016/0025-5408(80)90081-1
G. Fernández, I. Rey, S. Lasanta, M.P. Mato, F..J.. Pérez. Brady, Corrosion of alumina-forming austenitic steel in molten nitrate salts by gravimetric analysis and impedance spectroscopy. Mater. Corros. 65, 267–275 (2014). https://doi.org/10.1002/maco.201307422
D. Kumar, K. Pandey, Optimization of the process parameters in generic thermal barrier coatings using the Taguchi method and grey relational analysis. Proc. Ins. Mech. Eng. Part. L. J. Mat. Des. Appl. 231, 600–610 (2017). https://doi.org/10.1177/1464420715602727
ICDD. Powder diffraction file -2, Int. Cent. Diffr. Data Database. (n.d.)
J.T. Guo, L.Y. Sheng, Y. Xie, Z.X. Zhang, V.E. Ovcharenko, H.Q. Ye, Microstructure and mechanical properties of Ni3Al and Ni 3Al-1B alloys fabricated by SHS/HE. Intermetallics 19, 137–142 (2011). https://doi.org/10.1016/j.intermet.2010.08.027
E. Liu, J. Jia, Y. Bai, W. Wang, Y. Gao, Study on preparation and mechanical property of nanocrystalline NiAl intermetallic. Mater. Des. 53, 596–601 (2014). https://doi.org/10.1016/j.matdes.2013.07.052
J. Swaminathan, R. Singh, M.K. Gunjan, B. Mahato, Sensitization induced stress corrosion failure of AISI 347 stainless steel fractionator furnace tubes. Eng. Fail. Anal. 18, 2211–2221 (2011). https://doi.org/10.1016/j.engfailanal.2011.07.015
Y. Behnamian, A. Mostafaei, A. Kohandehghan, B.S. Amirkhiz, D. Serate, W. Zheng, D. Guzonas, M. Chmielus, W. Chen, J.L. Luo, Characterization of oxide scales grown on alloy 310S stainless steel after long term exposure to supercritical water at 500 °C. Mater. Charact. 120, 273–284 (2016). https://doi.org/10.1016/j.matchar.2016.09.013
A.G. Fernández, H. Galleguillos, F.J. Pérez, Thermal influence in corrosion properties of Chilean solar nitrates. Sol. Energy. 109, 125–134 (2014). https://doi.org/10.1016/j.solener.2014.07.027
M. Konieczny, Mechanical properties and deformation behavior of laminated Ni-(Ni2Al3+NiAl3) and Ni-(Ni3Al+NiAl) composites. Mater. Sci. Eng. A. 586, 11–18 (2013). https://doi.org/10.1016/j.msea.2013.08.002
G.S. Cho, K.R. Lee, K.H. Choe, K.W. Lee, A, Ikenaga (2006) Fabrication of Ni-Al intermetallic compounds on the Al casting alloy by SHS process, Inst Cast Met Eng - 67th World Foundry Congr Wfc06 Cast. Future 1, 361–370 (2006)
J. Cao, X.G. Song, L.Z. Wu, J.L. Qi, J.C. Feng, Characterization of Al/Ni multilayers and their application in diffusion bonding of TiAl to TiC cermet. Thin Solid Films 520, 3528–3531 (2012). https://doi.org/10.1016/j.tsf.2012.01.001
Funding
Funding for this work came from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant number 645725 (Framework of Innovation for Engineering of New Durable Solar Surfaces — FRIENDS2) project. SY received studentship from Centre for Doctoral Training in Sustainable Materials and Manufacturing with funding from the Engineering and Physical Research Council grant number EP/L016389/1.
Author information
Authors and Affiliations
Contributions
Sarah Yasir: Methodology, investigation, visualization, writing—original draft, data curation, formal analysis.
Jose Luis Endrino: Supervision, writing — review and editing.
Elena Guillen: Conceptualization, writing — review and editing.
Adrianus Indrat Aria: Methodology, conceptualization, validation, visualization, supervision, formal analysis, writing — review and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yasir, S., Endrino, J.L., Guillén, E. et al. Suppression of molten salt corrosion by plasma sprayed Ni3Al coatings. emergent mater. 4, 1583–1593 (2021). https://doi.org/10.1007/s42247-021-00334-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42247-021-00334-y