
Abstract
Gentry’s forest transect data have been frequently used to assess global patterns of plant diversity and plant species compositional changes along environmental and geographical gradients. Based on the worldwide woody plant abundance records from Gentry’s 197 localities/plots (each consisting of ten 2 m × 50 m subplots/transects), we apply a recently developed sampling-model-based standardization method (iNEXT.beta3D standardization) to examine how beta diversity among subplots varies with latitude. Beta diversity quantifies the extent of among-subplot differentiation which represents the interacting effect of species abundance distribution and spatial aggregation. Here beta diversity is obtained by a multiplicative decomposition scheme based on the framework of Hill numbers of any order q ≥ 0. Under statistical sampling models, data in nearly all of the 197 localities were incomplete, i.e., there were species present in the assemblage but undetected in the data. For Gentry’s data collected along narrow transects, the dependence among sampled individuals due to spatial aggregation is generally weak. The observed beta diversity depends on the among-subplot differentiation and sampling effort/completeness, which in turn induce dependence of the observed beta diversity on alpha and gamma diversity. To control for sampling effort/completeness, the iNEXT.beta3D method standardizes both alpha and gamma diversity at the same level of sample coverage to formulate coverage-based beta diversity. The resulting standardized beta diversity provides a statistical solution to remove the dependence of beta diversity on both alpha and gamma diversities, and thus reflects the pure among-subplot differentiation. The coverage-based standardization reveals latitudinal beta diversity patterns/trends not only for richness-based, but also for abundance-sensitive beta diversity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Alwyn Howard Gentry (1945−1993) was a renowned American botanist and taxonomist; he developed an innovative sampling design for a quick inventory of woody species diversity in species-rich tropical forests, which was also later applied to other regions. His sampling design, conducted in a selected forest locality, involved placing ten contiguous subplots, each in the shape of a 2-m narrow and 50-m long transect, which allowed for a fast census (Gentry, 1988; Miller et al., 1996; Phillips & Miller, 2002). After Gentry’s tragic death (in a flight accident on the way to Ecuador), the well-known dataset was made available on a website (see Data Accessibility) and also published in a printed version (Phillips & Miller, 2002). As James Miller indicated in a tribute to Gentry, “… he worked tirelessly, was ambitious, conducted groundbreaking and insightful scientific studies at a tremendous rate, and was passionate in his quest for understanding and conservation of the most diverse natural areas in the world” (Miller et al., 1996).
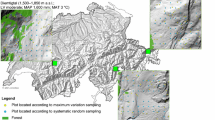
Gentry’s global forest transect data have been frequently examined in the literature to assess global patterns of plant diversity and plant species compositional changes along environmental and geographical gradients; see the next paragraph for a brief review. The original data consisted of woody plant abundance records from 225 localities/plots. Four contextual variables (latitude, longitude, elevation, precipitation) are available for each locality. Most previous authors used the data collected from 197 localities, each consisting of ten 2 m × 50 m subplots/transects; within each subplot, all woody individuals with diameter at breast height larger than 2.5 cm were determined and measured. Figure 1 shows the distribution of the 197 localities around the world.

Distribution of Gentry’s 197 localities around the world (from 40.7° S to 60.6° N and from 127.8° W to 166.8° E), each consisting of ten 2 m × 50 m subplots/transects; color indicates the magnitude of observed species richness in each locality
Based on the data from the 197 localities, the conclusions regarding the latitudinal pattern of beta diversity among the ten subplots are divergent. Kraft et al. (2011) applied an individual-based null-model approach to control for the species pool dependence of beta diversity and proposed the use of a statistic called beta deviation. They demonstrated that the previously well-documented negative trend of species-richness-based beta diversity with increasing absolute latitude disappears after species pool size is controlled for. That is, variation in species pool size is the main driver of the latitudinal differences in beta diversity. Kraft et al.’s paper has sparked intense debate (e.g., Bennett & Gilbert, 2016; Qian et al., 2012, 2013; Tuomisto & Ruokolainen, 2012; Xing & He, 2021) and stimulated new concepts and developments in the ecological literature (Chase et al., 2011, 2018; Engel et al., 2021; McGlinn et al., 2019, 2021; Ulrich et al., 2018; Xing & He, 2021 among others). Engel et al. (2021) obtained a similar conclusion as Kraft et al. (2011) based on a different method. By contrast, Qian et al. (2013) used the same null-model approach as Kraft et al. (2011) but found that standardized beta deviation is still significantly negatively related to increasing absolute latitude. Xing and He (2021) derived an analytic beta deviation formula and also showed a significant latitudinal beta diversity pattern under the assumption that the species abundance distribution follows a log-series distribution.
In this paper, we adopt a recently developed sampling-model-based approach (Chao et al., 2023) to examine how beta diversity among subplots varies with latitude using the same set of 197 localities. The approach treats the data in each locality as a representative sample from an assemblage (i.e., forest area for which the data collected in a locality can be regarded as a representative sample). Like all statistical inference models, a sampling-model-based approach makes a clear distinction between the theoretical assemblage level (unknown theoretical properties and parameters for assemblages) and the sampling data level (empirical/observed statistics computed from data). This distinction enables researchers to transparently see the estimating target at the assemblage level and pinpoint model assumptions at the data level. Below we elaborate on the two distinct levels along with statistical inferences separately for a single assemblage and for multiple assemblages.
1.1 Diversity in a single assemblage
For a single assemblage, the entire assemblage is characterized by the species abundance distribution (SAD) and how all the individuals are distributed over space (spatial aggregation or clustering). These assemblage-level properties are generally unknown to samplers. Species diversity (Hill numbers, reviewed in Sect. 2.1) at the theoretical level is formulated to purely reflect the information in the SAD. At the data level, the observed species diversity for area-based sampling data depends on three factors: the SAD, spatial aggregation, as well as sampling effort/completeness. To disentangle the effects of the three factors from any diversity measure, one possible statistical strategy to compare diversity of assemblages involves: (i) conducting an independent sampling in each assemblage, and (ii) standardizing or controlling for sampling effort/completeness across samples; see reasons below. Classic rarefaction with species richness (Gotelli & Colwell, 2011) and the iNEXT (interpolation and extrapolation) standardization with Hill numbers (Chao et al., 2014) represent two methodologies of this strategy. In the iNEXT standardization, all samples are standardized to a fixed sample size or a fixed level of sample coverage (or simply “coverage”, an objective measure of sample completeness); see below for the definition of sample coverage.
A critical assumption for classic rarefaction and the iNEXT standardization is that individuals are independently selected from the assemblage. Loosely speaking, the outcome of any sampled individual under an independent sampling, e.g., by randomly sampling mobile organisms or by sampling sedentary organisms (like plants) in small quadrats or along narrow transects, does not affect or reveal any information about the outcome of the other sampled individuals. When a sample of individuals (or other sampling units) is independently selected, statistical sampling theory implies that the assemblage-level spatial aggregation will not affect empirical data and any diversity measure at the data level; see Appendix S1 of Chao et al. (2023) for pertinent simulation results. Thus, the need to control for aggregation effects at the data level can be obviated. They further showed that the iNEXT methodology is quite robust to departures from fully independent sampling. Consequently, under independent or weakly-dependent samples, spatial aggregation on standardized diversity is almost negligible, thus only sampling effort/completeness should be standardized or controlled for.
An objective measure of sample completeness is sample coverage (or simply coverage), which is defined as the fraction of the individuals in the assemblage (including individuals of undetected species) belonging to species detected in the sample. The concept of sample coverage was originally developed by Alan Turing in his cryptographic work during WWII (Good, 1953). Under independent sampling, by controlling for sample coverage in data, the standardized diversity for rarefied and extrapolated samples purely reflects the information in the SAD, not confounded by spatial aggregation. Chao and Jost (2012) showed that rarefaction and extrapolation to a given degree of sample coverage were better able to judge the magnitude of the differences in diversity among assemblages, and ranked assemblages more efficiently, compared to traditional rarefaction and extrapolation to equal sample sizes. Therefore, the coverage-based rarefaction and extrapolation sampling curves via the iNEXT standardization can be used to meaningfully compare assemblage diversity across datasets.
Note that another assumption often made for classic rarefaction is that individuals should be randomly placed in the assemblage (i.e., the assumption of no aggregation or random placement of individuals). Under independent sampling, as explained above, this unrealistic assemblage-level assumption of random replacement is unnecessary and superfluous (Smith & Grassle, 1977; Tipper, 1979).
When individuals are not independently sampled from an assemblage (e.g., counting all individuals of plant species in relatively large quadrats), it is generally difficult at the data level to disentangle the three components (SAD, aggregation, and sampling completeness) unless additional random sampling can be carried out (Chao et al., 2023; Ulrich et al., 2017). In such dependent-sampling cases, traditional rarefaction requires the additional random-placement assumption to validate the standardization. This random-placement assumption is unrealistic because spatial aggregation exists in nearly all natural assemblages. To avoid this unrealistic assumption in the analysis, the iNEXT method for dependent data adopts a different approach which involves converting abundance to incidence data in proper sampling units; such a sampling strategy was first proposed by Shinozaki (1963), and theoretically proved by Smith et al. (1985) and by Chao et al. (2014, their Appendix A). The incidence data in sampling units are generally weakly dependent and less sensitive to aggregation. Then the iNEXT method can be applied to replicated incidence data; see Colwell et al. (2012) and Chao and Colwell (2017) for further justifications.
1.2 Beta diversity among N assemblages
When there are N assemblages, for presentation purposes, we refer to the species abundance distribution in the pooled assemblage as the regional SAD. Beta diversity quantifies the extent of among-assemblage differentiation or species compositional differentiation among assemblages. At the assemblage level, beta diversity reflects the interacting effect of the regional SAD and spatial aggregation over the N assemblages. In our study, beta diversity is based on Whittaker’s original richness-based multiplicative decomposition scheme, but it uses Hill numbers for any diversity order q ≥ 0. Species compositional differences thus refer to the change/turnover of species identities/abundances among assemblages. Richness-based (q = 0) beta diversity quantifies the extent of species identity shift, whereas abundance-based (q > 0) beta diversity also quantifies the extent of difference among assemblages in species abundance.
At the data level, as with the sampling models in the single-assemblage case (Sect. 1.1), we also require that the data represent an approximate independent sample taken from the pooled assemblage so that the spatial aggregation does not affect empirical data and statistics. Under independent sampling, statistical theory implies that the observed beta diversity depends on the among-assemblage differentiation and sampling effort/completeness, which in turn induce the dependence of beta diversity on alpha and gamma diversity (Kraft et al. 2011; Chase et al., 2011). Therefore, observed beta diversity computed from sampling data cannot be used to compare among-assemblage differentiation across datasets, and standardization of sampling effort/completeness is required. Previous authors mentioned earlier (e.g., Bennett & Gilbert, 2016; Kraft et al., 2011; Qian et al., 2013; Xing & He, 2021) mainly focused on controlling for the dependence of richness-based beta diversity on species pool size (gamma diversity of order q = 0) based on Gentry’s 197 localities data. The meaning of “dependence” or “independence” of beta diversity on its alpha/gamma component was an issue of contentious debate in an Ecology Forum (Ellison, 2010, and papers following it), and we discuss the details in Sect. 3.2.
1.3 The iNEXT.beta3D approach
Recently, Chao et al. (2023) extended the iNEXT standardization with Hill numbers for an assemblage to the iNEXT.beta3D method for beta diversity. In this extension, Chao et al. adapted the iNEXT standardization to alpha and gamma diversities. That is, alpha and gamma diversity are both assessed at a standardized level of the same sample coverage to formulate a standardized, coverage-based beta diversity. After standardizing sample coverage to control for sampling completeness, the resulting beta diversity provides a statistical solution to remove the dependence of beta diversity on both gamma and alpha values, and thus reflects pure among-assemblage differentiation. The freeware R package iNEXT.beta3D (an expanded version of iNEXT software) has been developed to facilitate all computations and graphics. For readers without R background, the online software ‘iNEXT.beta3D’ is available from https://chao.shinyapps.io/iNEXT_beta3D/ to facilitate all computation and graphics.
We show in the later text (Sect. 4.2) that under sampling models, Gentry’s sampling data in nearly all of the 197 localities were incomplete in the sense that there were species present in the assemblage but undetected in the sampled data. As indicated earlier, the observed beta diversity among subplots when there are undetected species cannot be used directly to compare species compositional differentiation across datasets. Typically, when sedentary organisms such as trees are surveyed in large quadrats, dependency among individual trees may be present due to spatial aggregation. From a statistical sampling perspective, one advantage of Gentry’s data using a narrow transect sampling design is that the sampled individuals are generally weakly dependent or nearly independent. Thus, the application of the iNEXT.beta3D standardization to Gentry’s abundance-based data is justified.
In this paper, we apply the iNEXT.beta3D standardization to Gentry’s data in 197 localities to reveal latitudinal beta diversity patterns. Because the iNEXT standardization forms the foundation of the iNEXT.Beta3D method, we briefly review the necessary background in Sect. 2. Then diversity decomposition (alpha, beta, and gamma) and the iNEXT.beta3D procedures are reviewed in Sect. 3. In Sect. 4.1, we first demonstrate the complete iNEXT.beta3D procedures for two selected localities, then in Sect. 4.2, the latitudinal beta diversity patterns of all 197 localities are examined. Our perspective on the widely used beta deviation statistics based on a null model is elaborated in the Discussion (Sect. 5). We also used simulated assemblage data to compare the performance of the beta deviation approach and the iNEXT.beta3D standardization.
2 A single assemblage: Hill numbers and the iNEXT standardization
In Sect. 2.1 we present species diversity formulas at the assemblage level for a single assemblage. Sections 2.2 reviews the iNEXT standardization at the data level.
2.1 Species diversity (Hill numbers)
Hill numbers have been increasingly used to quantify species diversity in an assemblage. Assume that there are S species in an assemblage, indexed by i = 1, 2,…, S. Let zi represent the absolute abundance (number of individuals) of species i, or other metrics of species importance. Then the abundance vector \((z_{1} ,z_{2} ,...,z_{S} )\) characterizes the SAD of the assemblage. Hill (1973) integrated species richness and species relative abundance into a continuum of diversity measures, referred to as Hill numbers, defined for a diversity order q ≥ 0, q ≠ 1, as
where \(z_{ + } = \sum\nolimits_{i = 1}^{S} {z_{i} }\) denote the total abundance in the assemblage, or assemblage size, and \(p_{i} = z_{i} /z_{ + }\) denotes the relative abundance of species i so that \(\sum\nolimits_{i = 1}^{S} {p_{i} } = 1\). The Hill number of order q is interpreted as the effective number of species.
The parameter q determines the sensitivity of the measure to the relative abundance of species. Hill numbers of orders q = 0, 1 and 2 unify three well-established indices of biodiversity: species richness (q = 0), Shannon diversity (q = 1, the exponential of Shannon entropy) and Simpson diversity (q = 2, the inverse of the one-complement of the Gini-Simpson index). MacArthur was the first to convert the two complexity measures (Shannon entropy and the Gini-Simpson index) to the concept of effective number of species, i.e., the number of equally abundant species that would be needed to yield the same value of the diversity measure (MacArthur, 1965).
2.2 The iNEXT standardization at the data level
Assume a reference sample of n individuals is independently selected from an assemblage with SAD characterized by the abundance vector \((z_{1} ,z_{2} ,...,z_{S} )\). At the data level, let \((X_{1} ,X_{2} ,...,X_{S} )\) denote the corresponding sample abundance vector in the reference sample; only species with Xi > 0 are observed. When sampling is not complete, as indicated in the Introduction (Sect. 1.1), observed species diversity under independent sampling depends on both the SAD and sampling effort/completeness, but is independent of spatial aggregation. To control for sampling effort/completeness when comparing diversity across more than one assemblage, the iNEXT method was developed (Chao et al., 2014), through interpolation (rarefaction) and extrapolation with Hill numbers, to standardize samples by sample size or sample coverage. The iNEXT software (Hsieh et al., 2016) features the following two types of rarefaction and extrapolation sampling curves along with asymptotic diversity estimates:
(1) Sample-size-based (or simply size-based) rarefaction and extrapolation curve up to double the reference sample size: For Hill numbers of q = 0, 1 and 2, assemblages are compared based on standardized diversity at the same sample sizes, which can be smaller than a particular observed sample (traditional rarefaction) or larger than an observed sample (extrapolation). The iNEXT method features such comparisons for a continuum of sample sizes by plotting the standardized diversity, for each assemblage, as a function of sample size up to 2n (twice the reference sample size).
This size-based sampling curve can be used to visually determine whether data are sufficient to provide accurate asymptotic diversity estimates (Chao et al., 2020). In most applications, the sampling curve becomes stable and levels off for Shannon diversity (q = 1) and Simpson diversity (q = 2). In these two cases, the bias of the asymptotic estimator is limited or negligible and thus the true diversity can be inferred from the sampling data. In contrast, the curve for species richness (q = 0) is typically still increasing at double the reference sample size, signifying that the asymptotic species-richness estimator represents only a lower bound.
(2) Coverage-based rarefaction and extrapolation curve up to a maximum coverage. Chao and Jost (2012) proposed comparing assemblages by plotting their diversities as a function of sample completeness, which is measured by sample coverage, i.e., the fraction of individuals in the entire assemblage that belong to detected species. This measure can be very efficiently and accurately estimated directly from sampling data. When data do not contain sufficient information to infer the diversity of an entire assemblage, i.e., when asymptotic estimators are subject to bias, we can infer and compare diversity for a standardized coverage value, i.e., a standardized fraction of the assemblage’s individuals.
The coverage-based sampling curve includes rarefaction and extrapolation (depending on if the standardized coverage level is less than or greater than the coverage of the reference sample), join smoothly at the reference sample point. The confidence intervals based on the bootstrap method also join smoothly. For the rarefaction and extrapolation curves with q = 1 and q = 2, if data are not sparse, the extrapolation can often be extended to the coverage of 100% to attain the estimated asymptote. However, the curve for species richness can be extended only up to a maximum value (i.e., the coverage value of an extrapolated sample with twice the reference sample size).
3 Multiple assemblages: diversity decomposition and the iNEXT.beta3D standardization
In this section, we briefly review the diversity decomposition (alpha, gamma and beta) at the assemblage level in Sect. 3.1, and the iNEXT.beta3D standardization at the data level in Sect. 3.2.
3.1 Gamma, alpha and beta diversity in N assemblages
When there are N assemblages, denote the species by assemblage abundance matrix by \([z_{ik} ] \ge 0\), i = 1, 2,…, S, k = 1, 2,…, N. Here zik represents species raw abundance or the true number of individuals or other abundance proxies of the i-th species in the k-th assemblage. Define the pooled assemblage as the assemblage in which the abundance of any species is obtained by directly pooling its abundances over the N assemblages. Let \(z_{i + } = \sum\nolimits_{k = 1}^{N} {z_{ik} }\) denote the total abundance of species i in the entire pooled assemblage. The SAD in the pooled assemblage (referred to as the regional SAD, as defined in Sect. 1.2) is characterized by the species abundances \(\{ z_{i + } ;\;i = 1,\;2,...,\;S\}\). Gamma diversity is computed as Hill numbers in the pooled assemblage based on the regional SAD:
where \(z_{ + + } = \sum\nolimits_{k = 1}^{N} {\sum\nolimits_{i = 1}^{S} {z_{ik} } }\) denotes the total abundance in the matrix, and \(p_{i + } = z_{i + } /z_{ + + }\) denotes the species relative abundance vector of species i in the pooled assemblage. Gamma diversity is interpreted as the effective number of species in the pooled assemblage.
For alpha diversity, Chao et al. (2023) showed that only with the Chiu et al. (2014) alpha formula can a simple and mathematically tractable rarefaction and extrapolation with alpha diversity for all q ≥ 0 be formulated; conventional alpha diversity (i.e., a weighted mean of the diversities of individual assemblages) does not work. In Chiu et al.’s (2014) approach, each individual is associated with two classifications, namely species identity and assemblage affiliation. The S × N cells of the raw abundance matrix are treated as if each cell were a “species” in the framework of Hill numbers. Each cell represents a species-assemblage combination.
We define a joint assemblage that consists of S × N species-assemblage combinations (as if they were “species” in the framework of Hill numbers). Although all combinations are arranged in a two-dimensional species × assemblage matrix, for alpha diversity, they are treated as if they form a single assemblage—the joint assemblage. For simplicity, we refer to the joint abundance distribution of species-assemblage combinations as the joint SAD in the joint assemblage, although “species” refer to cells/combinations. The joint SAD is characterized by the “species” abundances \((z_{11} ,z_{21} ,...,z_{S1} ,z_{12} ,z_{22} ,...,z_{S2} ,...,z_{SN} )\). Some zik may be 0, i.e., non-existing combinations which do not have any effect on diversity computations. The relative “species” abundance in the joint assemblage is defined as \(p_{ik} \equiv z_{ik} /z_{ + + }\). The joint diversity, a term first proposed by Gregorius (2010), is defined as the effective number of species-assemblage combinations in the joint assemblage, i.e., the Hill numbers for the joint assemblage based on the joint SAD:
Chiu et al. (2014) proposed the following alpha formula in terms of mean joint diversity:
Chiu et al.’s alpha diversity quantifies the effective number of species-assemblage combinations per assemblage. Then beta diversity is defined as the ratio of gamma diversity (based on the regional SAD) to alpha diversity (based on the joint SAD):
In other words, the among-assemblage differentiation is characterized by the regional SAD and the joint SAD. Beta diversity is interpreted as the effective number of assemblages based on the raw abundance matrix [zik]. For the special case of q = 0, richness-based beta diversity reduces to Whittaker’s (1960, 1972) original formula \(^{0} D_{\beta } = S/\overline{S}\), a ratio of species richness to the simple mean species richness of individual assemblages. The richness-based Jaccard and Sørensen dissimilarity indices are monotonic functions of beta diversity: Jaccard dissimilarity = \((1 - 1/{}^{0}D_{\beta } )/(1 - 1/N)\); Sørensen dissimilarity = \(({}^{0}D_{\beta } - 1)/(N - 1)\). In the special case of N = 2, these two dissimilarity measures reduce to the classic Jaccard and Sørensen two-assemblage dissimilarity indices.
3.2 The iNEXT.beta3D standardization at the data level
Assume that independent sampling was conducted in the pooled assemblage with the true species-by-assemblage abundance matrix \([z_{ik} ]\) as defined in Sect. 3.1. Suppose a sample abundance matrix \([X_{ik} ]\) (the reference sample) was recorded, where Xik denotes the observed raw abundance of the i-th species in the k-th assemblage, i = 1, 2,…, S, k = 1, 2,…, N.
In the iNEXT.beta3D standardization developed in Chao et al. (2023), the standardized beta diversity for any given level of sample coverage is expressed as the ratio between gamma and alpha diversity, where both alpha and gamma diversity are standardized at the same level of sample coverage.
(1) Rarefaction and extrapolation for gamma diversity in the pooled assemblage: Consider the regional SAD characterized by the true abundance vector \((z_{1 + } ,z_{2 + } ,...,z_{S + } )\) in the pooled assemblage, and apply the iNEXT standardization based on the gamma reference sample \((X_{1 + } ,X_{2 + } ,...,X_{S + } )\). Then the size- and coverage-based rarefaction and extrapolation curves, along with the estimated asymptote for gamma diversity, can be formulated.
(2) Rarefaction and extrapolation for alpha diversity in the joint assemblage: Consider the joint SAD characterized by the true abundance vector \((z_{11} ,z_{21} ,...,z_{S1} ,z_{12} ,z_{22} ,...,z_{S2} ,...,z_{SN} )\) in the joint assemblage, and apply the iNEXT standardization to this single joint assemblage based on the alpha reference sample \((X_{11} ,X_{21} ,...,X_{S1} ,X_{12} ,X_{22} ,...,X_{S2} ,...,X_{SN} )\). Then the size- and coverage-based rarefaction and extrapolation curves, along with the estimated asymptote for alpha diversity, can be formulated.
For any specified coverage value C, both alpha and gamma diversity are first assessed at the same given level of sample coverage. The resulting gamma and alpha standardized diversity estimates are denoted as \({}^{q}\hat{D}_{\gamma } (C)\) and \({}^{q}\hat{D}_{\alpha } (C)\) respectively. Let this common coverage value correspond to sample size mγ(C) in the pooled assemblage and sample size mα(C) in the joint assemblage. These two sample sizes vary with coverage value C, but for notational simplicity, we suppress the use of C in the notation of mγ(C) and mα(C) and simply use mγ ≡ mγ(C) and mα ≡ mα(C) assuming that C is the same for both sample sizes. The two sizes generally differ for a given common coverage, as will be shown in the next section. We define the standardized beta diversity at the coverage level of C > 0 as
Under independent sampling, as indicated in Sect. 1.1, the standardized gamma diversity purely reflects the information in the regional SAD, and the standardized alpha diversity purely reflects the information in the joint SAD. It follows from Eq. (4) that the standardized beta diversity then purely reflects the among-assemblage differentiation. A bootstrap method is used to approximate the uncertainties of both rarefied and extrapolated beta diversity estimates and to construct the associated confidence intervals (Chao et al., 2023).
Except for the special case in which no species are shared among assemblages, the sample sizes needed to standardize to a common coverage for gamma and alpha diversities generally differ. This difference in the two sizes implies that a standardization to a common size for gamma and alpha diversities is not a statistically legitimate procedure. Generally, size-based standardization for beta diversity fails to satisfy essential requirements for a differentiation measure (Chao et al., 2023).
In the iNEXT.beta3D method, the coverage-based rarefaction and extrapolation curve of beta diversity depicts \({}^{q}\hat{D}_{\beta } (C)\) as a function of sample coverage C > 0. As with the iNEXT standardization, for gamma and alpha diversity of orders q = 1 and q = 2, the coverage-based extrapolation generally can be reliably extended to asymptotes (i.e., coverage = 1) if data are not too sparse. Therefore, we can compare the standardized beta diversity obtained from Hill numbers of q = 1 and q = 2 across datasets at any standardized level of coverage up to 100%.
However, for richness-based (q = 0) gamma and alpha diversity, the extrapolation often can be extended only up to the coverage value for samples extrapolated to twice the reference sample size; here the sample size for both gamma and alpha reference samples is \(n = X_{ + + }\). We denote the sample coverages of the alpha and gamma observed (reference) sample as Cobs,α and Cobs,γ, respectively; also denote the two coverage values for twice the reference sample size as C2n,α and C2n,γ. The four coverage values typically satisfy either Cobs,α < C2n,α < Cobs,γ < C2n,γ or Cobs,α < Cobs,γ < C2n,α < C2n,γ.
Chao et al. (2023) suggested the sampling curve for beta diversity of q = 0 be extended up to the sample coverage value of C2n,α (i.e., the extrapolation limit for beta diversity is the same as alpha diversity) to avoid substantial bias. In other words, C2n,α represents the maximum coverage value that we can reliably infer for beta diversity of q = 0 for a given dataset. See Sect. 4 for numerical examples.
At the data level, Chao et al. (2023) showed that the expected standardized beta diversity at any fixed level of sample coverage satisfies all properties of a differentiation measure listed in Legendre and De Cáceres (2013). Among the properties, the following three essential properties assure the independence of the expected standardized beta diversity on alpha and gamma diversity (Chao & Chiu, 2016).
-
(1)
The fixed minimum criterion If all N assemblages are identical in terms of species identity and raw abundance, the expected standardized beta diversity at any fixed level of sample coverage should approach a fixed minimum value, regardless of gamma and alpha values. (In our case, the fixed minimum is one, i.e., effectively, there is only one assemblage.)
-
(2)
The fixed maximum criterion When no species are shared among N assemblages (i.e., complete species turnover), the expected standardized beta diversity at any fixed level of sample coverage should attain a fixed maximum value of N, regardless of gamma and alpha values. (In our case, the fixed maximum is N, i.e., effectively, N assemblages are needed to detect all species.)
-
(3)
The replication invariance principle: When K copies of an original species-by-assemblage (S × N) matrix are merged to obtain an expanded (KS × N) matrix, where all copies have identical abundance matrices (and thus identical beta values) but no species are shared among copies, the expected standardized beta value should remain unchanged for any value of K.
4 Application of the iNEXT.beta3D standardization to Gentry’s data
4.1 The complete iNEXT.beta3D standardization for two selected localities (Fig. 2)
We first selected two localities from Gentry’s data to illustrate how the complete iNEXT.beta3D standardization procedure can be applied to assess beta diversity among the ten subplots for each locality. The two localities are: the Madden Forest in Panama (site number 54, 9°6´N, 79°36´W, elevation 50 m a.s.l., annual precipitation 2433 mm) and the Río Jejuimi Forest in Paraguay (site number 145, 24°8´S, 55°32´W, elevation 150 m a.s.l., annual precipitation 1800 mm). Throughout this presentation, these two localities are referred to as the Madden and Jejuimi localities, respectively. Based on Gentry’s abundance data, here we aim to demonstrate of the iNEXT.beta3D procedures without considering the contextual variables. In the next subsection, the latitude will be considered for all 197 localities.

iNEXT.betaDiv standardization for two localities in Gentry’s data. a Sample-size-based rarefaction (solid curves) and extrapolation (dashed curves) with gamma diversity and alpha diversity, and b coverage-based rarefaction (solid curves) and extrapolation (dashed curves) with gamma, alpha, and beta diversity based on the woody plant abundance data of the Madden locality (blue curves) and the Jejuimi locality (orange curves) for diversity orders q = 0 (left panels), q = 1 (middle panels), and q = 2 (right panels). Here, size-based beta diversity is not a legitimate differentiation measure and thus is omitted. The two gamma reference samples are denoted by solid circles and alpha reference samples by hollow circles. The two extrapolated gamma and alpha samples with double the reference sample size (2n) are denoted, respectively, as solid triangles and hollow triangles. In (a), sample size in each locality is extrapolated up to 2n. In (b), the coverage values in the Madden locality for observed alpha, extrapolated alpha with size 2n, observed gamma, and extrapolated gamma with size 2n are, respectively: Cobs,α = 57.93%, C2n,α = 75.01%, Cobs,γ = 81.51%, and C2n,γ = 92.33%; in the Jejuimi locality, the corresponding four coverage values are: Cobs,α = 63.90%, C2n,α = 79.26%, Cobs,γ = 91.82%, and C2n,γ = 97.18%. In (b), for q = 1 and q = 2, the extrapolation for alpha, beta, and gamma can be extended to their asymptotes (i.e., coverage value of 100%). For q = 0, the extrapolation for gamma and alpha can be extended to the coverage value corresponding to size 2n; the extrapolation limit for beta diversity is the same as alpha diversity. All 95% confidence intervals (shaded areas) were obtained by a bootstrap method based on 200 replications. The comparisons of beta diversity between the two localities must be based on a standardized value of sample coverage
In the Madden locality, there were 131 species and a total of 356 individuals in 10 subplots. In the Jejuimi locality, there were 87 species and a total of 390 individuals in 10 subplots. Within each locality, our goal was to assess species compositional differentiation among the 10 subplots. Here, beta diversity attains a minimum value of one when the 10 subplots are identical in terms of species identity and raw abundance, and a maximum value of 10 when there are no shared species among the 10 subplots. Some summary statistics and numerical results are provided in Table S1 of Supplementary Information. The sample coverage values in the Madden locality for the alpha and gamma reference samples are, respectively, Cobs,α = 57.93%, and Cobs,γ = 81.51%; in the Jejuimi locality, the corresponding coverage values are: Cobs,α = 63.90%, and Cobs,γ = 91.82%. The observed beta diversity (in Table S1) cannot be used for comparing the two localities because, within each locality, gamma and alpha reference samples have different coverage values; between the two localities, the Jejuimi locality has higher gamma (and alpha) coverage values than the Madden locality. To make fair comparisons between the two localities, gamma and alpha sample coverage should be standardized not only within individual localities but also between localities. The complete iNEXT.beta3D standardization comprises the following two procedures:
-
(1)
Assessment of the size-based rarefaction and extrapolation curves for gamma and alpha diversity up to double the reference sample size (Fig. 2a)
Figure 2a shows the size-based rarefaction and extrapolation curves for gamma and alpha diversity for each locality. These curves can be used to determine whether data contain sufficient information to infer true beta diversity. For q = 0, both the gamma and alpha sampling curves for each locality do not level off, revealing that sampling data do not contain sufficient information to accurately infer true gamma and alpha diversity; fair comparisons of richness-based beta diversity thus can only be made by standardizing the sample coverage up to a limited coverage value described later.
For Shannon-diversity-based beta (q = 1) and Simpson-diversity-based beta (q = 2), both the alpha and gamma sampling curves in Fig. 2a tend to stabilize, implying that the bias of our asymptotic gamma (and alpha) estimator is limited for q = 1 and 2. The resulting asymptotic beta diversity (for complete coverage) is compared in the next procedure on coverage-based standardization. Note that in Fig. 2a the plot for size-based beta diversity (in which both gamma and alpha are assessed at the same sample size) is not shown, because the size-based approaches for beta diversity lack theoretical justification and do not lead to legitimate differentiation measures (Chao et al., 2023).
-
(B)
Assessment of coverage-based rarefaction and extrapolation curves for alpha, gamma, and beta diversity up to complete coverage (i.e., coverage = 1) for q = 1 and 2, and to a limited coverage value for q = 0 (Fig. 2b)
Figure 2b shows the coverage-based rarefaction and extrapolation curves for gamma, alpha, and beta diversity. For gamma and alpha diversities of q = 1 and q = 2, extrapolation can be extended to complete coverage. Thus, the corresponding beta diversity can be computed for any level of coverage up to 100%. For example, the asymptotic beta diversities of q = 1 and q = 2 are, respectively, 3.21 and 3.16 (in the Madden locality) and 1.70 and 1.37 (in the Jejuimi locality). These two values are lower than the corresponding observed beta values to some extent (Table S1), implying that observed beta diversity is likely to over-estimate the true parameters.
For q = 0, as shown in Fig. 2a, data are not sufficient for extrapolation to extend to asymptotes. In this case, the extrapolation for gamma and alpha can be only extended to the coverage value corresponding to double of each reference sample size. As discussed in Sect. 3.2, the rarefaction and extrapolation curve for beta diversity can be extended to the same limit as alpha diversity, which is C2n,α = 75.01% in the Madden locality and 79.26% in the Jejuimi locality. Therefore, extrapolation for beta diversity of q = 0 can be extended only up to the lower coverage value of the two extrapolated samples, i.e., only up to about 75%. At the common coverage level of 75%, the standardized beta value of q = 0 in the Madden locality is 3.22 and the corresponding beta in the Jejuimi locality is 1.63. See Table S1 for more numerical details.
In summary, for any specified coverage value up to 75% (for q = 0) and to 100% (for q = 1 and 2), Fig. 2b reveals that the two localities have approximately the same alpha diversity, whereas the gamma diversity in the Madden locality is significantly higher than that in the Jejumi locality. This leads to a higher beta diversity in the Madden locality. The difference in beta diversity is statistically significant because the confidence intervals for the two localities do not overlap. Within each locality, beta diversity stays roughly at the same level, regardless of orders q = 0, 1 and 2. This result implies that species compositional differentiation among the 10 subplots in each locality was due to changes in both species identity and species abundance distributions.
4.2 Latitudinal beta diversity pattern based on abundance data in 197 localities (Figs. 4 and 5)
We first show that under a statistical sampling model, the data of nearly all 197 localities were incomplete. Figure 3 depicts the distribution of four types of sample coverage values as defined in Sect. 3.2: Cobs,α (observed sample coverage value for the alpha reference sample), C2n,α (the coverage value for the extrapolated alpha sample with double the reference sample size) and the corresponding coverage values for gamma Cobs,γ, and C2n,γ. A clear pattern revealed by Fig. 3 is that each type of sample coverage greatly varied among the localities, and localities around the equator had substantially lower coverage values and thus more severe under-sampling than the others. Note that for gamma reference samples, nearly all of the 197 sample coverage values are less than 100%, signifying that the data in nearly all localities were incomplete. Thus, standardization of sample coverage among datasets is required to discover any sensible beta diversity patterns.

The distribution of four sample coverage values for Gentry’s 197 localities. a Cobs,α, observed sample coverage value for the alpha reference sample; b C2n,α, the coverage value for the extrapolated alpha sample with double the reference sample size; c Cobs,γ, observed sample coverage value for the gamma reference sample; and d C2n,γ, the coverage value for the extrapolated gamma sample with double the reference sample size
The same iNEXT.beta3D standardization procedures as those presented in Sect. 4.1 for two localities were also performed for each of the 197 localities. For each locality, we obtained coverage-based rarefaction and extrapolation curves for alpha, gamma, and beta diversity, just as we did for the Madden and Jejuimi localities, shown in Fig. 2b. Since it is not feasible to simultaneously compare 197 curves, we extracted standardized beta diversity for six levels of standardized coverage values (from 50 to 100% with increments of 10%); in this framework, standardized diversity corresponding to a coverage value of 100% represents the asymptotic estimate. Figure 4 shows the latitudinal patterns of beta diversity of orders q = 0, 1 and 2 for the selected six coverage values (rows 2–7), along with the observed patterns (row 1, where all localities are shown as grey solid points). Figure 5 shows the corresponding patterns with respect to absolute latitude based on the fitting of linear trends. (The fittings for quadratic and linear trends with respect to absolute latitude almost coincide. Thus, only linear trends are shown in Fig. 5.)

Latitudinal beta diversity gradient based on the iNEXT.beta3D standardization under six coverage values for Gentry’s 197 localities, each with 10 subplots/transects. For any standardized coverage level, all localities are classified into three categories: an “interpolated” locality (black solid circle; the standardized alpha diversity was computed from an interpolated sample), “short-range extrapolated” locality (red solid triangle; the standardized alpha diversity was computed from an extrapolated sample size less than twice the reference sample), or “long-range extrapolated” locality (green hollow circle; the standardized alpha diversity was computed from an extrapolated sample size greater than twice the reference sample). Row 1 shows observed beta diversity, and rows 2 to 7 show the standardized beta diversity under six coverage levels from 50 to 100% in increments of 10% for beta diversity of order q = 0 (column 1, 197 localities), q = 0 (column 2, restricted to those localities in the first two categories), q = 1 (column 3) and q = 2 (right panels). From rows 2 to 7, the number of localities restricted to the first two categories are 172, 141, 115, 82, 33, and 0. The fitted curves were made using a quadratic polynomial (the solid purple curves) and all fits are significant (P < 5%)

Latitudinal beta diversity gradient based on the iNEXT.beta3D standardization under six coverage values for Gentry’s 197 localities, each with 10 subplots/transects. See Fig. 4 for legend; the only difference here is that the x-axis is absolute latitude
Within each locality, we first computed two coverage values, Cobs,α and C2n,α, defined earlier. The performance of the estimated beta diversity within each locality depends on the specified standardized coverage value C; we thus classify any locality into one of the three categories according to the following criteria:
-
(i)
if C ≤ Cobs,α, the locality is classified as an “interpolated” locality (denoted as a black solid circle in Figs. 4 and 5), because standardized alpha diversity is obtained by rarefaction;
-
(ii)
if Cobs,α < C ≤ C2n,α, the locality is classified as a “short-range extrapolated” locality (denoted as a red solid triangle), because standardized alpha is obtained by extrapolating to a sample size less than or equal to double the reference sample size;
-
(iii)
if C2n,α < C, the locality is classified as a “long-range extrapolated” locality (denoted as a green hollow circle), because standardized alpha is obtained by extrapolating to a sample size greater than double the reference sample size.
Such classification is important for richness-based (q = 0) beta diversity, as indicated in Sect. 3.2, and will be used in assessing the latitudinal trends below. Based on Figs. 4 and 5, we offer the following conclusions and comparisons separately for two cases:
(1) Abundance-sensitive beta diversity patterns (q = 1 and 2, columns 3 and 4 in Figs. 4 and 5)
If we focus on abundant species (q = 1) and very abundant species (q = 2), beta diversity of these two orders typically can be extrapolated to asymptotes, and thus all localities can be used to assess latitudinal patterns for any standardized coverage value. Figure 4 clearly shows that standardized beta diversity exhibits a significant (at P < 0.05) quadratic pattern with respect to latitude for any coverage up to 100% (asymptote); Fig. 5 shows a significant linear decreasing pattern with respect to absolute latitude. That is, for abundance-sensitive beta diversity, Gentry’s data are sufficient to support latitudinal patterns up to complete coverage.
(2) Richness-based beta diversity patterns (q = 0, columns 1 and 2 in Figs. 4 and 5)
In the first column of Figs. 4 and 5, we fitted a quadratic curve (in Fig. 4) and a linear trend (in Fig. 5) to all localities for each standardized coverage level (rows 2 to 7). All fits were significant (at P < 0.05), revealing that richness-based beta diversity declines from the tropical regions with higher latitudes. Since the standardized beta diversity estimates for those localities classified into the third category (“long-range extrapolation”) may be subject to some bias, we conservatively restricted the quadratic fit to the localities classified into the first two categories, as shown in column 2 of Figs. 4 and 5. For complete coverage (100%), none of the localities are classified in the first two categories. For a coverage value of 90%, only 33 localities (no localities in the tropics) are classified in the first two categories. When the standardized coverage level is decreased from 80 to 50%, the number of localities classified in the first two categories are, respectively, 82 localities (for 80%), 115 localities (for 70%), 141 localities (for 60%), 172 localities (for 50%). In other words, when coverage is up to 80%, we have at least 82 localities to justify the resulting significant quadratic (in Fig. 4) and linear (in Fig. 5) latitudinal trends for beta diversity based on species richness.
In comparison with observed beta diversity (row 1 in Figs. 4 and 5), the gradient steepness (the rate of change in species diversity) in the quadratic (in Fig. 4) and linear (in Fig. 5) patterns in standardized beta diversity for all three orders q = 0, 1 and 2 is less steep and less pronounced. A plausible explanation is that the observed gradient steepness is overestimated, due to the positive bias of observed beta diversity (Chao et al., 2005), especially in the tropics. That is, the observed beta value based on incomplete data typically exceeds the “true” beta diversity that would be obtained from complete data.
In conclusion, if we focus on common/abundant (q = 1) or very abundant (q = 2) species, data based on Gentry’s 197 localities provide evidence for a latitudinal pattern of beta diversity, under any coverage value up to 100%. For species richness (q = 0), the latitudinal pattern is valid for coverage values only up to 80%, due to insufficient information for the rarest 20% fraction of individuals in some localities. The plots and fits based on the iNEXT.beta3D standardization in Figs. 4 and 5 clearly reveal that latitudinal beta diversity trends exist not only for richness-based beta diversity, but also for abundance-sensitive beta diversity. The iNEXT.beta3D standardization provides a theoretically rigorous and statistically sound approach to demonstrating the latitudinal beta diversity patterns.
5 Discussion
Latitudinal beta diversity patterns revealed by statistical sampling-model-based approach We have shown that beta diversity calculated from alpha and gamma diversity standardized to the same level of coverage creates a hump-shaped pattern along latitude, with the highest values close to the Equator; this pattern consistently appears for a wide range of coverage values. This pattern clearly contrasts with the results of Kraft et al. (2011), whose beta diversity based on beta deviation showed no latitudinal pattern (see below). Beta diversity among assemblages reflects an intersection between local and regional processes, in which the former includes environmental filtering, biotic interactions, and dispersal, while the latter includes large-scale biogeographical and evolutionary patterns (Vellend, 2016). Kraft et al. (2011) explained the lack of latitudinal beta diversity patterns in their study as a consequence of different large-scale processes in each latitudinal zone on one side (influencing the size of the species pool), and comparable local processes in each latitudinal zone on the other side. We argue that the presence of latitudinal beta diversity patterns indicate that both regional and local processes are, in some systematic manner, changing along the latitudinal gradient, although the more detailed ecological explanation of why this may be the case goes beyond the scope of our methodological study.
We used simulated assemblage data and a set of different sampling scenarios to numerically examine the performance of the iNEXT.beta3D standardization. Simulation results in Appendix S2 clearly reveal that the coverage-standardized beta diversity obeys the three essential properties required for a differentiation measure (the minimum and maximum criteria and the replication invariance principle; see Sect. 3.2), thus removing the dependence on gamma and alpha diversities. We therefore can confirm that the iNEXT.beta3D standardization is a rigorous and meaningful approach to measure beta diversity and quantify species compositional differences among assemblages.
Individual-based, null model approach to beta diversity Kraft et al. (2011) developed an individual-based randomization procedure, under a null model, to remove species pool dependence of beta diversity. They proposed the use of a statistic called beta deviation to quantify species compositional turnover among assemblages. Kraft et al. and subsequent followers focused only on the richness-based (q = 0) “beta” measure \(\beta_{p} = 1 - \overline{S}/S\) (i.e., proportional species turnover), where S denotes the species richness in the pooled assemblage (i.e., richness-based gamma) and \(\overline{S}\) denotes the simple average of the species richness of individual assemblages (i.e., richness-based alpha). Note that this measure is a monotonic transformation of our beta diversity (in Eq. 4, q = 0). Beta deviation takes values between 0 and \(1 - (1/N)\), where N denotes the number of assemblages. Normalizing the proportional “beta” to the range of [0, 1] leads to the classic richness-based Jaccard dissimilarity; see Sect. 3.1.
In Kraft et al.’s approach, beta deviation is defined as the difference between the observed “beta” and the average of expected “beta” values, divided by the standard deviation of expected values; see Appendix S2 for details. The expected “beta” values are calculated by randomly distributing all individuals in the observed pool to all assemblages, while preserving the species abundance distribution in the observed pool and the number of individuals in the data from each assemblage. Using Gentry’s 197 localities data with 10 subplots in each locality, Kraft et al. (2011) demonstrated that the empirical decreasing pattern of beta diversity along absolute latitude disappears based on empirical beta-deviation values; but see Qian et al. (2013) for a different pattern and perspective.
Here we focus our discussion on whether the empirical beta deviation can remove species pool dependence (Bennett & Gilbert, 2016; Qian et al., 2013; Ulrich et al., 2017, 2018), i.e., whether beta deviation is invariant to species replications. In Appendix S2, our simulation results show that only in the special case that all the N assemblages have nearly identical species abundance distribution can beta deviation remove species pool dependence. This special case was the simulation scenario that Kraft et al. presented (in their Fig. S4) to justify the use of beta deviation to remove species pool dependence. However, in general cases where some species are shared among assemblages and some are non-shared, our simulation results (Figs. S1 and S2) demonstrate that beta deviation is strictly monotonic with increasing species pool size, and thus fails to remove species pool dependence. In addition, when no species are shared among assemblages, beta deviation values are not a fixed maximum, violating the fixed maximum criterion (Sect. 3.2). Consequently, our simulation results prove that beta deviation cannot be used to quantify species compositional differences among assemblages.
As Kraft et al. indicated, beta deviation provides a statistic for testing the null hypothesis (random spatial distribution), i.e., whether the observed beta diversity can be obtained by random sampling from the observed pool of individuals. From a statistical perspective, the inference one can extract from this test is: if the magnitude of beta deviation exceeds the critical value, one can reject the above randomness assumption. Otherwise, data are not sufficient to support non-randomness. The critical values can be assessed from a sophisticated bootstrap resampling method, but in most applications, the quantiles of a standard normal distribution can be used; see Scenario A in Figs. S1 and S2, where nearly all beta deviation under the randomness null model takes values between − 2 and 2 (i.e., approximate lower and upper critical values based on a standard normal distribution for a significance level of 5%). From Fig. 3c of Kraft et al.’s paper, the beta deviation values for a vast majority of localities are outside the range [− 2, 2], revealing that the null hypothesis of random spatial distribution is rejected (P < 0.05). Statistical inference implies only non-random spatial distribution for most localities, i.e., the observed beta in most localities cannot be obtained from a random sampling of the observed pool of individuals. However, from our perspective, the lack of any systematic pattern for beta deviation along latitudinal gradients (as shown in Fig. 3c of Kraft et al.’s paper) does not imply that there is no latitudinal beta diversity trend, because beta deviation is not a proper measure of species compositional differentiation based on our simulation results in Appendix S2.
Engel et al.’s (2021) coverage-based standardization approach Engel et al. (2021) applied the coverage-based rarefaction and extrapolation method to analyze the same dataset of Gentry’s 197 localities. They focused only on the species-richness-based (q = 0) beta diversity. Although Engel et al. used a coverage-based approach, their standardization is different from the iNEXT.beta3D approach. Their procedures only lead to standardized richness-based beta diversity for a single targeted value of sample coverage; our procedures lead to standardized beta diversity for a range of coverage values. For example, their method compared Gentry’s localities based only at a single target coverage level of 10% (Engel et al., 2021, their Fig. 5) and found no evidence for latitudinal beta diversity patterns. Under such a low coverage value, a large amount of data in each locality were discarded, and only a few species were involved in the comparison across localities. Thus, data have lower power to detect any systematic changes.
For our method, with extrapolation to twice the reference sample size in each locality, we can infer beta diversity for a range of coverage values up to a maximum. The maximum values for the 197 localities are shown in Fig. 3 (upper right panel). From the legend of Fig. 4, we can reliably compare beta diversity for 82 localities up to coverage of 80%, 115 localities up to 70%, 141 localities up to 60%, and 172 localities up to 50%. Data then have sufficient power to reveal latitudinal beta diversity patterns, as shown in Figs. 4 and 5.
A unified framework for three dimensions of beta diversity In Gentry’s data, only species abundances and contextual variables are available. Thus, we have focused mainly on taxonomic beta diversity, and do not consider species phylogenetic relations and species trait differences. One advantage of using Hill numbers (see Sect. 2.1) is that they can be generalized to integrate the three dimensions of biodiversity (taxonomic, phylogenetic, and functional diversity) in a unified framework called attribute diversity or Hill-Chao numbers (Chao et al., 2019, 2021). The three dimensions of biodiversity are all in the same units of species/lineages/functional-groups equivalents and can be meaningfully compared. Based on sampling data, the iNEXT methodology (Sect. 2.2) and the iNEXT.beta3D standardization (Sect. 3.2) have been also extended to phylogenetic and functional versions; see Chao et al., (2021, 2023). Thus, a unified framework is also established for three-dimensional beta diversity and pertinent standardization based on sampling data.
Data availability
Gentry’s transect data used in this study are available in the book by Phillips and Miller (2002), and raw data are at http://www.mobot.org/MOBOT/Research/gentry/transect.shtml; we downloaded the 197 localities data from David Zelený’s webpage at https://www.davidzeleny.net/anadat-r/doku.php/en:data:gentry.
Change history
31 March 2024
A Correction to this paper has been published: https://doi.org/10.1007/s42081-024-00246-1
References
Bennett, J. R., & Gilbert, B. (2016). Contrasting beta diversity among regions: How do classical and multivariate approaches compare? Global Ecology and Biogeography, 25, 368–377.
Chao, A., Chazdon, R. L., Colwell, R. K., & Shen, T. J. (2005). A new statistical approach for assessing similarity of species composition with incidence and abundance data. Ecology Letters, 8, 148–159.
Chao, A., & Chiu, C.-H. (2016). Bridging the variance and diversity decomposition approaches to beta diversity via similarity and differentiation measures. Methods in Ecology and Evolution, 7, 919–928.
Chao, A., Chiu, C.-H., Villéger, S., Sun, I.-F., Thorn, S., Lin, Y.-C., Chiang, J. M., & Sherwin, W. B. (2019). An attribute-diversity approach to functional diversity, functional beta diversity, and related (dis)similarity measures. Ecological Monographs, 89, e01343.
Chao, A., & Colwell, R. K. (2017). Thirty years of progeny from Chao’s inequality: Estimating and comparing richness with incidence data and incomplete sampling. SORT (Statistics and Operations Research Transactions), 41, 3–54.
Chao, A., Gotelli, N. G., Hsieh, T. C., Sander, E. L., Ma, K. H., Colwell, R. K., & Ellison, A. M. (2014). Rarefaction and extrapolation with Hill numbers: A framework for sampling and estimation in species biodiversity studies. Ecological Monographs, 84, 45–67.
Chao, A., Henderson, P. A., Chiu, C.-H., Moyes, F., Hu, K. H., Dornelas, M., & Magurran, A. E. (2021). Measuring temporal change in alpha diversity: A framework integrating taxonomic, phylogenetic and functional diversity and the iNEXT.3D standardization. Methods in Ecology and Evolution, 12, 1926–1940.
Chao, A., & Jost, L. (2012). Coverage-based rarefaction: Standardizing samples by completeness rather than by size. Ecology, 93, 2533–2547.
Chao, A., Kubota, Y., Zelený, D., Chiu, C.-H., Li, C.-F., Kusumoto, B., Yasuhara, M., Thorn, S., Wei, C.-L., Costello, M. J., & Colwell, R. K. (2020). Quantifying sample completeness and comparing diversities among assemblages. Ecological Research, 35, 292–314.
Chao, A., Thorn, S., Chiu, C.-H., Moyes, F., Hu, K.-H., Chazdon, R. L., Wu, J., Magnago, L. F. S., Dornelas, M., Zelený, D., Colwell, R. K., & Magurran, A. E. (2023). Rarefaction and extrapolation with beta diversity under a framework of Hill numbers: the iNEXT.beta3D standardization. Ecological Monographs. https://doi.org/10.1002/ecm.1588
Chase, J. M., Kraft, N. J., Smith, K. G., Vellend, M., & Inouye, B. D. (2011). Using null models to disentangle variation in community dissimilarity from variation in α-diversity. Ecosphere, 2(2), art24.
Chase, J. M., McGill, B. J., McGlinn, D. J., May, F., Blowes, S. A., et al. (2018). Embracing scale-dependence to achieve a deeper understanding of biodiversity and its change across communities. Ecology Letters, 21, 1737–1751.
Chiu, C.-H., Jost, L., & Chao, A. (2014). Phylogenetic beta diversity, similarity, and differentiation measures based on Hill numbers. Ecological Monographs, 84, 21–44.
Colwell, R. K., Chao, A., Gotelli, N. J., Lin, S. Y., Mao, C. X., Chazdon, R. L., & Longino, J. T. (2012). Models and estimators linking individual-based and sample-based rarefaction, extrapolation, and comparison of assemblages. Journal of Plant Ecology, 5, 3–21.
Ellison, A. M. (2010). Partitioning diversity. Ecology, 91, 1962–1963.
Engel, T., Blowes, S. A., McGlinn, D. J., May, F., Gotelli, N. J., McGill, B. J., & Chase, J. M. (2021). Using coverage-based rarefaction to infer non-random species distributions. Ecosphere, 12(9), e03745.
Gentry, A. H. (1988). Changes in plant community diversity and floristic composition on environmental and geographical gradients. Annals of the Missouri Botanical Garden, 75, 1–34.
Good, I. J. (1953). The population frequencies of species and the estimation of population parameters. Biometrika, 40, 237–264.
Gotelli, N. J., & Colwell, R. K. (2011). Estimating species richness. In A. Magurran & B. McGill (Eds.), Biological diversity: Frontiers in measurement and assessment (pp. 39–54). Oxford University Press.
Gregorius, H.-R. (2010). Linking diversity and differentiation. Diversity, 2, 370–394.
Hill, M. (1973). Diversity and evenness: A unifying notation and its consequences. Ecology, 54, 427–432.
Hsieh, T. C., Ma, K. H., & Chao, A. (2016). iNEXT: An R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods in Ecology and Evolution, 7, 1451–1456.
Kraft, N. J. B., Comita, L. S., Chase, J. M., Sanders, N. J., Swenson, N. G., Crist, T. O., et al. (2011). Disentangling the drivers of β diversity along latitudinal and elevational gradients. Science, 333, 1755–1758.
Legendre, P., & De Cáceres, M. (2013). Beta diversity as the variance of community data: Dissimilarity coefficients and partitioning. Ecology Letters, 16, 951–963.
MacArthur, R. H. (1965). Patterns of species diversity. Biological Reviews, 40, 510–533.
McGlinn, D. J., Engel, T., Blowes, S. A., Gotelli, N. J., Knight, T. M., McGill, B. J., et al. (2021). A multiscale framework for disentangling the roles of evenness, density, and aggregation on diversity gradients. Ecology, 102, e03233.
McGlinn, D. J., Xiao, X., May, F., Gotelli, N. J., Engel, T., Blowes, S. A., et al. (2019). Measurement of Biodiversity (MoB): A method to separate the scale-dependent effects of species abundance distribution, density, and aggregation on diversity change. Methods in Ecology and Evolution, 10, 258–269.
Miller, J. S., Barkley, T. M., Iltis, H. H., Lewis, W. H., Forero, E., Plotkin, M., Phillips, O., Rueda, R., & Raven, P. H. (1996). Alwyn Howard Gentry, 1945–1993: A tribute. Annals of the Missouri Botanical Garden, 83, 433–460.
Phillips, O. L., & Miller, J. S. (2002). Global patterns of plant diversity: Alwyn H. Gentry’s forest transect data set. Missouri Botanical Garden Press.
Qian, H., Chen, S., Mao, L., & Ouyang, Z. (2013). Drivers of β-diversity along latitudinal gradients revisited. Global Ecology and Biogeography, 22, 659–670.
Qian, H., Wand, X., & Zhang, Y. (2012). Comment on “Disentangling the drivers of β diversity along latitudinal and elevational gradients.” Science, 335, 1573.
Shinozaki, K. (1963). Notes on the species-area curve. In: Abstracts of 10th Annual Meeting of Ecological Society of Japan.
Smith, E. P., Stewart, P. M., & Cairns, J. (1985). Similarities between rarefaction methods. Hydrobiologia, 120, 167–170.
Smith, W., & Grassle, J. F. (1977). Sampling properties of a family of diversity measures. Biometrics, 33, 283–292.
Tipper, J. C. (1979). Rarefaction and rarefiction—the use and abuse of a method in paleoecology. Paleobiology, 5, 423–434.
Tuomisto, H., & Ruokolainen, K. (2012). Comment on “Disentangling the drivers of β diversity along latitudinal and elevational gradients.” Science, 335, 1573.
Ulrich, W., Baselga, A., Kusumoto, B., Shiono, T., Tuomisto, H., & Kubota, Y. (2017). The tangled link between β-and γ-diversity: A Narcissus effect weakens statistical inferences in null model analyses of diversity patterns. Global Ecology and Biogeography, 26, 1–5.
Ulrich, W., Kubota, Y., Kusumoto, B., Baselga, A., Tuomisto, H., & Gotelli, N. J. (2018). Species richness correlates of raw and standardized co-occurrence metrics. Global Ecology and Biogeography, 27, 395–399.
Vellend, M. (2016). The theory of ecological communities. Princeton University Press.
Whittaker, R. H. (1960). Vegetation of the Siskiyou mountains, Oregon and California. Ecological Monographs, 30, 279–338.
Whittaker, R. H. (1972). Evolution and measurement of species diversity. Taxon, 12, 213–251.
Xing, D., & He, F. (2021). Analytical models for β-diversity and the power-law scaling of β-deviation. Methods in Ecology and Evolution, 12, 405–414.
Acknowledgements
We thank two anonymous reviewers for providing very helpful comments. We also thank Drs. Osamu Komori and Yasuhiro Kubota for organizing this theme issue and for inviting us to submit a paper. This work is supported by the Taiwan National Science and Technology Council under Contracts NSTC-110-2118M-007-006 and NSCT-111-2118M-007-007 (for AC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised due to a retrospective Open Access order.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chao, A., Chiu, CH., Hu, KH. et al. Revisiting Alwyn H. Gentry’s forest transect data: latitudinal beta diversity patterns are revealed using a statistical sampling-model-based approach. Jpn J Stat Data Sci 6, 861–884 (2023). https://doi.org/10.1007/s42081-023-00214-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42081-023-00214-1