
Abstract
Effecting oxidation reactions electrochemically dispenses with the need for reactive and potentially toxic reagents but barriers remain towards wide adoption of the technique, in part because of negative prior experiences with batch-mode reactions. Electrochemical flow set-ups fix the electrodes to maintain a uniformly narrow gap, and can operate continuously until a desired quantity of substrate has been processed. We describe the fabrication of an electrochemical flow cell and its application in the transformation of furfuryl alcohols into hydroxypyrones. The cell is simple to operate with inexpensive equipment under a constant current regime, flow rate being controlled by a standard laboratory syringe pump. With the addition of a trace of NaClO4 as electrolyte to provide a stable current flow, the oxidations proceed routinely with a current efficiency of around 60%.

ᅟ
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Oxidative rearrangements of furans are widely employed in the synthesis of natural products [1]. Among these, the Achmatowicz reaction converts furfuryl alcohols into hydroxypyrones en route to cyclic ethers, carbohydrates, and precursors for oxidopyrylium cycloadditions [2]. The overall process comprises the reaction of furfuryl alcohols 1 (Scheme 1) with bromine in methanol (the Clauson-Kaas reaction) [3,4,5], then acidic hydrolysis of the intermediate 2,5-dimethoxy dihydrofuran derivatives 2 during which the hydroxyl group cyclises onto the distal carbonyl group. Subsequently, other oxidants were reported to effect the overall reaction directly; for example: pyridinium chlorochromate [6], N-bromosuccinimide [7], peroxy acids [8], dimethyldioxirane [9], t-butyl hydroperoxide / vanadyl acetylacetonate [10], singlet oxygen followed by hydrolysis [11], and more recently biocatalysts [10,11,12,13,14,15].

Achmatowicz reaction of furfuryl alcohols yielding hydroxypyrones
The application of electrochemistry to generate 2,5-dimethoxy dihydrofurfuryl alcohols had been described nearly 20 years before Achmatowicz’s report [16,17,18,19,20]. The mechanism proposed [21] for the anodic oxidation of furans (A) in methanol depicted in Scheme 2 involves one-electron oxidation of the furan at the anode (→ B), trapping by a molecule of solvent (→ C), then further oxidation (→ D), and capture by a second solvent molecule (→ E); in parallel, reduction at the cathode generates hydrogen gas. Electrochemically-generated 2,5-dimethoxy-dihydrofurfuryl alcohols were later employed in Achmatowicz reactions for the synthesis of maltol and analogues [22,23,24,25,26], a process effected on multi-tonne scale by Otsuka Chemical Company. Applications of this process more widely have not been reported, perhaps a reflection of the relatively low adoption of electrochemical methods within the mainstream synthesis community. This, in turn, may be due to a perceived need for specialised equipment, and lack of familiarity with some of the less tangible experimental parameters that are often crucial to success in synthetic electrochemistry.

Anodic oxidation of furans in methanol → dimethoxydihydrofurans
Advances in reaction technology have made electrochemical synthetic methodologies more accessible, and recent high-profile reviews will help the approach to gain traction among mainstream synthetic chemists [27,28,29,30,31]. In this context, flow chemistry has emerged as a major enabling technology in organic synthesis [32], and the construction of electrochemical flow cells [33,34,35,36,37] allows the reactions to be performed in the absence of an added electrolyte by virtue of the very close proximity of the two electrodes [38,39,40]. Furthermore, reactions may be performed using simple equipment under a constant current regime, without the need for a considerably more expensive potentiostat. Synthetic electrochemical transformations have been reported in which the power source is a lantern battery [41], a D-cell battery [42], a mobile phone charger [43], or a photovoltaic cell [44, 45], among others.
Our group has worked extensively on oxidative dearomatisation reactions of furan derivatives en route to mostly spirocyclic natural products. Of most relevance is a method based on stepwise electron- and proton-transfer steps mediated by DDQ [46]. The extended alkylidene spiroacetals generated in this reaction were found to be poorly stable in the presence of the oxidant and acceptable results could only be obtained by running the reaction at −90 °C and keeping the reaction time short; the reactions were then clean but incomplete. An electrochemical flow variant of this process could, in principle, achieve complete oxidation by fine control of the flow rate / residence time. In related work [47], we described the formation of butenolide spiroacetals from 2-(4-hydroxybutyl)furan derivatives either directly, using an excess of MCPBA, or in two steps, using sequential oxidations with MCPBA and PDC, with overall yields of 60–65%. Again, an electrochemical variant of this reaction would remove the need to employ reactive chemical oxidants and then separate the spent reagent from the desired product. This paper describes a preliminary project to fabricate a suitable device, that could be deployed in any organic chemistry laboratory, and illustrate its effectiveness in a transformation of value to synthetic chemists. The aim was to develop a flow method for the anodic oxidation of furfuryl alcohol derivatives under constant current conditions, employing a commercial power supply, and then determine the synthetic potential of the approach for accessing side-chain functionalised hydroxypyrone derivatives.
Results and discussion
Cell design and construction
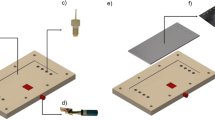
The electrochemical cell was designed using Vectorworks 2015 (Nemetschek/Techlimits), and constructed using a Roland MDX-40 desktop milling machine (Roland DG) using VCarvePro 7.5 CAM software (Vectric). The cell, depicted in Fig. 1, consists of two polyether ether ketone (PEEK) mounts. The upper mount (Fig. 1a) supports the carbon anode (Fig. 1b) in a slot akin to the one that hosts the platinum cathode (Fig. 1c) in the lower mount (Fig. 1d) [48]. This slot (Fig. 1e) features a series of grooves to facilitate adhesion of the electrodes to the PEEK using Araldite® epoxy resin glue. Once the electrodes had been fastened into the sockets they were polished flush into the mounts using diamond polishing suspensions down to 50 nm in a metallographic grinder. Both electrodes were polished to a mirror finish.

Flow cell schematic. a) PEEK mount; b) glassy carbon anode; c) platinum cathode; d) PEEK mount showing a recess e) for the electrodes; f) port for electrical connection; g) and h) the inlet ports for the reaction solution; i) gasket; j) the complete assembly
Electrical connection to the electrodes was achieved using three spring-loaded pins (Preci-dip) with a standard pitch separation of 2.54 mm, and hidden in a straight socket (Fig. 1f) to facilitate the attachment of a cable. The upper mount also includes fluidic threaded ports (Fig. 1g) to match M660 1/16″ super flangeless fittings (Upchurch Scientific). Solutions were driven in and out the cell through 1/16″ outer diameter PTFE tubing (Cole Parmer). Inside the cell, the inlet and outlet are 1 mm in diameter (Fig. 1h). Suitable gaskets (Fig. 1i) were manufactured in thin sheets (25–100 μm) from a variety of materials – including PEEK, PTFE, FEP, and Kapton – using a Roland GX-24 CAMM cutter plotter (Roland DG). For operation, the cell is held together by ten M3 screws. Detailed 2D and 3D diagrams are available as Supporting Information, and the original Vectorworks file will be provided by the authors upon request.
Cell testing and optimisation
The flow cell was tested by effecting anodic oxidation of a 20 mM solution of furfuryl alcohol 4 in methanol. A Keithley 2200-72-1 programmable power supply was used as the power source, and all runs were performed under constant current conditions. In contrast to Atobe’s report [48], initial runs in the absence of supporting electrolyte were plagued by inconsistent results, with the solution conductivity being too low for useful current densities to be reached. Decreasing the electrode distance from 100 μm to 25 μm via use of a thinner gasket provided no improvement. It was found subsequently that addition of a trace of NaClO4 to the substrate solution (ca. 0.05 mM, corresponding to 0.25% with respect to the substrate) considerably increased the conductivity and rendered all subsequent runs perfectly reproducible. Table 1 summarises the results of a brief examination of the conversions obtained with varying current densities and flow rates. Increasing the current density up to 5.8 mA cm−2 provided an improvement in conversion, while further increases made no difference. A flow rate of 165 μL min−1 was deemed ideal, balancing substrate throughput and conversion; decreasing the rate to 118 μL min−1 gave only a marginal increase in conversion.
Application to substituted furfuryl alcohols
Table 2 summarises the results of applying these parameters in the electrolysis of representative furfuryl alcohols, with side-chains bearing common simple functionality. Alkanes, alkenes, and alkynes are well tolerated; phenyl rings are compatible with the oxidation but entry 4 reveals a low yield that we attribute to a competing decomposition pathway resulting in the extrusion of benzaldehyde (Scheme 3), evident in the 1H NMR spectrum of the crude electrolysis mixture. Moving the phenyl substituent to a position where it cannot interfere mechanistically in this way largely obviated this problem (entry 5).

Loss of benzaldehyde during the oxidation of substrate 1d may occur either immediately following loss of one electron (Z = •) or after capture by methanol and loss of a second electron (Z = OMe)
In general, separate oxidations were conducted back-to-back over the course of a day without the need to disassemble the cell and clean the electrodes. To begin, the electrodes were wiped in a figure-of-eight motion with a paper towel, the cell was assembled and the various substrates were oxidised sequentially, typically in two runs of 20 min for each substrate. For cleaning between substrates, methanol was flowed through the cell via syringe. No drop in performance of the cell was noted during these daily runs, which required 2.0–2.5 h in total. At the end of a set of oxidations, the cell was disassembled, the electrodes were wiped over with methanol, and the cell assembled with a blank gasket (no channel) between the electrodes for storage. All the oxidations described in this article were performed without the need to re-polish the electrode surfaces and no passivation was observed by adhering to this regime.
Based on previous studies [47, 49], it was expected that the process could be extended to form spirocycles by siting the alcohol group further from the furan ring, as in compound 6 (→ 7, Fig. 2). Formation of the spirocycle 7 was indeed successful and an approximately equimolar mixture with dimethoxylated compound 8 was obtained from the electrolysis reaction; however, as a solution in CDCl3 (NMR sample), this product mixture converted over 6 days into just the spirocycle 7.

Spirocyclisation substrate (6) and products (7 and 8)
The current efficiency of the process is calculated to be ~60%, based on substrate conversion measured by GC analysis of the crude product solution. Loss of efficiency may result from methanol oxidation to form formaldehyde and/or formic acid; for comparison, Atobe’s flow oxidation method demonstrated an efficiency of <10% due to solvent oxidation [48].
Compounds 2a–e were converted directly into hydroxypyrones 3a–e by acidic hydrolysis of the residue obtained from the crude electrolysis mixture following evaporation of the methanol and dissolution in THF (Table 3). In most cases the yield over the two steps paralleled that for the oxidation step. An attempt was made to perform the hydrolysis and rearrangement of 2 → 3 in flow mode. When an aqueous methanol solution of the product mixture from the electrolysis of compound 1a was passed through a short column of Dowex 50 W-X8 or Amberlyst 15, a mixture of unidentified products was obtained. These results indicate that strongly acidic sulfonic acid resins may not be ideal for this transformation, and alternatives should be investigated [50].
Conclusions
In conclusion, an electrochemical flow cell has been constructed and its utility demonstrated by the anodic oxidation of furfuryl alcohols 1, with the resulting compounds affording hydroxypyrones 3 following acidic treatment. This method works well when the substrates lack features that promote fragmentation (cf. Scheme 3). Applications of the cell to oxidations more broadly are being trialled within the group.
References
Recent reviews: (a) Montagnon T, Kalaitzakis D, Triantafyllakis M, Stratakis M, Vassilikogiannakis G (2014). Furans and singlet oxygen – why there is more to come from this powerful partnership. Chem. Commun. 50:15480–15498; b Palframan MJ, Pattenden G (2014). The versatility of furfuryl alcohols and furanoxonium ions in synthesis. Chem. Commun. 50:7223–7242; c Merino P, Tejero T, Delso JI, Matute R (2007). Furan oxidations in organic synthesis: recent advances and applications. Curr. Org. Chem. 11:1076–1091
Reviews: (a) Mahajan PS, Humne VT, Mhaske SB (2017). Achmatowicz reaction: a versatile tool in bioactive natural product synthesis. Curr. Org. Chem. 21:503–545; b Deska J, Thiel D, Gianolio E (2015). The Achmatowicz rearrangement – oxidative ring expansion of furfuryl alcohols. Synthesis 47:3435–3450; Original reports: (c) Cavill GWK, Laing DG, Williams PJ (1969). A synthesis of juvenile hormone, methyl cis-10,11-epoxy-7-ethyl-3,11-dimethyltrideca-trans-2,trans-6-dienoate. Aust. J. Chem. 22:2145–2160; d Achmatowicz Jr O, Bukowski P, Szechner B, Zwierzchowska Z, Zamojski A (1971). Synthesis of methyl 2,3-dideoxy-DL-alk-2-enopyranosides from furan compounds – a general approach to the total synthesis of monosaccharides. Tetrahedron 27:1973–1996
Jones DG (1947) Substitution products of 2,5-dihydrofuran. GB patent 595041 19471125, Nov 25, 1947; (1947). Chem. Abstr. 1948:13786
Clauson-Kaas N (1947). Reactions of the furan nucleus. 2,5-Dialkoxy-2,5-dihydrofurans and 2,5-diacetoxy-2,5-dihydrofuran. Kgl. Danske Videnskab. Selskab. Mat.-fys. Medd. 24(6)
Clauson-Kaas N, Limborg F, Fakstorp J (1948) The alkoxylation of simple furans and related reactions. Acta Chem. Scand. 2:109–115
Piancatelli G, Scettri A, D’Auria M (1977) The oxidation of furan derivatives with pyridinium chlorochromate: a novel synthesis of 6-hydroxy-2H-pyran-3(6H)-ones. Tetrahedron Lett. 18:2199–2200
Georgiadis MP, Couladouros EA (1986) Products from furans. 4. Selective oxidation of 2-furfuryl alcohol derivatives, in the presence of aryl thioethers, with N-bromosuccinimide (NBS). A new procedure for the preparation of 2H-pyran-3(6H)-ones. J. Org. Chem. 51:2725–2727
Lefebvre Y (1972) Oxidation of furans - I. synthesis of 6-hydroxy-2H-pyran-3(6H)-ones. Tetrahedron Lett. 13:133–136
Adger BM, Barrett C, Brennan J, McKervey MA, Murray RW (1991) Oxidation of furans with dimethyldioxirane. J. Chem. Soc. Chem. Commun.:1553–1554
Ho T-L, Sapp SG (1983) Sharpless epoxidation of 2-furancarbinols. Mass spectra of 6-hydroxy-3(2H)pyranones. Synth. Commun. 13:207–211
Sammes PG, Street LJ (1983) Total synthesis of (±)-β-bulnesene, (±)-cryptofauronol, (±)-fauronyl acetate, and (±)-valeranone. J. Chem. Soc. Chem. Commun.:666–668
Asta C, Schmidt D, Conrad J, Förster-Fromme B, Tolasch T, Beifuss U (2013). The first enzymatic Achmatowicz reaction: selective laccase-catalyzed synthesis of 6-hydroxy-(2H)-pyran-3(6H)-ones and (2H)-pyran-2,5(6H)-diones. RSC Adv. 3:19259–19263
Thiel D, Doknić D, Deska J (2014) Enzymatic aerobic ring rearrangement of optically active furylcarbinols. Nat. Commun. 5:5278
Blume F, Sprengart P, Deska J (2018) Lipase-induced oxidative furan rearrangements. Synlett 29:1293–1296
Thiel D, Blume F, Jäger C, Deska J (2018) Chloroperoxidase-catalyzed Achmatowicz rearrangements. Eur. J. Org. Chem.:2717–2725
Clauson-Kaas N, Limborg F, Glens K (1952) Electrolytic methoxylation of furan. Acta Chem. Scand. 6:531–534
Clauson-Kaas N, Limborg F, Dietrich P (1952) Electrolytic methoxylation of some α-substituted furans. Acta Chem. Scand. 6:545–550
Clauson-Kaas N, Limborg F (1952) The alkoxylation of furans with a negative α-substituent; preparation of 2,5-dimethoxy-2,5-dihydro-2-furoic acid methyl ester and 2,5-dimethoxytetrahydro-2-furoic acid methyl ester. Acta Chem. Scand. 6:551–555
Nedenskov P, Elming N, Nielsen JT, Clauson-Kaas N (1955) New examples of electrolytic methoxylation of furans. Acta Chem. Scand. 9:17–22
Baggaley AJ, Brettle R (1968) Anodic oxidation. Part IV. Some reactions with furans. J. Chem. Soc. (C):969–974
Weinberg NL, Weinberg HR (1968) Electrochemical oxidation of organic compounds. Chem. Rev. 68:449–523
Torii S, Tanaka H, Anoda T, Simizu Y (1976) A convenient preparation of maltol, ethylmaltol, and pyromeconic acid from 2-alkyl-6-methoxy-2H-pyran-3(6H)-ones. Chem. Lett. 5:495–498
Shono T, Matsumura Y (1976) Novel syntheses of maltol and related compounds. Tetrahedron Lett. 17:1363–1364
Matsumura Y, Shirai K, Maki T, Itakura Y, Kodera Y (1998) Facile synthesis of allixin and its related compounds. Tetrahedron Lett. 39:2339–2340
Sun J-J, Hao M, Wang L, Cui L-H (2013) Research on the synthesis process of ethylmaltol by electrochemistry. Appl. Mech. Mater. 284–287:380–384
Nad S, Breinbauer R (2004) Electroorganic synthesis on the solid phase using polymer beads as supports. Angew. Chem. Int. Ed. 43:2297–2299
Möhle S, Zirbes M, Rodrigo E, Gieshoff T, Wiebe A, Waldvogel SR (2018) Modern electrochemical aspects for the synthesis of value-added organic products. Angew. Chem. Int. Ed. 57:6018–6041
Wiebe A, Gieshoff T, Möhle S, Rodrigo E, Zirbes M, Waldvogel SR (2018) Electrifying organic synthesis. Angew. Chem. Int. Ed. 57:5594–5619
Yan M, Kawamata Y, Baran PS (2018) Synthetic organic electrochemistry: calling all engineers. Angew. Chem. Int. Ed. 57:4149–4155
Yan M, Kawamata Y, Baran PS (2017) Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 117:13230–13319
Horn EJ, Rosen BR, Baran PS (2016) Synthetic organic electrochemistry: an enabling and innately sustainable method. ACS Cent. Sci. 2:302–308
Leading reviews: (a) Plutschack MB, Pieber B, Gilmore K, Seeberger PH (2017). The hitchhiker’s guide to flow chemistry. Chem. Rev. 117:11796–11893; b Porta R, Benaglia M, Puglisi A (2016). Flow chemistry: recent developments in the synthesis of pharmaceutical products. Org. Process. Res. Dev. 20:2–25; c McQuade DT, Seeberger PH (2013). Applying flow chemistry: methods, materials, and multistep synthesis. J. Org. Chem. 78:6384–6389; d Pastre JC, Browne DL, Ley SV (2013). Flow chemistry syntheses of natural products. Chem. Soc. Rev. 42:8849–8869; e Wegner J, Ceylan S, Kirschning A (2012). Flow chemistry – a key enabling technology for (multistep) organic synthesis. Adv. Synth. Catal. 354:17–57; f Wegner J, Ceylan S, Kirschning A (2011). Ten key issues in modern flow chemistry. Chem. Commun. 47:4583–4592
Watts K, Gattrell W, Wirth T (2011) A practical microreactor for electrochemistry in flow. Beilstein J. Org. Chem. 7:1108–1114
Watts K, Baker A, Wirth T (2014) Electrochemical synthesis in microreactors. J. Flow Chem. 4:2–11
Folgueiras-Amador AA, Wirth T (2017) Perspectives in flow electrochemistry. J. Flow Chem. 7:94–95
Folgueiras-Amador AA, Philipps K, Guilbaud S, Poelakker J, Wirth T (2017) An easy-to-machine electrochemical flow microreactor: efficient synthesis of isoindolinone and flow functionalization. Angew. Chem. Int. Ed. 56:15446–15450
Walsh FC, Ponce de León C (2018) Progress in electrochemical flow reactors for laboratory and pilot scale processing. Electrochim. Acta 280:121–148
Paddon CA, Pritchard GJ, Thiemann T, Marken F (2002) Paired electrosynthesis: micro-flow cell processes with and without added electrolyte. Electrochem. Commun. 4:825–831
Horcajada R, Okajima M, Suga S, J-i Y (2005) Microflow electroorganic synthesis without supporting electrolyte. Chem. Commun.:1303–1305
Amemiya F, Horii D, Fuchigami T, Atobe M (2008) Self-supported paired electrosynthesis using a microflow reactor without intentionally added electrolyte. J. Electrochem. Soc. 155:E162–E165
Frey DA, Wu N, Moeller KD (1996) Anodic electrochemistry and the use of a 6-volt lantern battery: a simple method for attempting electrochemically based synthetic transformations. Tetrahedron Lett. 37:8317–8320
Finney EE, Ogawa KA, Boydston AJ (2012) Organocatalyzed anodic oxidation of aldehydes. J. Am. Chem. Soc. 134:12374–12377
Frankowski KJ, Liu R, Milligan GL, Moeller KD, Aubé J (2015) Practical electrochemical anodic oxidation of polycyclic lactams for late stage functionalization. Angew. Chem. Int. Ed. 54:10555–10558
Nguyen BH, Redden A, Moeller KD (2014) Sunlight, electrochemistry, and sustainable oxidation reactions. Green Chem. 16:69–72
Nguyen BH, Perkins RJ, Smith JA, Moeller KD (2015) Photovoltaic-driven organic electrosynthesis and efforts toward more sustainable oxidation reactions. Beilstein J. Org. Chem. 11:280–287
Robertson J, Naud S (2008) Synthesis of spiroacetal enol ethers by oxidative activation of furan derivatives. Org. Lett. 10:5445–5448
Robertson J, Meo P, Dallimore JWP, Doyle BM, Hoarau C (2004) Stereoselective synthesis of the lituarine tricyclic spiroacetal. Org. Lett. 6:3861–3863
Horii D, Atobe M, Fuchigami T, Marken F (2005) Self-supported paired electrosynthesis of 2,5-dimethoxy-2,5-dihydrofuran using a thin layer flow cell without intentionally added supporting electrolyte. Electrochem. Commun. 7:35–39
The electrochemical Clauson-Kaas procedure was applied to 2-(3-hydroxypropyl)furans in pioneering studies on furan oxidative spirocyclisation reactions: (a) Ponomarev AA, Markushina IA (1959). Synthesis of 1,6-dioxaspiro[4.4]-3-nonene derivatives in electrolytic alkoxylation of γ-furylalkanols. Dokl. Akad. Nauk SSSR 126:99–102; b Ponomarev AA, Markushina IA (1961). Furan compounds. XIII. Formation of derivatives of 1,6-dioxaspiro[4.4]-3-nonene in electrolytic methoxylation of γ-furylalkanols. Zh. Obshch. Khim. 31:554–60
An aqueous suspension of Dowex 50 has been used to effect analogous transformations [13b]
Acknowledgements
The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007–2013) under REA grant agreement no 316955.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Syntrivanis, LD., Javier del Campo, F. & Robertson, J. An electrochemical flow cell for the convenient oxidation of Furfuryl alcohols. J Flow Chem 8, 123–128 (2018). https://doi.org/10.1007/s41981-018-0016-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41981-018-0016-3